Figure 1.1 Classification of solids.
1.2 Crystalline solids: structure and properties
1.2.1 Crystal structure
Crystals contain highly ordered arrays of molecules and atoms held together by non-covalent interactions. We can consider as a simple example the unit cell of an inorganic salt, sodium chloride. Figure 1.2 shows the ordered arrangement of Cl− ions and Na+ ions that make up the sodium chloride crystal structure. We can draw a square on one side connecting the chloride ions. Similar squares could be drawn on all the sides to form a cubic repeating unit, which we call the unit cell. Within a specific crystal, each unit cell is the same size and contains the same number of molecules or ions arranged in the same way. It is usually most convenient to think of the atoms or molecules as points and the crystal as a three-dimensional array of these points, or a crystal lattice.
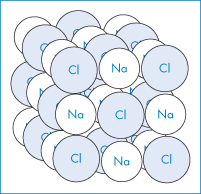
Figure 1.2 Space lattice of the sodium chloride crystal. Each sodium ion is octahedrally surrounded by six chloride ions and each chloride ion is octahedrally surrounded by six sodium ions.
For all possible crystals there are seven basic or primitive unit cells, which are shown in Fig. 1.3. We will represent the lengths of the sides as a, b and c and the angles as
α (between sides b and c)
β (between sides a and c)
γ (between sides a and b)
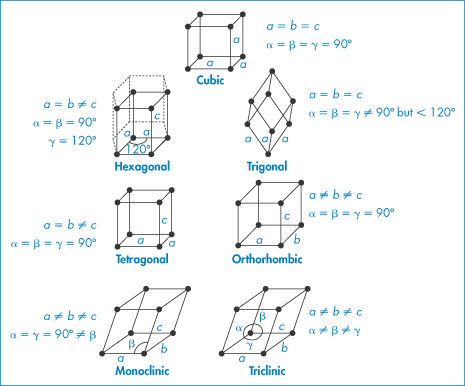
Figure 1.3 The seven possible primitive unit cells with atoms or molecules only at each corner of the unit cell. Drug molecules will typically form triclinic, monoclinic and orthorhombic unit cells.
Figure 1.3 shows the characteristic side lengths and angles for these ‘primitive’ unit cells. The structures in Fig. 1.3 have atoms or molecules only at each corner of the unit cell. It is possible to find unit cells with atoms or molecules also at the centre of the top or bottom faces (end-centred), at the centre of every face (face-centred) or with a single atom in the centre of the cell (body-centred), as in Fig. 1.4.
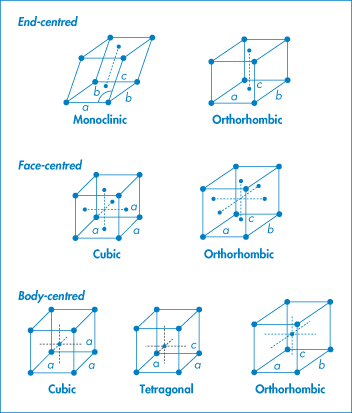
Figure 1.4 Variations on primitive cells.
Note that these variations do not occur with every type of unit cell: we find
- end-centred monoclinic and orthorhombic
- face-centred cubic and orthorhombic
- body-centred, cubic, tetragonal and orthorhombic.
Altogether there are 14 possible types of unit cell and we call these the Bravais lattices. For drugs there are three common types of unit cell: triclinic, monoclinic and orthorhombic.
|
|

Miller indices
We can identify the various planes of a crystal using the system of Miller indices. To understand how this system is used, let us consider the plane drawn through the cubic crystal shown in Fig. 1.5a. The plane cuts the a axis at one unit length and also the c axis at one unit length. It does not, however, cut the b axis, and hence the intercept to this axis is infinity. One way we could label planes is to denote each set by the distances along the axes to the point where the plane crosses the axis. So, for example, the planes marked in Fig. 1.5a would have intercept lengths of a = 1, b = ∞, c = 1. This system of labelling the faces is inconvenient because of the appearance of ∞. A way around this problem is to take the reciprocals of the numbers (since the reciprocal of ∞ = 0). The plane shown then becomes 1/1, 1/∞, 1/1 for the a, b and c axes, i.e. 1, 0, 1. The Miller indices for this plane are then written as (101).
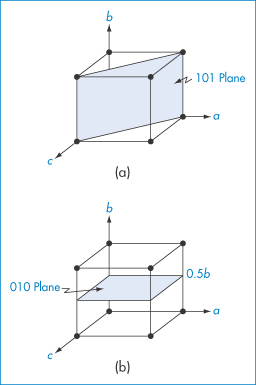
Figure 1.5 Cubic crystal showing planes with Miller indices of (a) (101) and (b) (010).
A second example is illustrated in Fig. 1.5b. This plane does not cut the a axis: it cuts the b axis at a unit cell length of ½, and does not cut the c axis. The intercept lengths are therefore a = ∞, b = 1/2, c = ∞, which on taking reciprocals become 0, 2, 0. A second rule of Miller indices is now applied, that is, to reduce the numbers to the lowest terms – in this case by dividing them all by 2. The Miller indices for this plane are therefore (010).
Other rules for applying Miller indices are shown by the following examples, which for ease of illustration are shown using a two-dimensional array (the c axis can be imagined to be at right angles to the page). None of the sets of planes we will consider crosses the c axis, i.e. we consider them to intersect it at ∞. The plane X in Fig. 1.6 has a, b and c intercepts of 3, 2 and ∞, giving reciprocals of ⅓, ½ and 0. The procedure is now to clear the fractions, in this case by multiplying each term by 6, giving 2, 3 and 0. It is not possible to reduce these further, and the Miller indices are therefore (230). The plane Y in Fig. 1.6 shows an example of a negative intercept where the a axis is crossed. The reciprocals of the a, b and c intercepts are −1, 1 and 0. The procedure that is now used is to write the negative number using a bar above it, giving Miller indices for this plane of (1̄10). We should notice that the smaller the number in the Miller index for a particular axis, the more parallel is the plane to that axis – a zero value indicates a plane exactly parallel to that axis. The larger a Miller index, the more nearly perpendicular a plane is to that axis.
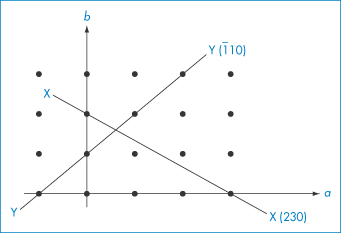
Figure 1.6 Planes in a two-dimensional array.
|
The general rules for expressing planes using the system of Miller indices:
|

|
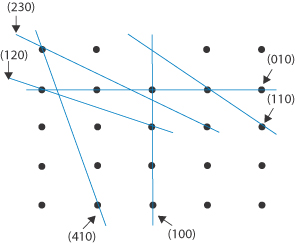
Draw a two-dimensional lattice array and indicate the planes with the following Miller indices: (i) (100); (ii) (010); (iii) (110); (iv) (120); (v) (230); and (vi) (410). Answer
|

1.2.2 Crystal form
The solid state is important for a variety of reasons, summarised in Fig. 1.7: morphology, particle size, polymorphism, solvation or hydration can affect filtration, flow, tableting, dissolution and bioavailability. These are described below. In addition, crystallisation can sometimes occur in vivo, often, as in the case of gout and urinary stones (see below), with painful consequences.
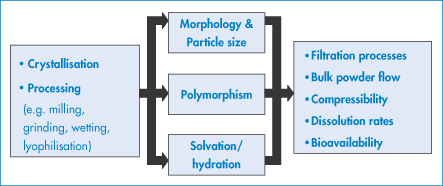
Figure 1.7 The solid state in pharmaceutical science: potential causes and effects of structural change (after AJ Florence).
The crystals of a given substance may vary in size, the relative development of the given faces and the number and kind of the faces (or forms) present; that is, they may have different crystal habits. The habit describes the overall shape of the crystal in rather general terms and includes, for example, acicular (needle-like), prismatic, pyramidal, tabular, equant, columnar and lamellar types. Figure 1.8 shows the crystal habits of a hexagonal crystal.
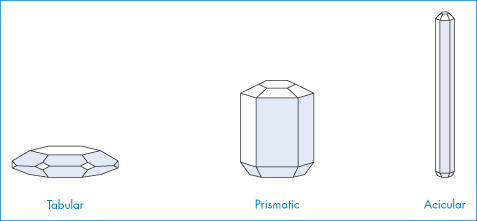
Figure 1.8 Crystal habits of a hexagonal crystal.
Although there may not be significant differences in the bioavailability of drugs with different crystal habits, the crystal habit is of importance from a technological point of view. The ability to inject a suspension containing a drug in crystal form will be influenced by the habit: plate-like crystals are easier to inject through a fine needle than are needle-like crystals. The crystal habit can also influence the ease of compression of a powder and the flow properties of the drug in the solid state. The plate-like crystals of tolbutamide, for example, cause powder bridging in the hopper of the tablet machine and also capping problems during tableting. Neither of these problems occurs with tolbutamide in other crystal habits. The habits acquired depend on the conditions of crystallisation, such as the solvent used, the temperature, the concentration and presence of impurities. Ibuprofen crystallises from hexane as elongated needle-like crystals, which have been found to have poor flow properties; crystallisation from methanol produces equidimensional crystals with better flow properties and compaction characteristics, making them more suitable for tableting. The crystal morphology of the excipients (such as powdered cellulose) included in tablet formulations can also have a significant influence on the strength and disintegration time of tablets.
Crystallisation and factors affecting crystal form2
Crystallisation from solution can be considered to be the result of three successive processes:
1. supersaturation of the solution
2. formation of crystal nuclei
3. crystal growth round the nuclei.
|
Gout usually manifests itself as a sudden excruciating pain in the big toe (usually of men), although other joints such as the ankle, heel, instep, knee, wrist, elbow, fingers or spine may be affected. It is a consequence of the precipitation of needle-like crystals of uric acid, in the form of monosodium urate, on the articular cartilage of joints when the levels of uric acid in blood serum exceed a critical solubility level (approximately 6.7 mg dL–1); the crystals inside the joint cause intense pain whenever the affected area is moved. Uric acid is a normal component of blood serum and is a product of the metabolism of purines, which are generated by the body via breakdown of cells in normal cellular turnover, and also are ingested as part of a normal diet in foods such as liver, sardines, anchovies and dried peas and beans. The uric acid is normally filtered out of the blood by the kidneys and excreted in the urine. Sometimes, however, too much uric acid is produced by the body or the kidneys are not sufficiently efficient at removing it and it accumulates in the blood, a condition known as hyperuricaemia. Precipitation of uric acid is also markedly enhanced when the blood pH is low (acidosis), a consequence of reduced solubility under such conditions. Patients with long-standing hyperuricaemia can have uric acid crystal deposits called tophi in other tissues, such as the helix of the ear. Urinary stone formation (urolithiasis) is a common disorder affecting 1–5% of the population in industrialised countries. Although it is a consequence of complex physicochemical processes, the major driving force behind the formation of many stones is the state of saturation of urine with crystal-forming substances such as oxalic or uric acid. For example, urinary uric acid solubility is limited to 9.6 mg dL–1. Since uric acid excretion in humans generally exceeds 600 mg day–1, this poses a great risk for its precipitation. Uric acid is a weak acid (pKa of 5.35 at 37°C) with a solubility of 158 mg dL–1 at pH 7.0. Reducing the urinary pH to 5.0 decreases the solubility of uric acid to less than 8.0 mg dL–1, leading to supersaturation of the urine and precipitation of sparingly soluble uric acid in the kidney or bladder in the form of stones. |

Supersaturation can be achieved by cooling, by evaporation, by the addition of a precipitant or by a chemical reaction that changes the nature of the solute. Supersaturation itself is insufficient to cause crystals to form; the crystal embryos must form by collision of molecules of solute in the solution, or sometimes by the addition of seed crystals, or dust particles, or even particles from container walls. Deliberate seeding is often carried out in industrial processes; seed crystals do not necessarily have to be of the substance concerned but may be isomorphous substances (i.e. of the same morphology). As soon as stable nuclei are formed, they begin to grow into visible crystals.
Crystal growth can be considered to be a reverse dissolution process and the diffusion theories of Noyes and Whitney, and of Nernst, consider that matter is deposited continuously on a crystal face at a rate proportional to the difference of concentration between the surface and the bulk solution. Thus an equation for crystallisation can be proposed in the form:

where m is the mass of solid deposited in time t, A is the surface area of the crystal, cs is the solute concentration at saturation and css is the solute concentration at supersaturation. As km = D/δ (D being the diffusion coefficient of the solute and δ the diffusion layer thickness; see Fig. 1.19), the degree of agitation of the system, which affects δ, also influences crystal growth. Crystals generally dissolve faster than they grow, so growth is not simply the reverse of dissolution. It has been suggested that there are two steps involved in growth in addition to those mentioned earlier, namely, transport of the molecules to the surface and their arrangement in an ordered fashion in the lattice. Equation (1.1) turns out to be better written in a modified form:

kg being the overall crystal growth coefficient and n the ‘order’ of the crystal growth process. For more details, reference 2 should be consulted.
Precipitation
Precipitation may be induced by altering the pH of the solution so that the saturation solubility is exceeded. Precipitation may be made to occur from a homogeneous solution by slowly generating the precipitating agent by means of a chemical reaction, a process likely to occur, for example, in intravenous infusion fluids and liquid pharmaceuticals. Precipitation by direct mixing of two reacting solutions sometimes does not bring about immediate nucleation and, as a result, the mixing stage may be followed by an appreciable lag time. The rate of precipitation is an important factor in determining habit, as might be imagined with a dynamic process such as crystallisation, involving nucleation and subsequent crystal growth. The form of phenylsalicylate, for example, depends on rate of crystal growth. Transition to an acicular shape occurs when the rate of growth increases. At low rates of growth, crystals of a more regular shape are obtained. In studies of the effect of solvents on habit it is generally found that less viscous media favour the growth of coarse and more equidimensional crystal forms.
Habit modification
Crystal habit can be modified by adding impurities or ‘poisons’; for example, sulfonic acid dyes alter the crystal habit of ammonium, sodium and potassium nitrates.
Surfactants in the solvent medium used for crystal growth (or, for example, in stabilisation or wetting of suspensions) can alter crystal form by adsorbing on to growing faces during crystal growth. This is best illustrated by the effect of anionic and cationic surfactants on the habit of adipic acid crystals.3 X-ray analysis showed that the linear six-carbon dicarboxylic acid molecules were aligned end to end in a parallel array in the crystal, with their long axis parallel to the (010) faces, so that the (001) face is made up entirely of −COOH groups while the (010) and (110) faces contain both −COOH and hydrocarbon (HC) portions of the molecule (Fig. 1.9). The cationic surfactant trimethyldodecylammonium chloride is twice as effective in hindering the growth of the (001) face as that of the (110) and (010) faces. In high concentrations it causes the formation of very thin plates or flakes. Conversely, the anionic surfactant sodium dodecylbenzene sulfonate at 55 parts per million (ppm) is three times as effective in reducing the growth rates of the (110) and (010) faces as of the (001) face. Higher levels of sodium dodecylbenzene sulfonate cause extreme habit modification, producing not hexagonal plates but long, thin rods or needles. The crystallographic faces whose growth rates are depressed most are those upon which surfactant adsorption is the greatest. Cationic additives adsorb on the face composed of carboxylic groups (001), and anionic additives on the (110) and (010) faces, which are hydrophobic. A coulombic interaction of the cationic head groups and the −COO− groups on the (001) faces has been suggested. The adsorption of the anionic surfactant, repelled from the anionic (001) faces, takes place amphipathically on the hydrophobic (110) faces and (010) faces (Fig. 1.9).
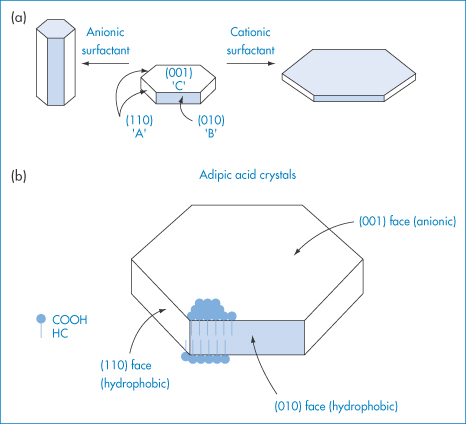
Figure 1.9 (a) Effect of anionic and cationic surfactants on the habit of adipic acid crystals. (b) A diagrammatic (not to scale) representation of the arrangement of molecules at the crystal surface.
|
|

1.2.3 Polymorphism4
As we have seen, compounds can crystallise out of solution in a variety of different habits depending on the conditions of crystallisation. These crystal habits usually have the same internal structure and so have the same X-ray diffraction patterns. A more fundamental difference in properties may be found when the compounds crystallise as different polymorphs. When polymorphism occurs, the molecules arrange themselves in two or more different ways in the crystal: either they may be packed differently in the crystal lattice or there may be differences in the orientation or conformation of the molecules at the lattice sites. These variations cause differences in the X-ray diffraction patterns of the polymorphs and this technique is one of the main methods of detecting the existence of polymorphs. The polymorphs have different physical and chemical properties; for example, they may have different melting points and solubilities and they also usually exist in different habits.
We will consider two drugs that exhibit this phenomenon. Spironolactone (I), which is a diuretic steroidal aldosterone agonist, crystallises as two polymorphic forms and also as four solvated crystalline forms depending on the solvents and methods used for crystallisation.5 We will consider the occurrence of solvated forms in section 1.2.4; at the moment we will concentrate on the two polymorphs only. Form 1 is produced when spironolactone powder is dissolved in acetone at a temperature very close to the boiling point and the solution is then cooled within a few hours down to 0°C. Form 2 is produced when the powder is dissolved in acetone, dioxane or chloroform at room temperature and the solvent is allowed to evaporate spontaneously over a period of several weeks. In both polymorphs the steroid nuclei (A, B, C and D rings) are almost planar and perpendicular to the E ring and to the 7α-acetothio side-chain. The packing of the molecules in the two polymorphs is compared in Fig. 1.10. Both unit cells are orthorhombic but they differ in their dimensions. The a, b and c axes of Form 1 were found to be 0.998, 3.557 and 0.623 nm, respectively, compared with equivalent lengths for Form 2 of 1.058, 1.900 and 1.101 nm. There are also differences in the crystal habits: Form 1 crystals are needle-like, while those of Form 2 are prisms (Fig. 1.11). The melting points are slightly different: Form 1 melts at 205°C whereas Form 2 has a melting point of 210°C.
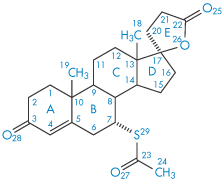
Structure I Spironolactone
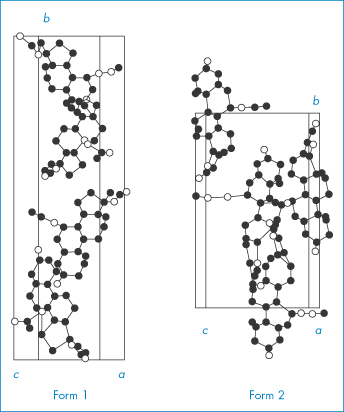
Figure 1.10 Unit cells of spironolactone.
Reproduced from Agafonov V et al. Polymorphism of spironolactone. J Pharm Sci 1991;80:181–185. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
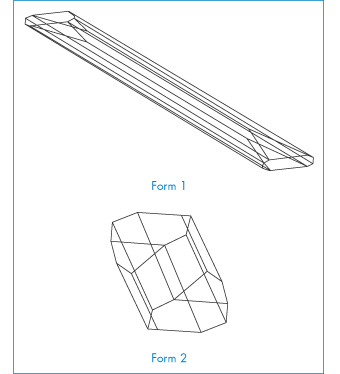
Figure 1.11 Crystal forms of spironolactone.
Reproduced from Agafonov V et al. Polymorphism of spironolactone. J Pharm Sci 1991;80:181–185. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission..
Our second example of a drug exhibiting polymorphism is paracetamol (II). This drug is known to exist in two polymorphic forms, monoclinic (Form 1) and orthorhombic (Form 2), of which Form 1 is the more thermodynamically stable at room temperature and is the commercially used form.6 However, this form is not suitable for direct compression into tablets and has to be mixed with binding agents before tableting, a procedure that is both costly and time consuming. In contrast, Form 2 can readily undergo plastic deformation upon compaction and it has been suggested that this form may have distinct processing advantages over the monoclinic form. Monoclinic paracetamol is readily produced by crystallisation from aqueous solution and many other solvents; production of the orthorhombic form has proved more difficult but may be achieved, at least on a laboratory scale, by nucleating a supersaturated solution of paracetamol with seeds of Form 2 (from melt-crystallised paracetamol). Figure 1.12 shows scanning electron micrographs of the two polymorphic forms when crystallised from industrial methylated spirit. Form 1 is described as having a prismatic to plate-like habit that is elongated in the direction of the c-axis, while Form 2 crystallises as prisms that are elongated along the c-axis.
Structure II Paracetamol

Figure 1.12 Scanning electron micrographs showing the crystal habit of (a) Form 1 and (b) Form 2 of paracetamol grown from supersaturated industrial methylated spirit. Note different scales.
Reproduced from Nichols G, Frampton CS. Physicochemical characterization of the orthorhombic polymorph of paracetamol crystallized from solution. J Pharm Sci 1998;87:684–693. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Polymorphism is common with pharmaceutical compounds. Although we do not yet understand the process sufficiently well to predict which drugs are likely to exhibit this phenomenon, it is clear that certain classes of drug are particularly susceptible. Eight crystal modifications of phenobarbital have been isolated, but 11 have been identified, with melting points ranging from 112 to 176°C. Of the barbiturates used medicinally, about 70% exhibit polymorphism. The steroids frequently possess polymorphic modifications, testosterone having four: these are cases of true polymorphism and not pseudopolymorphism, in which solvent is the cause (see section 1.2.4). Of the commercial sulfonamides, about 65% are found to exist in several polymorphic forms. Examples of the differing solubilities and melting points of polymorphic sulfonamides and steroids are given in Table 1.1.
Table 1.1 Melting points of some polymorphic forms of steroids, sulfonamides and riboflavin
Compound | Form and/or melting point (°C) | |||
Polymorphic steroids | (I) | (II) | (III) | (IV) |
Corticosterone | 180–186 | 175–179 | 163–168 | 155–160 |
β-Estradiol | 178 | 169 |
|
|
Estradiol | 225 | 223 |
|
|
Testosterone | 155 | 148 | 144 | 143 |
Methylprednisolone | I (205, aqueous solubility 0.075 mg cm−3) II (230, aqueous solubility 0.16 mg cm−3) | |||
Polymorphic sulfonamides | ||||
Sulfafurazole | 190–195 | 131–133 |
|
|
Acetazolamide | 258–260 | 248–250 |
|
|
Tolbutamide | 127 | 117 | 106 |
|
Others |
|
|
|
|
Riboflavin | I (291, aqueous solubility 60 mg cm−3) II (278, aqueous solubility 80 mg cm−3) III (183, aqueous solubility 1200 mg cm−3) | |||
Reproduced with permission from Kuhnert-Brandstatter M. Thermomicroscopy in the Analysis of Pharmaceuticals. New York: Pergamon Press, 1971.
Predictability of the phenomenon is difficult except by reference to past experience. Its pharmaceutical importance depends very much on the stability and solubility of the forms concerned. It is difficult, therefore, to generalise, except to say that where polymorphs of insoluble compounds occur there are likely to be biopharmaceutical implications. Table 1.2 is a partial listing of the drugs for which polymorphic and pseudopolymorphic states have been identified or for which an amorphous state has been reported.
Table 1.2 Polymorphic and pseudopolymorphic drugs
Compound | Number of forms | ||
Polymorphs | Amorphous | Pseudopolymorphs | |
Ampicillin | 1 | − | 1 |
Beclometasone dipropionate | − | − | 2 |
Betamethasone | 1 | 1 | − |
Betamethasone 21-acetate | 1 | 1 | − |
Betamethasone 17-valerate | 1 | 1 | − |
Caffeine | 1 | − | 1 |
Cefaloridine | 4 | − | 2 |
Chloramphenicol palmitate | 3 | 1 | − |
Chlordiazepoxide HCl | 2 | − | 1 |
Chlorthalidone | 2 | − | − |
Dehydropregnenolone | 1 | − | 7 |
Dexamethasone acetate | 3 | − | 1 |
Dexamethasone pivalate | 4 | − | 7 |
Digoxin | − | 1 | − |
Erythromycin | 2 | − | − |
Fludrocortisone acetate | 3 | 1 | − |
Fluprednisolone | 3 | − | 2 |
Glutethimide | 1 | − | 1 |
Hydrocortisone TBA | 1 | − | 3 |
Indometacin | 3 |
|
|
Mefenamic acid | 2 | − | − |
Meprobamate | 2 | − | − |
Methyl p-hydroxybenzoate | 6 | − | − |
Methylprednisolone | 2 | − | − |
Novobiocin | 1 | 1 | − |
Prednisolone | 2 | − | − |
Prednisolone TBA | 2 | − | 2 |
Prednisolone TMA | 3 | − | − |
Prednisolone acetate | 2 | − | − |
Prednisone | 1 | − | 1 |
Progesterone | 2 | − | − |
Sorbitol | 3 | − | − |
Testosterone | 4 | − | − |
Theophylline | 1 | − | 1 |
Triamcinolone | 2 | − | − |
Modified with permission from Bouché R, Draguet-Brughmans M. [Recent developments of thermal analysis.] J Pharm Belg 1977;32:347, with additions.
HCl, hydrochloride; TBA, tertiary butyl acetate (tebutate); TMA, trimethyl acetate.
Pharmaceutical implications of polymorphism
We have already considered the problems in tableting and injection that may result from differences in crystal habit (see section 1.2.2). Since polymorphs frequently have different habits, they too will be subject to these same problems. However, polymorphs also have different crystal lattices and consequently their energy contents may be sufficiently different to influence their stability and biopharmaceutical behaviour.
As the different polymorphs arise through different arrangement of the molecules or ions in the lattice, they will have different interaction energies in the solid state. Under a given set of conditions the polymorphic form with the lowest free energy will be the most stable, and other polymorphs will tend to transform into it. The rate of conversion is variable and is determined by the magnitude of the energy barrier between the two polymorphs – the higher the energy barrier and the lower the storage temperature, the slower is the conversion rate. Occasionally, the most stable polymorph appears only several years after the compound was first marketed. We can determine which of two polymorphs is the more stable by a simple experiment in which the polymorphs are placed in a drop of saturated solution under the microscope. The crystals of the less stable form will dissolve and those of the more stable form will grow until only this form remains. Figure 1.13 shows this process occurring with the two polymorphs of paracetamol, discussed earlier. Figure 1.13a shows the presence of both forms of paracetamol at room temperature in saturated benzyl alcohol. Over a time interval of 30 minutes the less stable of the two forms, the orthorhombic Form 2, has completely converted to the more stable monoclinic Form 1 (Fig. 1.13b). For drugs with more than two polymorphs we need to carry out this experiment on successive pairs of the polymorphs of the drug until we eventually arrive at their rank order of stability.

Figure 1.13 Photomicrographs showing the solution-phase polymorphic conversion of orthorhombic paracetamol (needles) to monoclinic paracetamol (prisms and plates). Micrograph (a) was taken at t = 0 and (b) was taken at t = 30 minutes. Scale bars = 250 μm.
Reproduced from Nichols G, Frampton CS. Physicochemical characterization of the orthorhombic polymorph of paracetamol crystallized from solution. J Pharm Sci 1998;87:684–693. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Transformations
The transformation between polymorphic forms can lead to formulation problems. Phase transformations can cause changes in crystal size in suspensions and their eventual caking. Crystal growth in creams as a result of phase transformation can cause the cream to become gritty. Similarly, changes in polymorphic forms of vehicles, such as theobroma oil used to make suppositories, could cause products with different and unacceptable melting characteristics.
Analytical issues
For analytical work it is sometimes necessary to establish conditions whereby different forms of a substance, where they exist, might be converted to a single form to eliminate differences in the solid-state infrared spectra that result from the different internal structures of the crystal forms. As different crystal forms arise through different arrangements of the molecules or ions in a three-dimensional array, this implies different interaction energies in the solid state. Hence, one would expect different melting points and different solubilities (and of course different infrared spectra). Changes in infrared spectra of steroids due to grinding with potassium bromide have been reported; changes in the spectra of some substances have been ascribed to conversion of a crystalline form into an amorphous form (as in the case of digoxin), or into a second crystal form. Changes in crystal form can also be induced by solvent extraction methods used for isolation of drugs from formulations prior to examination by infrared spectroscopy. Difficulties in identification arise when samples that are thought to be the same substance give different spectra in the solid state; this can happen, for example, with cortisone acetate, which exists in at least seven forms, or dexamethasone acetate, which exists in four. Therefore, where there is a likelihood of polymorphism it is best where possible to record solution spectra if chemical identification only is required. The normal way to overcome the effects of polymorphism is to convert both samples into the same form by recrystallisation from the same solvent, although obviously this technique should not be used to hide the presence of polymorphs.
Consequences
The most important consequence of polymorphism is the possible difference in the bioavailability of different polymorphic forms of a drug, particularly when the drug is poorly soluble. The rate of absorption of such a drug is often dependent upon its rate of dissolution. The most stable polymorph usually has the lowest solubility and slowest dissolution rate and consequently often a lower bioavailability than the metastable polymorph. Fortunately, the difference in bioavailability of different polymorphic forms of a drug is usually insignificant. It has been proposed that when the free energy differences between the polymorphs are small there may be no significant differences in their biopharmaceutical behaviour as measured by the blood levels they achieve. Only when the differences are large may they affect the extent of absorption. For example, ΔGB→A for the transition of chloramphenicol palmitate Form B to Form A is −3.24 kJ mol−1; ΔH is −27.32 kJ mol−1. For mefenamic acid ΔGII→I is −1.05 kJ mol−1 and ΔH is −4.18 kJ mol−1. Whereas differences in biological activity are shown by the palmitate polymorphs, no such differences are observed with the mefenamic acid polymorphs. When little energy is required to convert one polymorph into another, it is likely that the forms will interconvert in vivo and that the administration of one form in place of the other will be clinically unimportant.
Particle size reduction may lead to fundamental changes in the properties of the solid. Grinding of crystalline substances such as digoxin can lead to the formation of amorphous material (see section 1.3) that has an intrinsically higher rate of solution and therefore apparently greater activity. Such is the importance of the polymorphic form of poorly soluble drugs that it has to be controlled. For instance, there is a limit on the inactive polymorph of chloramphenicol palmitate. Of the three polymorphic forms of chloramphenicol palmitate, Form A has a low biological activity because it is so slowly hydrolysed in vivo to free chloramphenicol.7 We can see from Fig. 1.14 that the maximum blood levels attained with 100% Form B polymorph are about seven times greater than with 100% Form A polymorph, and that with mixtures of A and B the blood levels vary in proportion to the percentage of B in the suspension.8
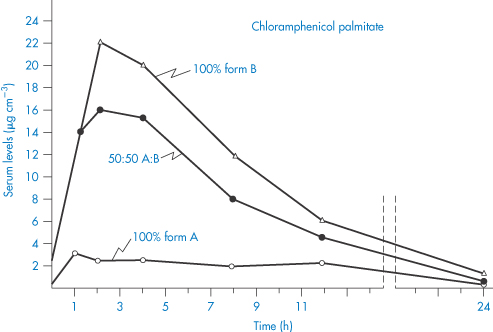
Figure 1.14 Comparison of serum levels (μg cm−3) obtained with suspensions of chloramphenicol palmitate after oral administration of a dose equivalent to 1.5 g of chloramphenicol.
Redrawn from Aiguiar AJ, Zelmer JE. Dissolution behaviour of polymorphs of chloramphenicol palmitate and mefanamic acid. J Pharm Sci 1969;58:983–987. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
During formulation development it is vital that sufficient care is taken to determine polymorphic tendencies of poorly soluble drugs. This is so that formulations can be designed to release drug at the correct rate and so that intelligent guesses can be made before clinical trial about possible influences of food and concomitant therapy on drug absorption. As will be seen later, particle characteristics (of nitrofurantoin, for example) can affect drug interaction as well as drug absorption. Above all, it is important that during toxicity studies care is given to the characterisation of the physical state of the drug, and that during development the optimal dosage form is attained. It is insufficient that drug is ‘available’ from the dosage form; on both economic and biological grounds, the maximum response must be achieved with the minimum amount of drug substance.
|
|

1.2.4 Multicomponent crystals
Crystal hydrates and solvates
When some compounds crystallise they may entrap solvent in the crystal. Crystals that contain solvent of crystallisation are called crystal solvates, or crystal hydrates when water is the solvent of crystallisation. Crystals that contain no water of crystallisation are termed anhydrates.
Crystal solvates exhibit a wide range of behaviour depending on the interaction between the solvent and the crystal structure. With some solvates the solvent plays a key role in holding the crystal together; for example, it may be part of a hydrogen-bonded network within the crystal structure. These solvates are very stable and are difficult to desolvate. When these crystals lose their solvent they collapse and recrystallise in a new crystal form. We can think of these as polymorphic solvates. In other solvates, the solvent is not part of the crystal bonding and merely occupies voids in the crystal. These solvates lose their solvent more readily and desolvation does not destroy the crystal lattice. This type of solvate has been called a pseudopolymorphic solvate.
By way of illustration of this phenomenon, we return to the case of spironolactone which we considered earlier. As well as the two polymorphs, this compound also possesses four solvates, depending on whether it is crystallised from acetonitrile, ethanol, ethyl acetate or methanol. Each of these solvates is transformed to the polymorphic Form 2 on heating, indicating that the solvent is involved in the bonding of the crystal lattice.
The stoichiometry of some of the solvates is unusual. Fludrocortisone pentanol solvate, for example, contains 1.1 molecules of pentanol for each steroid molecule, and its ethyl acetate solvate contains 0.5 molecules of ethyl acetate per steroid molecule. A succinylsulfathiazole solvate appears to have 0.9 moles of pentanol per mole of drug. Beclometasone dipropionate forms solvates with chlorofluorocarbon propellants.
Infrared measurements show that cefaloridine exists in α, β, δ, ε, ζ and μ forms (that is, six forms after recrystallisation from different solvents).9 Proton magnetic resonance spectroscopy showed that, although the μ form contained about 1 mole of methanol and the ε form about 1 mole of dimethyl sulfoxide, ethylene glycol or diethylene glycol (depending on the solvent), the α, β, anhydrous δ and ε forms contained less than 0.1 mole, that is, non-stoichiometric amounts of solvent. The α form is characterised by containing about 0.05 mole of N,N-dimethylacetamide. This small amount of ‘impurity’, which cannot be removed by prolonged treatment under vacuum at 10−5–10−6 Torr, is apparently able to ‘lock’ the cefaloridine molecule in a particular crystal lattice.
Pharmaceutical consequences of solvate formation
Modification of the solvent of crystallisation may result in different solvated forms. This is of particular relevance because the hydrated and anhydrous forms of a drug can have melting points and solubilities sufficiently different to affect pharmaceutical behaviour. For example, glutethimide exists in both an anhydrous form (m.p. 83°C, solubility 0.042% at 25°C) and a hydrated form (m.p. 68°C, solubility 0.026% at 25°C). Other anhydrous forms show similar higher solubilities than the hydrated materials and, as expected, the anhydrous forms of caffeine, theophylline, glutethimide and cholesterol show correspondingly higher dissolution rates than their hydrates.
One can assume that, as the hydrate has already interacted intimately with water (the solvent), then the energy released for crystal break-up, on interaction of the hydrate with solvent, is less than for the anhydrous material. The non-aqueous solvates, on the other hand, tend to be more soluble in water than the non-solvates. The n-amyl alcohol solvate of fludrocortisone acetate is at least five times as soluble as the parent compound, while the ethyl acetate solvate is twice as soluble.
The equilibrium solubility of the non-solvated form of a crystalline organic compound that does not dissociate in the solvent (for example, water) can be represented as:

where Ks is the equilibrium constant. This equilibrium will of course be influenced by the crystal form, as we have seen, as well as by temperature and pressure. For a hydrate A·xH2O, we can write:

Ksh is then the solubility of the hydrate. The process of hydration of an anhydrous crystal in water is represented by an equation of the type:
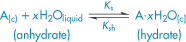
and the free energy of the process is written:

ΔGtrans can be obtained from the solubility data of the two forms at a particular temperature, as for theophylline and glutethimide in Table 1.3.
Table 1.3 Solubility of theophylline and glutethimide forms at various temperatures
| Temperature (°C) | Solubility | |
Hydrate (mg cm−3) | Anhydrate (mg cm−3) | ||
Theophylline | 25 | 6.25 | 12.5 |
35 | 10.4 | 18.5 | |
45 | 17.6 | 27.0 | |
55 | 30 | 38 | |
Glutethimide |
| (%w/v) | (%w/v) |
25 | 0.0263 | 0.042 | |
32 | 0.0421 | 0.0604 | |
40 | 0.07 | 0.094 | |
Reproduced with permission from Eriksen SP. Am J Pharm Educ 1964;28:47.
The dissolution rates of solvates can vary considerably. Table 1.4 shows the range of intrinsic dissolution rates reported for solvates of oxyphenbutazone into a dissolution medium containing a surface-active agent (to avoid wetting problems). The superior dissolution rates of the benzene and cyclohexane solvates (B and C respectively) are apparent but, of course, the possible use of these solvates is prohibited because of their likely toxicity.
Table 1.4 Intrinsic dissolution rates of the crystal forms of oxyphenbutazone
Sample | Intrinsic dissolution ratea (μg min−1 cm−2) |
Solvate C | 21.05 ± 0.02 |
Solvate B | 18.54 ± 0.47 |
Anhydrate | 14.91 ± 0.47 |
Hemihydrate | 17.01 ± 0.78 |
Monohydrate | 9.13 ± 0.23 |
Reproduced with permission from Stoltz M, Lotter AP, van der Walt JG. Physical characterization of two oxyphenbutazone pseudopolymorphs. J Pharm Sci 1988;77:1047.
aMean ± range of uncertainty of two determinations.
Differences in solubility and dissolution rate between solvates can lead to measurable differences in their bioavailabilities. You can see in Table 1.5 the differences in in vivo absorption rates of solvates of prednisolone t-butyl acetate and hydrocortisone t-butyl acetate after implantation of pellets of these compounds. Note, for example, that the monoethanol solvate of prednisolone has an absorption rate in vivo that is nearly five times greater than that of the anhydrous t-butyl acetate. Differences in the absorption of ampicillin and its trihydrate can be observed (Fig. 1.15), but the extent of the difference is of doubtful clinical significance. The more soluble anhydrous form appears at a faster rate in the serum and produces higher peak serum levels.
Table 1.5 Absorption rate of hydrocortisone tertiary butyl acetate and prednisolone tertiary butyl acetate (mg h−1 cm−2)
Compound | Absorption rate (mg h−1 cm−2) |
Prednisolone tertiary butyl acetate | |
Anhydrous | 1.84 x 10−3 |
Monoethanol solvate | 8.7 x 10−3 |
Hemiacetone solvate | 2.2 x 10−1 |
Hydrocortisone tertiary butyl acetate | |
Anhydrous | 4.74 x 10−3 |
Monoethanol solvate | 1.83 x 10−3 |
Hemichloroform solvate | 7.40 x 10−1 |
Modified with permission from Ballard BE, Biles J. Effect of crystallizing solvent on absorption rates of steroid implants. Steroids 1964;4:273.
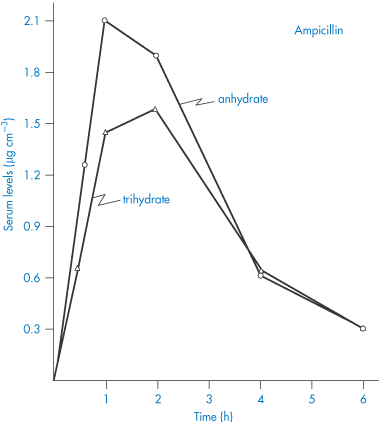
Figure 1.15 Serum levels (μg cm−3) obtained after oral administration of a suspension containing 250 mg ampicillin as the anhydrate and as the trihydrate.
Reproduced with permission from Poole JW et al. Physicochemical factors influencing the absorption of the anhydrous and trihydrate forms of ampicillin. Curr Ther Res 1968;10:292.
Cocrystals
Cocrystals are an important class of pharmaceutical materials that have the potential to enhance solubility, dissolution and consequent bioavailability of poorly water-soluble drugs. A pharmaceutical cocrystal is a multicomponent crystal comprising two or more compounds (at least one of which is a drug molecule) that are solids under ambient conditions, present in a stoichiometric ratio and interact by non-covalent interactions such as hydrogen bonding. They are similar to crystal solvates in that the crystal is composed of the drug and another molecule; in cocrystals, however, the other molecule (the coformer) is a pharmaceutically acceptable compound that is a solid under ambient conditions rather than a liquid as in crystal solvates. An important advantage of cocrystals compared to solvates and hydrates is that it is easier to control the ratio of components (i.e. the stoichiometry) and therefore the physicochemical properties of the cocrystal.
Cocrystals differ from solid dispersions (section 1.9), which are physical mixtures of drug and a highly water-soluble carrier molecule in microcrystalline or molecular form. The aim with each of these solid-state dosage forms is nevertheless the same – to alter the physicochemical properties of the drug, for example, to increase solubility or reduce hygroscopicity. In this respect cocrystallisation provides a means of presenting very different physicochemical properties compared to the parent drug without covalent modification of its molecular structure, whereas it is only possible to achieve marginal changes in solubility with solvates, hydrates and polymorphs. The potential for improvement of solubility and dissolution profile is similar to that which can be achieved by forming more soluble salts of the drug. Cocrystal development is, however, particularly attractive when salt formation is not feasible or when existing salts fail to exhibit suitable properties for use in a drug product. In addition, cocrystals of pharmaceutical salts, namely ionic cocrystals, can also be prepared and evaluated. These potentially offer more scope for solubility enhancement since both the counterion and the coformer can be varied to achieve optimum solubility.
In selecting a suitable molecule to cocrystallise with the drug, consideration is made of the potential for hydrogen bonding between the two molecules. Figure 1.16 shows the molecular packing within the 1 : 1 cocrystals of carbamazepine and saccharin. The carbamazepine molecules hydrogen bond to each other to form a dimer and saccharin molecules form double hydrogen bonds to both N−H donor and O acceptor atoms on this dimer. Other drugs forming cocrystals include caffeine and theophylline, both of which form cocrystals with a range of dicarboxylic acids that have improved stability to high humidity.
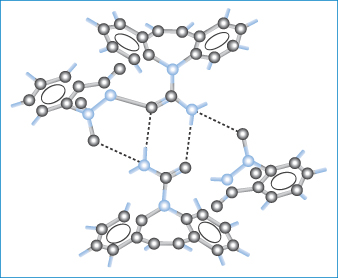
Figure 1.16 Packing of molecules in cocrystals of carbamazepine and saccharin. The dashed lines indicate hydrogen bonding of carbamazepine molecules to each other to form a dimer and the formation of double hydrogen bonds between saccharin and N−H donor and O acceptor atoms on the dimer.
Reprinted with permission from Fleischman SG et al. Crystal engineering of the composition of pharmaceutical phases: multi-component crystalline solids involving carbamazepine. Cryst Growth Des 2003;3:909. Copyright 2003, American Chemical Society.
It is interesting to note that not only is cocrystal formation a means of increasing the intrinsic solubility of a poorly soluble drug, but it also provides an opportunity to alter its pH-solubility profile through the choice of a suitable ionisable coformer. Figure 1.17 shows that cocrystals of a non-ionisable drug (carbamazepine) can exhibit very different pH-solubility profiles depending on the coformer ionisation properties. A diprotic coformer such as succinic acid leads to an increase of solubility with pH; an amphoteric coformer such as 4-aminobenzoic acid will, however, result in a U-shaped curve with a solubility minimum in a pH range between the two pKa values.
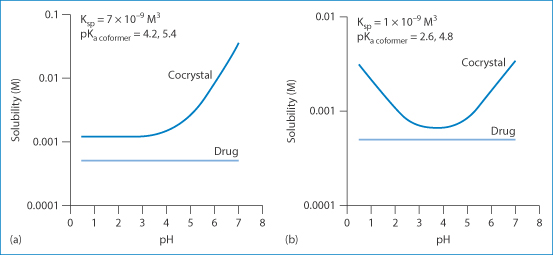
Figure 1.17 Manipulation of the pH-solubility profile of carbamazepine by forming cocrystals with (a) succinic acid and (b) 4-aminobenzoic acid hydrate.
Reproduced with permission from Thakuria R et al. Pharmaceutical cocrystals and poorly soluble drugs. Int J Pharm 2013;453:101. Copyright Elsevier 2013.
Despite the successful application of cocrystallisation in manipulation of the physical properties of a drug, there are currently no marketed products utilising cocrystals. One of the difficulties encountered in the design of possible cocrystals for a particular drug is that, although cocrystal solubility often correlates strongly with coformer solubility, there are insufficient data to predict the solubility enhancement with certainty. A more important concern, however, is that these metastable crystal forms may change their form during storage. However, a recent review10 discusses potential developments in cocrystal technology and concludes that ‘the systematic commercialisation of cocrystal drug substances into drug products is likely just a matter of time’.
|
|

1.3 Amorphous solids
Solids in which there is no long-range ordering of the molecules are said to be amorphous. These disordered systems differ in solubility, stability, dissolution properties and compression characteristics from the more traditionally used crystalline counterparts and provide attractive alternatives to them in drug delivery formulations. In principle, most classes of material can be prepared in the amorphous state if the rate at which they are solidified is faster than that at which their molecules can align themselves into a crystal lattice with three-dimensional order. It is also possible to convert crystalline material to amorphous inadvertently when supplying mechanical or thermal energy, for example, during grinding, compression and milling the solid or during drying processes. Some materials, notably polymers such as poly(lactic acid), polyvinylpyrrolidone and polyethylene glycol, are inherently amorphous. Even at slow solidification rates, large molecules such as these are often unable to form perfect crystals because of the difficulty in arranging the chains of these flexible molecules in an ordered manner; such materials are frequently semicrystalline, with areas of a crystalline nature surrounded by amorphous regions, as shown in Fig. 1.18.
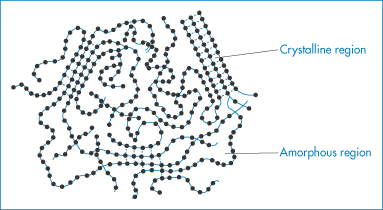
Figure 1.18 Diagrammatic representation of a solid polymer showing regions of crystallinity and regions that are amorphous.
Unlike crystals, amorphous or semicrystalline materials do not have sharp melting points, but instead there is a change in the properties of the material at a characteristic temperature, called the glass transition temperature, Tg. Below Tg, the material is said to be in its glassy state and is brittle; as the temperature is increased above Tg, the molecules become more mobile and the material is said to become rubbery. The transition temperature may be lowered by the addition of plasticisers, which are generally small molecules that are able to fit between the glassy molecules, so increasing their mobility. Water, for example, is used as a plasticiser for a wide range of polymers used in film coating.
Amorphous solids, because they exhibit a higher energy state than crystalline solids, are inherently less stable and have the potential for converting to the thermodynamically more stable crystalline form over time. In addition, because of their higher molecular mobility, they often show stronger chemical reactivity and hence a faster rate of chemical degradation. Nevertheless, the amorphous form of a drug often has a higher solubility than its crystalline form and the use of the amorphous form of a drug may provide an opportunity to enhance its bioavailability in the case of poorly water-soluble drugs.
1.4 Dissolution of solid drugs
Whether the solution process takes place in the laboratory or in vivo, there is one law that defines the rate of solution of solids when the process is diffusion controlled and involves no chemical reaction. This is the Noyes–Whitney equation, which may be written:

where k = D/δ. The equation is the analogue of equation (1.1), discussed previously. Figure 1.19 shows the model on which this equation is based. The terms of the equation are: dw/dt, the rate of increase of the amount of material in solution dissolving from a solid; k, the rate constant of dissolution; cs, the saturation solubility of the drug in solution in the diffusion layer; and c, the concentration of the drug in the bulk solution. A is the area of the solvate particles exposed to the solvent, δ is the thickness of the diffusion layer and D is the diffusion coefficient of the dissolved solute. The relevance of polymorphism and solid-state properties to this equation lies in the fact that A is determined by particle size. Particle size reduction, if it leads to a change in polymorph, results in a change in cs, and if dissolution is the rate-limiting step in absorption, then bioavailability is affected. In more general terms, one can use the equation to predict the effect of solvent change or other parameters on the dissolution rate of solid drugs. These factors are listed in Table 1.6.
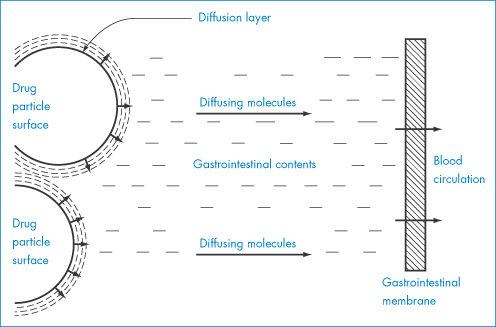
Figure 1.19 Schematic diagram of dissolution from a solid surface.
Table 1.6 How the parameters of the dissolution equation can be changed to increase (+) or decrease (−) the rate of solution
Equation parameter | Comments | Effect on rate of solution |
D (diffusion coefficient of drug) | May be decreased in presence of substances which increase viscosity of the medium | (−) |
A (area exposed to solvent) | Increased by micronisation and in ‘amorphous’ drugs | (+) |
δ (thickness of diffusion layer) | Decreased by increased agitation in gut or flask | (+) |
cs (solubility in diffusion layer) | That of weak electrolytes altered by change in pH, by use of appropriate drug salt or buffer ingredient | (−)(+) |
c (concentration in bulk) | Decreased by intake of fluid in stomach, by removal of drug by partition or absorption | (+) |
|
A preparation of drug granules weighing 5.5 g and having a total surface area of 2.8 × 103 cm2 dissolves in 500 mL of water at 25°C. The quantity of drug dissolved after the first minute is 0.76 g. The saturated solubility of the drug is 15 mg mL−1 and the diffusion layer thickness is 5 × 10−3 cm. Calculate: (a) the dissolution rate constant; (b) the diffusion coefficient; and (c) the effect on the dissolution rate of an increase of surface area to 5 × 103 cm2. Answer (a) Converting the mass of drug dissolved into mg gives the rate of dissolution at t = 60 s as:
The concentration of the drug in the bulk solution is
Therefore, from equation (1.4),
(b) The diffusion coefficient D can be calculated from:
(c) When the surface area is increased to 5 × 103 cm2,
i.e. the increase in surface area has caused an increase of the rate of dissolution from 12.67 to 22.65 mg s−1. Note the importance of ensuring that the parameters are converted to the appropriate units when substituting in equation (1.4) or indeed in any equation. If you are in doubt about units then it is easy to carry out a check to ensure that the units balance on both sides of the equation. In the particular example of equation (1.4) we have:
|

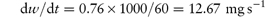
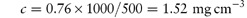
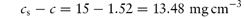
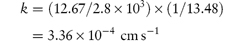
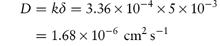
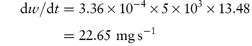
1.5 Biopharmaceutical importance of particle size
As discussed in Chapter 9, control of particle size is important in pulmonary delivery because only very fine particles are able to penetrate the alveolar regions of the respiratory tract. But there is an optimum. Indeed, if the particle size is reduced too far, particles may be exhaled and not deposited. The vital influence (for mainly aerodynamic reasons) of particle size and density in the activity of inhaled drug particles is discussed in Chapter 9, section 9.8.1. In this section we will consider the influence of particle size on the dissolution and absorption of drugs.
It has generally been believed that only substances in the molecularly dispersed form (that is, in solution) are transported across the intestinal wall and absorbed into the systemic circulation. This is the premise on which much thinking on bioavailability from pharmaceutical dosage forms is based. Although this is generally true, it has been shown that very small particles in the nanometre size range can also be transported through enterocytes by way of pinocytosis, and that solid particles of starch, for example, in the micrometre size range enter by a mechanism involving passage of particles between the enterocytes.11 Submicrometre particulate uptake by the M cells of the gut-associated lymphoid tissue is a phenomenon of increasing importance.12 Because of the much greater absorptive area available to molecules, however, the opportunity for molecules to penetrate the cell membrane is obviously higher than that for particles.
The rate of absorption of many slightly soluble drugs from the gastrointestinal tract and other sites is limited by the rate of dissolution of the drug. The particle size of a drug is therefore of importance if the substance in question has a low solubility. In some cases, notably that of griseofulvin, there is pharmacopoeial control of particle size; the British Pharmacopoeia (2010) specifies for this drug ‘a microfine powder, the particles of which generally have a maximum dimension of up to 5 μm, although larger particles that may exceed 30 μm may occasionally be present’. The control exercised over the particle size is due to its very low solubility; the experience is that if the solubility of a drug substance is about 0.3% or less then the dissolution rate in vivo may be the rate-controlling step in absorption.
The Noyes–Whitney equation demonstrates that solubility is one of the main factors determining rate of solution. When the rate of solution is less than the rate of absorption, the solution process becomes rate limiting. Generally speaking, it should become so only when the drug is of low solubility at the pH of the stomach and intestinal contents. The rate of absorption, the speed of onset of effect and the duration of therapeutic response can all be determined by particle size for most routes of administration. Figure 1.20 shows the effect of particle size of phenobarbital suspensions on the drug’s bioavailability after intramuscular injection compared with a solution of the drug, which probably precipitates in fine crystal form at the site of injection. The rate of solution of the drug crystals controls the extent of absorption from the intramuscular site.
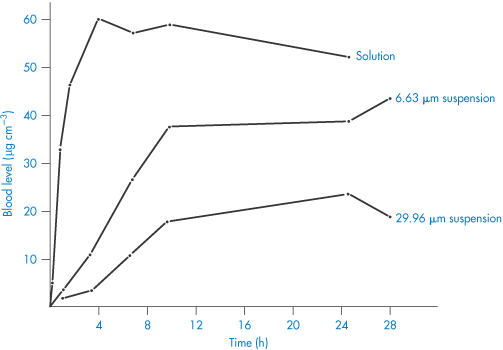
Figure 1.20 Blood levels (μg cm−3) of phenobarbital versus time after intramuscular injection of three formulations.
Redrawn from Miller LG, Fincher JH. Influence of drug particle size after intramuscular dosage of phenobarbital to dogs.J Pharm Sci 1971;60:1733, with permission from Wiley.
We have seen in the previous section that particle size has an important influence on dissolution rate, the larger surface exposed to the solvent increasing significantly the dissolution rate of smaller particles. The effect of particle size reduction on dissolution rate is one of exposure of increasing amounts of surface of the drug to the solvent. It is only when comminution reduces particle size below 0.1 μm that there is an effect on the intrinsic solubility of the substance (see Chapter 4), and thus on its intrinsic dissolution rate. Very small particles have a very high surface/bulk ratio. If the surface layer has a higher energy than the bulk, as is the case with these small particles, they will interact more readily with solvent to produce higher degrees of solubility.
It was with the action of phenothiazine that the importance of particle size was first recognised, in 1939, in relation to its toxicity to codling moth larvae, and in 1940 in relation to its anthelmintic effect, in both of which it was shown that reduction in particle size increased activity. The improvement in biological response to griseofulvin on micronisation is well known; similar blood levels of the drug were obtained with half the dose of micronised drug compared with those of non-micronised griseofulvin.13 The influence of particle size on the bioavailability of digoxin14 and dicoumarol (bishydroxycoumarin)15 has also been investigated. In both cases, plasma levels of drug are of high significance in clinical and toxic responses.
In the case of digoxin there is evidence that milling to reduce particle size can produce an amorphous modification of the drug with enhanced solubility and hence increased bioavailability. The possibility of changing the crystal structure during processing is therefore important: comminution, recrystallisation and drying can all affect crystal properties.
During the pharmacological and toxicological testing of drugs before formal formulation exercises have been carried out, insoluble drugs are frequently administered in suspension form, often routinely in a vehicle containing gum arabic or methylcellulose. Without adequate control of particle size or adequate monitoring, the results of these tests must sometimes be in doubt, as both pharmacological activities and toxicity generally result from absorption of the drug. In a few cases particle size influences side-effects such as gastric bleeding or nausea. Gastric bleeding may in part be the direct result of contact of acidic particles of aspirin or non-steroidal anti-inflammatory agents with the mucosal wall. The influence of drug form on the LD50 of pentobarbital in mice is shown in Table 1.7. A twofold range of LD50 values is obtained by the use of different, simple formulations of the barbiturate. Even in solution form, sodium carboxymethylcellulose affects the LD50 by mechanisms that are not confirmed. Adsorption of the polymer at the intestinal surface may retard absorption, or some of the drug may be adsorbed on to the polymer.
Table 1.7 Influence of formulation on the potency ratios of pentobarbital in the form of the sodium salt and the free acid
Pentobarbital form | Dosage form | Vehicle | Particle size (μm) | LD50 | Potency ratioa |
Sodium salt | Solution | Water | – | 132 | 1 |
Sodium salt |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


