Figure 12.1 Drug/human factors and dosage form factors in adverse events following medication administration.
Modified from Uchegbu IF, Florence AT. Adverse drug events related to dosage forms and delivery systems. Drug Saf 1996;14:39–67, with kind permission from Springer Science and Business Media.
The variety of adverse events that have occurred in the recent past is illustrated in Table 12.1. Historical or ‘classic’ cases are valuable as they can provide clues to adverse events occurring with new therapeutic agents or delivery systems.
Table 12.1 Some ‘classic’ adverse events as a result of use of formulations
Dosage form | Trade name | Adverse event |
Indometacin osmotic minipump tablets | Osmosin | Intestinal perforation |
Fluspirilene intramuscular injection | Redeptin | Tissue necrosis at site of injection |
Epidural injection of prednisolone injection containing benzyl alcohol as preservative | Depo Medrol | Mild paralysis |
Inhalation of nebulised ipratropium bromide solution containing benzalkonium chloride as preservative | Atrovent | Paradoxical bronchoconstriction |
Povidone-iodine solution for vaginal application |
| Anaphylactoid reaction |
Timolol eye drops | Timoptic | Bronchoconstriction |
Fentanyl transdermal patch | Duragesic | Respiratory depression and death |
Reproduced from Uchegbu IF, Florence AT. Adverse drug events related to dosage forms and delivery systems. Drug Saf 1996;14:39–67, with kind permission from Springer Science and Business Media.
The example of the indometacin osmotic tablets (see Chapter 7, section 7.7) was a case of three factors conspiring to produce the adverse effects: the nature of the drug itself, the osmotic core of potassium chloride and the localised release of the drug from the tablet. The formulation was withdrawn from the market.
12.2 Excipient effects
Excipients, as discussed elsewhere in this text, are components of formulations other than the drug or other active ingredient. Their functions are many, variously to aid processing, to aid dissolution of solid-dose forms or conversely, to retard release of the drug, to stabilise the formulation or to protect the drug from adverse environments both in vitro and in vivo. Excipients are the dominant material in many tablet and capsule formulations, as sometimes their main role is to provide sufficient bulk for a low-dose drug to be administered safely. This section deals with the potential of excipients to influence outcomes of medication.
Excipients are not always the inert substances that we presume. Some cases and reports of the adverse action of excipients are discussed here. While labelling requirements insist on the listing of ingredients in some products, the plethora of trade names (e.g. for surfactants, polymers, lipids and other excipients) can make accurate identification of materials difficult. The European Commission guidelines require that all excipients must be declared on the labelling if the product is an injectable, or a topical preparation (for skin, for inhalation, delivery to the vaginal, nasal or rectal mucosae) or an eye preparation.
Batch-to-batch variation of many excipient raw materials adds another layer of complexity in tracking down and comparing case histories. One problem faced is that products available in one national market may have different formulations from those marketed in another. The clinical literature does not always detail the formulations or product brands used in clinical studies.
12.2.1 Not always inert
Excipients are intended to be inert, but they are not always so in all patients.1 Excipients must be of high quality, safe and must have a high degree of functionality, that is, a total fitness for use.
Fitness for use in one application may not mean fitness for use in another, for example, by another route of administration. Even though adverse reactions to additives might be relatively rare, it is for this reason that the possibility that an excipient might be the cause of any adverse event should be kept in mind. Fillers like lactose bulk out a low dose (milligrams or micrograms) so that the resulting tablet or capsule can be manufactured and is of a sufficient size to be handled. Lactose is also used in dry-powder inhaler formulations as a carrier for the drug. There may be lubricants such as magnesium stearate to aid the flow of powders and their subsequent tableting or filling into capsules (see Chapter 1).
Preservatives and dyes used widely in formulation are listed in Tables 12.2 and 12.3, which is the result of surveys of these ingredients and their frequency of use,2 albeit not in the UK. These figures are used here to give an impression of the excipients most likely to be encountered.
Table 12.2 Preservatives found in liquid pharmaceutical formulations (n = 73)
Preservatives | % of formulations |
Methylparaben | 45.2 |
Propylparaben | 35.6 |
Sodium benzoate | 32.8 |
Sodium metabisulfite | 11 |
Benzoic acid | 8 |
Hydroxyparabenzoate | 4 |
Potasssium sorbate | 2 |
Hydroxyparabenzoic acid | 1 |
No preservatives | 2 |
Reproduced with permission from Balbani APS et al. Pharmaceutical excipients and the information on drug labels. Rev Bras Otorinolaringol 2006;72:400–406.
Table 12.3 Dyes found in pharmaceutical formulations (n = 73)
Dyes | % of formulations |
Dusk Yellow (FD&C #6) | 15 |
Tartrazine Yellow (FD&C #5) | 9.5 |
Erythrosine | 6.8 |
Ponceau 4R Red | 5.4 |
Caramel | 4.1 |
Red #40 | 4.1 |
Food Red | 4.1 |
Bordeaux S Red | 2.7 |
Quinoline Yellow | 2.7 |
Yellow #10 | 2.7 |
Blue #1 | 1.3 |
Red #10 | 1.3 |
Iron oxide | 1.3 |
Reproduced with permission from Balbani APS et al. Pharmaceutical excipients and the information on drug labels. Rev Bras Otorinolaringol 2006;72:400–406.
Relatively simple molecules are used as excipients in formulations, from substances such as lactose or magnesium stearate through surfactants, preservatives, colours and flavours to macromolecules. With small molecules, such as the para-aminobenzoic acid esters (parabens), it is possible to know whether the compound is pure, and thus exactly what is in the formulation. It is not possible to generalise about the effects of excipients with all their structural diversity and uses. Many polymeric materials are complex (see Chapter 7), not only because of the existence in any one sample of a range of molecular weights, but also because many contain plasticisers to adjust their physical properties or agents to aid production. Plasticisers such as diethyl phthalate leach out from plastic giving sets into infusions, particularly those that have ingredients (such as surfactants) that might aid the solubilisation of the phthalate. Dioctyl phthalate and dioctyl adipate have been found in some silicone tubing.
With polymers and other macromolecules one needs to know their molecular weight, or more likely, their molecular weight distribution, and about the presence of impurities, such as catalysts and peroxides, the latter occuring in polysorbate 80 (see Chapter 5). Cremophor EL, a castor oil polyoxyethylene surfactant derivative which is used in paclitaxel formulations, is ‘cleaned’ rather than pure.
Excipients are rarely produced to the extremely high standards of purity that apply to drug substances. Hence the presence of impurities rather than the material itself might be the cause of any adverse event, perhaps by inducing degradation of the drug. There may be batch differences or brand differences in excipients, which are confusing.
Problems with excipients to which the American Academy of Pediatrics have drawn attention are shown in Table 12.4. Note that the route of administration and sometimes the mode of administration (for example, a particular device such as a nebuliser) and the concentration will affect the appearance or severity of many adverse effects.
Table 12.4 Excipients that have caused problems in paediatric and adult medicinesa
Excipient or class | Selected observed reactions |
Sulfites | Wheezing, dyspnoea, anaphylactoid reactions |
Benzalkonium chloride | Paradoxical bronchoconstriction, reduced forced expiry volume |
Aspartame | Headache, hypersensitivity |
Saccharin (I) | Dermatological reactions; avoid in children with sulfa allergies |
Benzyl alcohol | In high concentrations can cause neonatal death |
Various dyes | Reactions to tartrazine, similar to aspirin intolerance; patients with the ‘classic aspirin triad’ reaction (asthma, urticaria, rhinitis) may develop similar reactions from other dyes such as amaranth, erthyrosin, indigo, carmine, Ponceau, Sunset Yellow, Brilliant Blue |
Lactose | Problem in lactose-sensitive patients (those with lactase deficiency) |
Propylene glycol | Localised contact dermatitis topically; lactic acidosis after absorption |
aData from American Academy of Pediatrics. Pediatrics 1997; 99:266–278.
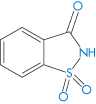
Structure I Saccharin, an o-toluene sulfonamide
12.3 E-numbers
From Table 12.4 it is clear that there are many ways of referring to additives. A classification system has been developed that codifies additives both in food and in pharmaceuticals in terms of E-numbers. Box 12.2 gives the general numbering system for several classes of ingredients. It is useful when trying to detect cross-reactivity and sensitivities to know the E-numbers of dyes, preservatives and other ingredients. Each additive has a number within the classifications shown in Box 12.2.
Box 12.2 General system of E-numbers |
E100–E199 Colours E200–E299 Preservatives E300–E399 Antioxidants, acidity regulators E400–E499 Thickeners, stabilisers, emulsifiers E500–E599 Acidity regulators, anti-caking agents E600–E699 Flavour enhancers |
Further deconstruction of E-numbers might be useful in identifying possible causes of adverse events.
- E100–109 are yellow dyes; for example, tartrazine (F&DC Yellow 5) is E102.
- E140-149 are green dyes. E430–439 are polyoxyethylene derivatives such as polysorbate 80, which is E433.
- E210–219 are benzoates.
- E230–239 are phenols and methanoates.
- E220–220 are sulfites (e.g. sodium metabisulfite has an E-number of 223).
During the 1999/2000 influenza outbreak, a 53-year-old man consulted because of a persistent productive cough that followed flu-like illness. The patient was examined and prescribed erythromycin (Erymax™, Elan). He made it clear that he had a previous history of aspirin allergy and was reassured that there was no known cross-sensitivity between erythromycin and aspirin. Two days later, the patient’s wife came into the surgery; she was angry and upset because shortly after taking the erythromycin capsules, her husband had developed some tingling and swelling of his fingers and feet similar to the symptoms he had previously experienced with aspirin. They were both disturbed to find the following warning in the patient information leaflet: ‘Capsules contain the colouring agent E110. This can cause allergic-like reactions including asthma. You are more likely to have a reaction if you are also allergic to aspirin’. This case raised many issues. Was the patient allergic to E110 (Sunset Yellow FCF, Orange Yellow S, FD&C Yellow 6), or was the patient allergic to erythromycin itself? The physician concerned was unaware both of the presence of E110 in the formulation and of the apparent cross-reactivity between aspirin and this colouring agent, there being no mention of either in the physician’s BNF [British National Formulary] or in the Pharmaceutical Data Sheet Compendium. To be able to foresee such interactions requires that the structures of the molecules concerned be known and this is a tall order. However, understanding after the event is a reasonable goal for avoiding future occurrences. Taken verbatim from Millar JS. Pitfalls of ‘inert’ ingredients. Br J Gen Pract 2001;51:570. |

Table 12.5 Adverse effects of dyes and colouring agents5
Compound | Structure | Adverse effects |
Sunset Yellow |
| Urticaria exacerbation |
Indigo carmine |
| Urticaria exacerbation |
Tartrazine |
| Headache, gastrointestinal disturbance, exacerbation of asthma, dangerous in aspirin-intolerant individuals |
Amaranth |
| Potential carcinogenicity (banned) |
Brilliant Blue |
| Hypersensitivity reactions |
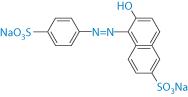
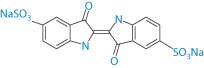
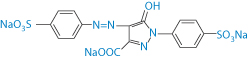
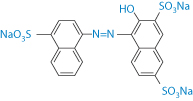
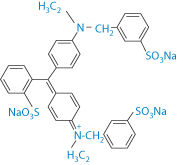
Reproduced from Pifferi G, Restani P. The safety of pharmaceutical excipients. Il Farmaco 2003;58:541–550. Copyright Elsevier 2003.
The structures of some of these dyes are quite complex and, perhaps not surprisingly, some do have pharmacological effects3, listed in Table 12.5. Another report4 discussed patients with reactions to E102 (tartrazine: F&DC Yellow 5) in oxytetracycline tablets and to E131 (Patent Blue V) in a doxycycline formulation.
Azo dyes (those with the –N=N– linkage) account for 60–70% of dyes used in food and textile manufacture. Their acute toxicity is low but some azo dyes have been banned from foods because of the toxicity of dye breakdown products rather than the dye itself. The mechanism by which tartrazine causes allergic reactions is not fully understood.
12.4 Cross-reactivity of drugs
Cross-reactivity can be defined as:
- a reaction to different compounds which may or may not have some structural similarity. Often the immune system is involved.
Cross-reactions between different azo dyes and para-amino compounds have been studied6 in azo dye-sensitive subjects, in the clinical aspects of azo dye dermatitis, and to attempt to relate the pattern of cross-sensitisations to the chemical structure of the different dyes. Out of 6203 consecutively tested patients, 236 were sensitised to at least one of six azo compounds employed as textile dyes. One hundred and seven subjects reacted to Disperse Orange 3 (DO3), 104 to Disperse Blue 124 (DB124), 76 to para-aminoazobenzene (PAAB), 67 to Disperse Red 1 (DR1), 42 to Disperse Yellow 3 (DY3) and 31 to p-dimethylaminoazobenzene (PDAAB). Co-sensitisations to para-phenylenediamine were present in most subjects sensitised to DO3 (66%) and PAAB (75%), in 27% and 36% of DR1 and DY3-sensitive subjects, and in only 16% of subjects sensitised to DB124. After the hands and the face, the neck and the axillae were the most frequently involved skin sites. Cross-sensitisations between azo dyes and para-amino compounds can partially be explained on the basis of structural similarities, as may be seen from structures II–IV.
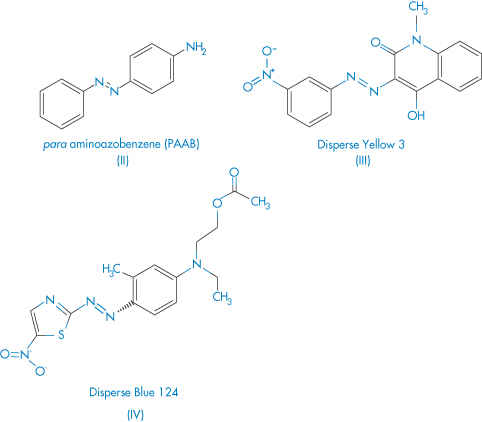
Structures II–IV Azo compounds used as dyes
12.5 Non-ionic surfactants
Non-ionic surfactants are widely used in formulations as wetting agents and solubilising agents. As discussed in Chapter 5, because surfactants are by structure and nature surface-active they will accumulate at interfaces, the air–water interface and the oil–water interface as well as the membrane–water interface. They can influence the fluidity of the membrane, and at higher concentrations, generally above their critical micelle concentration, they will cause membrane damage because they can solubilise structural membrane lipids and phospholipids.
While non-ionic surfactants allow poorly water-soluble drugs to be formulated as injectables, some have side-effects that are by now well known. The two most commonly used, and therefore cited as causing adverse events, are polysorbate 80 (Tween 80; E433) and a polyoxyethylated castor oil (Cremophor EL). Anaphylactic reactions are the most frequently cited, although they occur in a minority of patients. Hence it is essential, if such reactions to injections occur in new or experimental formulations, that their presence be recognised. Some products that contain, or have contained (see key point box below), Cremophor EL include teniposide, ciclosporin and paclitaxel formulations; docetaxel contains polysorbate 80, as does etoposide. The pharmacological effects of formulation vehicles have been discussed in detail elsewhere.7
|
Different formulations of these drugs exist in different countries under different trade names, hence the true composition of the product in question must be ascertained in assessing outcomes. |

12.6 Polyoxyethylene glycols
Polyoxyethylene glycols (PEGs) (macrogols) are not surface-active as they are completely hydrophilic with no hydrophobic domains, but they can exert an adverse effect when used as suppository bases through being hygroscopic. They absorb water from the rectal tissues and thus cause irritation, but this can be minimised by first moistening the PEG base before insertion. This affinity for water is the result of the interaction of water molecules with the oxygen of the repeating –CH2CH2O– units; a macrogol of molecular weight 44 500 Da has approximately 1000 such ethylene oxide units, each interacting with up to four H2O molecules. Macrogols 4000 and 3350 are used to sequester water in the bowels (Idrolax (Ipsen) or Laxido (Galen)). Note: ‘giving fluids with macrogols may reduce the dehydrating effect sometimes seen with osmotic laxatives.’ (British National Formulary 68).
Its presence in laxative formulations is most likely to aid the penetration of water into the faecal mass. Paediatric powder formulations of PEGs are available for faecal impaction and constipation (Movicol Paediatric, Norgine) with a dose of 6.56 g of Macrogol 3350. Sodium dodecyl (lauryl) sulfate is also an ingredient of an osmotic laxative, Relaxit Micro-enema. Usually it is found as a wetting agent in pharmaceuticals.
12.7 Adjuvants as therapeutic substances
This subsection is appropriate in this chapter as it emphasises again the fact that adjuvants can be biologically active.
12.7.1 Nonoxynol-9
Structure V Nonoxynol 9
Nonoxynol-9 (V) is similar to many non-ionic surfactants used as excipients. It is used as a spermicide because, being membrane-active, it interacts with spermatozoal membranes and reduces sperm mobility. It has also some activity against human immunodeficiency virus (HIV). However, it is also suspected to increase HIV access through the vaginal wall as, like other surfactants, it can damage biomembranes. It also has been shown to increase rectal infection by the herpes simplex virus. The microscopic evidence is clear that the compound damages the epithelial wall, as shown in Fig. 12.2 in the case of rectal tissue.8
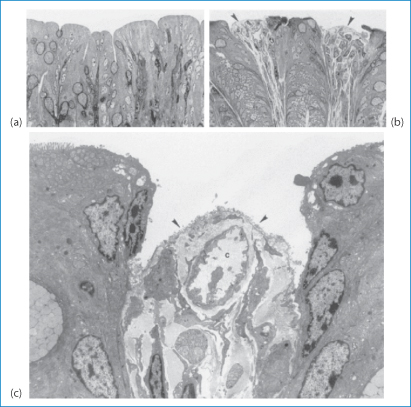
Figure 12.2 Light micrographs of the rectal epithelium and lamina propria of mice treated for 10 minutes with phosphate-buffered saline (PBS) (a) and nonoxynol-9. (b). Control (PBS-treated) tissue is characterised by a continuous epithelium of columnar and goblet cells. In tissue treated with nonoxynol-9, epithelial cells appear necrotic. In some areas the epithelium is missing and connective tissue is directly exposed to the rectal lumen (arrows). In the transmission electron micrograph shown in (c), connective tissue (arrows) appears to be exposed to the rectal lumen. Epithelial cells are missing microvilli. A capillary is shown in (c).
Reproduced from Phillips DM, Zacharopoulous VR. Nonoxynol-9 enhances rectal infection by herpes simplex virus in mice. Contraception 1998;57:341–348, with kind permission from Springer Science and Business Media.
12.7.2 Poloxamers
Poloxamers are ABA block copolymer surfactants (see Chapter 5, section 5.5.3); in designating them, A is the hydrophilic polyoxyethylene chain and B is the hydrophobic polyoxypropylene chain. The properties of the poloxamers depend on the length of each chain. ABA block copolymer surfactants such as the poloxamers have many pharmaceutical uses, as wetting, solubilising and emulsifying agents, but some, like poloxamer 188 (Pluronic F68) have some useful biological activity. It improves microvascular blood flow by reducing blood viscosity, particularly in low-shear conditions. Its mechanism of action is not clear, but it is suggested that the surfactant binds to cells via the hydrophobic B portion, leaving the polyoxyethylene chains to provide a hydrated barrier, reducing cell–cell, cell–protein and protein–protein interactions in the blood.9
Poloxamer 188 can have a protective effect on damaged heart muscle cells. Purified poloxamer 188 also has a beneficial effect on the treatment of sickle-cell disease,10 perhaps because it decreases the adhesion of sickle cells to the microvasculature.
It has been suggested that Poloxamer 188 may also operate as a cell repair agent or membrane sealant,11 as shown in Fig. 12.3.
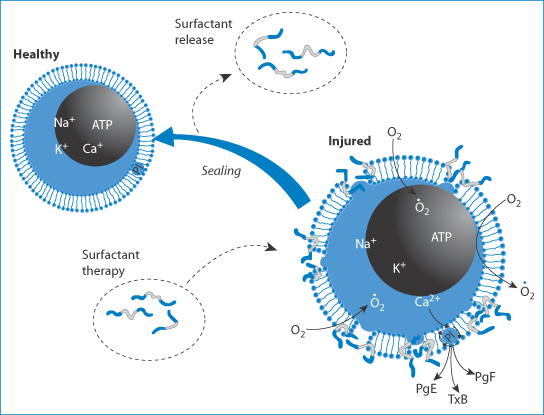
Figure 12.3 Suggested mechanism of action of Poloxamer 188, operating as a cell repair agent or membrane sealant. The surfactant, shown with its hydrophobic portion in light grey and hydrophilic part in blue, interacts with the injured cell, ‘sealing’ the membrane and returning it to normal. ATP, adenosine triphosphate; PgE, PgF, prostaglandins E and F, respectively; T B, thromboxane B.
B, thromboxane B.
Reproduced with permission from Agarwal J, Walsh AM, Lee RC. Ann NY Acd Sci 2005;1006:295–309.
12.7.3 Talc as therapeutic agent and excipient
Talc is hydrated magnesium silicate; there are variable amounts of calcium, magnesium and iron present in different samples of talc. Its particle size can vary considerably. Humble talc is used not only in baby care but also as a lubricant in the manufacture of tablets and as a lubricant for surgical gloves.
There is one less well-known use of talc, which is as an active agent rather than as an excipient. This is in the procedure of pleurodesis, when the membranes around the lung adhere. Talc prevents the build-up of fluid between the membranes. An irritant such as bleomycin, tetracycline or talc powder is instilled inside the pleural cavity to instigate an inflammatory response that ‘tacks the two pieces together’.12 Talc is one of the most effective agents to achieve the desired outcome, but there have been concerns about side-effects, especially acute respiratory distress syndrome (ARDS), assumed to be related to both the particle size and perhaps the shape of the talc particles.13 Particle size has been found to be key in determining the distribution of talc in the body,14 suggesting the possibility of migration of talc through the lymphatics.
In the USA, talc used in this procedure has a smaller particle size than in European samples, and it is in the USA that most cases of ARDS seem to occur. It has been shown15 in comparing two sizes of talc (normal and large) that they both elicit the same benefit in pleurodesis but the smaller sizes have greater pulmonary and systemic deposition of talc particles and cause greater pleural inflammation. This is another example where the physical properties of a material influence its biological effect. Small particles, once they gain entry, can in fact translocate throughout the body, depending on their size and surface properties, an issue discussed in Chapter 14 on pharmaceutical nanotechnology.
The smaller the particles of any substance, the farther they can travel, hence there is real concern about the migration of such particles, whether inhaled, swallowed or inadvertently placed in contact with open wounds, for example. There are similar concerns with cytotoxic drugs in powder form, which can be inhaled or absorbed through the skin (to be discussed).
12.8 Active excipients in multiple therapies
Box 12.3 When an excipient in one formulation affects, say, P-glycoproteins (P-gp), and it is administered along with another product whose absorption is P-gp-dependent, then interpretation of interactions can be obscured if the excipients in both products are not taken into account. |
Examples to illustrate the point made in Box 12.3 might include paclitaxel formulations with Cremophor EL and polysorbate 80 when used along with other formulations. Several non-ionic surfactants have been found to inhibit P-gp-mediated transport in vitro16; in one system the descending order of effect was Tocopheryl Polyethylene Glycol 1000 Succinate (TPGS) > Pluronic PE8100 > Cremophor EL > Pluronic PE6100 ≈ Tween 80. These surfactants had no effect on multiple drug resistance protein 2 function, suggesting specific action.
The well-known effects of surfactants on absorption enhancement have in the past been thought to be mainly due to direct interactions of the surfactant with biomembranes, at low concentrations causing increased fluidity and transport and at high concentrations causing solubilisation of key membrane components. Sometimes surfactants affect absorption in multiple ways, by interacting with the barrier membrane, by forming a microemulsion and thus aiding dispersal of lipophilic drugs or by affecting intestinal secretory transit.17
12.9 Influence of dosage form type
12.9.1 Adhesion of tablets
Oesophageal damage caused by tablets is discussed later, but it is important to consider here the following two case studies.
A tablet of pantoprazole (Protonix) was seen ‘perched’ in the oesophagus of a 57-year-old woman with oesophageal dysmotility; this patient with diffuse bronchiolar disease required hospitalisation.18 In another patient an enteric-coated aspirin tablet, its coating still intact, was seen in an ulcer in the gastric antrum of another patient.19 Adhesion or simply entrapment in diverticula may be an issue, In this case, as with others, a combination of pathology and dose form conspired to cause a problem. |

Adverse events are rarely simple. The manner in which dose forms behave in the presence of small or larger amounts of aqueous media, which may be found respectively in the oesophagus or later in the stomach, are varied. Simple in vitro studies can be illuminating, as Fig. 12.4 illustrates.20 This shows the unusual disintegration properties of two tablet formulations, the lower example being an extreme case typical of emopronium bromide (VI) tablets, once marketed as Cetiprin for the treatment of nocturnal enuresis.

Figure 12.4 Two tablets disintegrating in an aqueous medium at room temperature. Both show the splitting of the coating layer on the tablets, but (b) shows a now-discontinued tablet of emopronium bromide (Cetiprin) that is unravelling to expose the core of pure drug, irritant to the oesophageal lining.
Reproduced with permission from Florence AT, Salole EG (eds). Formulation Factors in Adverse Reactions. London: Wright; 1990.
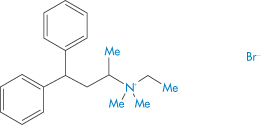
Structure VI Emopronium bromide, showing its surfactant-like nature
The Cetiprin tablet is seen to break apart, exposing a core of pure drug, which has surfactant properties. If this occurred in the oesophagus, pain and damage would result and this indeed was the case. Given its indication, many patients took the tablets with insufficient water and many cases of dysphagia were reported. The product was subsequently withdrawn from the market.
In this case the combination of adherence and the presence of a potentially irritant drug create the problem. Close connection between epithelia and product leads to high concentrations of drug and to damage. The oesophagus is a primary site for such adverse events, since tablets and capsules will generally not have disintegrated during their oesophageal transit and retain their bulk. Small uncoated tablets may also cause problems in the oesophagus because they can adhere firmly to the mucosal surface. A range of drugs have been reported to cause oesophageal injury, as shown in Table 12.6 and discussed in Box 12.4.
Table 12.6 Some drugs that have caused oesophageal injury
|
Box 12.4 What, if any, are the common features of the drugs listed in Table 12.6? What might be the main reason or reasons for each to cause injury? These include the acidity of the concentrated solutions of some of the drugs (aspirin, alendronate and risendronate); the proximity of high concentrations of drugs such as doxycycline which chelate calcium and hence disrupt the epithelial lining; the surface activity of agents such as emopronium bromide; or high electrolyte concentrations from the inorganic drugs. N.B.: If these agents are in dosage forms that are swallowed with sufficient water and do not lodge in the oesophagus, damage is less likely to be caused. |
Dosage forms have other effects and influences in modulating or causing adverse events, apart from the influence they have on pharmacokinetics and bioavailability. Precipitation of drugs from injection solutions is one prime example. Figure 12.1 reminds us of others:
- irritation from the adhesives of transdermal patches
- corneal irritation from eye drops
- rectal irritation from the use of suppository bases (such as PEGs) that, as discussed, extract water from the mucosa.
12.10 Tear films and eye drops
The formation and rupture of tear films were explained over 40 years ago by Holly.21 Eye drops can disrupt the natural tear film, either by the action of the drugs they contain or through additives such as benzalkonium chloride. The latter is a component of eye drops, but it can be toxic as it is a cationic surfactant. It is not a single compound, but comprises a mixture of alkyl chains† (see Chapter 5); the component molecules can adsorb on to the corneal surface in dry-eye syndrome, rendering that surface even more hydrophobic. The film can then de-wet the surface, thereby exposing the corneal epithelial cells to the air. The process may start at a given point on the corneal surface. A critical thickness for stability is breached and the film breaks, as shown in Fig. 12.5.
The film break-up time (Fig. 12.6) is measured in the clinic and is a useful measure to compare artificial tear fluids or severity of the syndrome of dry eye.
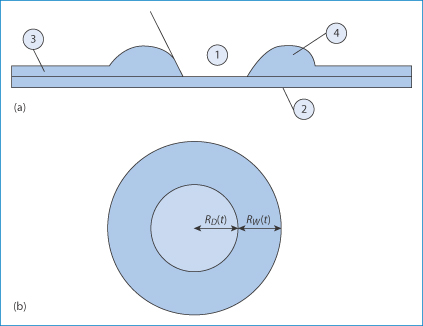
Figure 12.5 (a) Simplified diagram showing a thin liquid film (3) on a surface such as the cornea (2). When the film reaches a critical thickness, instability causes the film to rupture and surface forces pull the film away from that point, leaving an exposed surface (1). The liquid in the film is gathered in a rim (4). The contact angle between the broken film and the corneal surface will be reduced on addition of an agent that lowers surface tension. On the other hand, the adsorption of, say, benzalkonium chloride on the surface, with the onium group towards the negative cells, will render the surface in (1) more hydrophobic, exacerbating the situation. (b) Schematic diagram used to calculate the radial velocity of de-wetting.
Reproduced with permission from Njobuenwu DO. Spreading of trisiloxanes on thin water film: dry spot profile. Leonardo J Sci 2007;6:165–178.
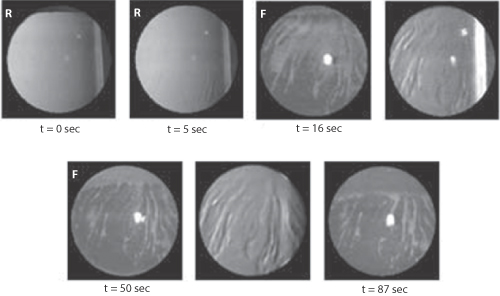
Figure 12.6 Time course and spatial topography of tear film break-up for subject AL obtained during a 90-second period of non-blinking. The time immediately after a blink is t = 0 sec. R = retro-illumination image; F = fluorescein image. The lack of a blink allows the drying out of the film, especially when the surface has been made more hydrophobic by adsorption of, say, benzalkonium chloride.
Reproduced with permission from Himebaugh NL et al. IUSO Visual Sciences Groups 1999 Indiana University. Available online at: www.opt.Indiana.edu (accessed 25 March 2015).
Tear film formation may be compromised, but not necessarily cause clinical problems unless the eyes are challenged with smoke or dust or certain drugs. Contact lenses may also of course affect tear film formation and stability.
As discussed in Chapter 10, neonates have normal tear fluid but low rates of blinking.23 This incomplete blinking24 allows time for the film to evaporate, as discussed above, and to cause the dry spots that lead to exposed points on the corneal epithelium. A low rate of tear fluid turnover is also believed to be the cause of reduced barriers to potential pathogens. The tear film in effect has a ‘washing function’, reducing the likelihood of bacterial adhesion to the corneal surface. This is similar to the case with saliva, which removes bacteria on teeth.
12.10.1 Fluoroquinolone eye drops
How do different fluoroquinolones behave in tears? In one study, three fluoroquinolone solutions were evaluated25: ciprofloxacin 0.3% (Ciloxan), norfloxacin (Chibroxin) and ofloxacin 0.3% (Ocuflox). The pH of the tear film for the first 15 minutes after instillation of each of the drugs is determined by the pH of its formulation. Rapid precipitation of ciprofloxacin was seen in a model system 8 minutes after dosing owing to the supersaturation of the drug in tears, while the tear concentration of ofloxacin and norfloxacin remained below saturation solubility at all pH values studied. These findings may explain the reports of precorneal deposits following the use of ciprofloxacin. The reasons for the precipitation of Ciloxan were discussed in Chapter 4 (section 4.2.4) and the relevant figure is shown again here. Figure 12.7 shows the solubility–pH plots for the three drugs, which explain the problem that occurs with Ciloxan in the model tear system when drug tear concentration exceeds solubility. One can see that the solubility line crosses the concentration line in the diagram for Ciloxan (Box 12.5).
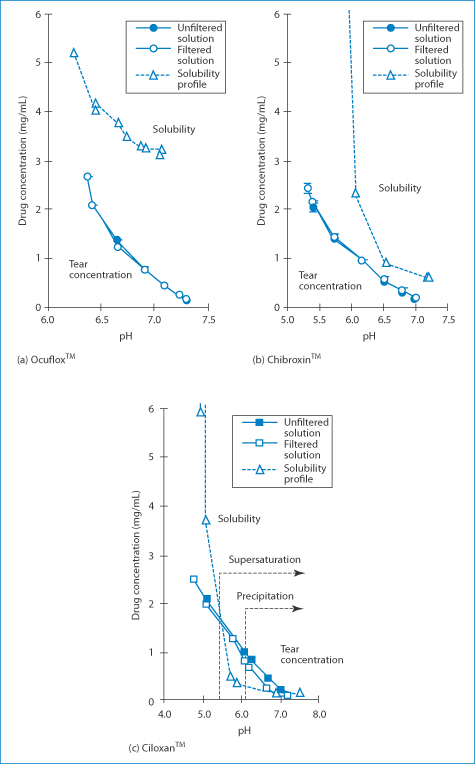
Figure 12.7 Relationship between the drug concentration in a tear turnover model and drug solubility (dashed lines).
Reproduced from Firestone BA et al. Solubility characteristics of three fluoroquinolone ophthalmic solutions in an in vitro tear model. Int J Pharm 1998;164:119–128. Copyright Elsevier 1998.
Box 12.5 The pH–solubility profiles of the fluoroquinolones up to pH 7.2 are typical of a base. Solubility decreases with increasing pH, with a minimum in solubility around pH 7. If the pH were to increase above 7.5, solubility would increase again as the compound possesses a carboxyl group. The pH of tear fluids is just over 6.8. When the solubility curve falls below the concentration curve, precipitation can be anticipated, a quick way of predicting the occurrence of precipitation. |
12.11 Reactions to impurities
Reactions to impurities generally refer to the effect of breakdown products of the active ingredient, which may be initiated by interactions with moisture or acidity or with excipients or materials from containers and packaging. The manufacturing process may contribute some impurities, although these will be limited by the marketing authorisation, but the validity of these requirements and limitations will depend on the same route of synthesis and production being adhered to.
12.11.1 Heparin
In January 2008, Baxter Healthcare Corporation withdrew batches of nine lots of heparin sodium in the USA26 because around 700 acute allergic-type adverse reactions had been reported after their use. The number of deaths was 19. The source of the active ingredient for this product was the Scientific Protein Laboratories (SPL) in Changhzou, China. The US Food and Drug Administration found that the heparin batches associated with the reactions contained 5–20% of a heparin-like compound as a contaminant. |

In the case above the contaminant was identified as over-sulfated chondroitin sulfate27 (Fig. 12.8). More than 80 adverse reactions were also reported in Germany with products where the heparin was also sourced from China, although from another company.
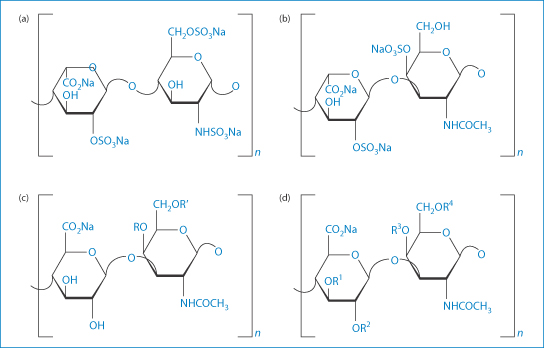
Figure 12.8 Structural formulae of (a) heparin; (b) dermatan sulfate; (c), chondroitin sulfate A and C; and (d) oversulfated chondroitin sulfate (OSCS). For chondroitin sulfate A, group R represents the sulfated moiety, as for chondroitin sulfate C the residual group R′ is sulfated. For OSCS, R1–R4 label possibly sulfated moieties. The dermatan in heparin preparations is a signal of poor purification methods.
Reproduced from Beyer T et al. Quality assessment of unfractionated heparin using 1H nuclear magnetic resonance spectroscopy. J Pharm Biomed Anal 2008;48:13–19. Copyright Elsevier 2008.
This example illustrates that the final product safety is compromised if the raw material has been sourced from an unreliable supplier. In-house testing of the incoming material should have detected that there was a problem, given the high percentage of the impurities, but it is difficult to cater for previously unknown impurities.
12.11.2 Hyaluronic acid
Hyaluronic acid (HA) is used in osteoarthritis as a synovial fluid supplement (see Chapter 7). It is also used in ophthalmic surgery. In vivo metabolic degradation of HA by hyaluronidase is one of the determinants of its biological half-life. HA can be sourced from bacteria, from rooster combs or from human umbilical cord, and the resulting HA products will contain different impurities.28 Several commercially available products in one study did not degrade on digestion with hyaluronidase, clearly indicating that biological half-life will not be the same. Vigilance is necessary and, where problems arise, the provenance of biological products must be determined. Sources matter, processing matters, purification matters and, of course, proving quality by the application of the highest-quality analytical technology is essential.
12.11.3 Oligomeric impurities in penicillins
Acute sensitivity to penicillins occurs in a minority of patients, but the reaction is well known in practice. Nevertheless, the source of at least some of the adverse reactions is not always appreciated. Impurities in penicillins have been blamed for sensitivities and allergies to this class of drug. The number of impurities found in samples of amoxicillin (VII) is quite remarkable.29
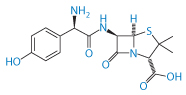
Structure VII Amoxicillin
Such impurities may include 2-hydroxy-3-(4-hydroxyphenyl)pyrazine, 4-hydroxyphenylglycine, 4-hydroxyphenylglyclamoxicillin, 6-aminopenicillanic acid, amoxicilloic acid and seven others, not including the dimer and the trimer (Fig. 12.9). These latter and other oligomers are possibly prime culprits in penicillin allergies as they are formed with peptide bonds and resemble peptides.
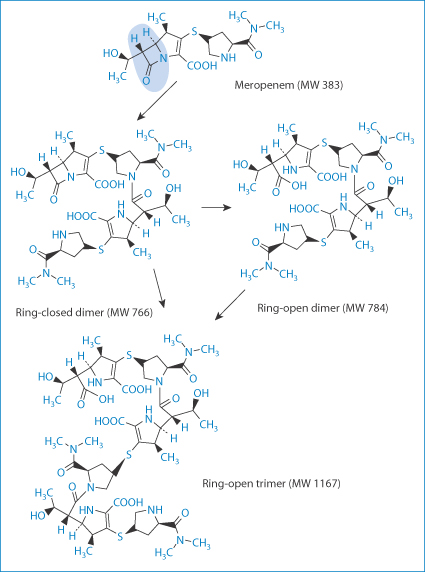
Figure 12.9 Ring-opened and ring-closed dimers and a ring-open trimer. The ring in question, the β-lactam ring, is highlighted.
12.11.4 Contamination from containers
Sensitisation of the skin to topical products may not always be the result of either the drug or of the excipients. There is the potential for contamination by substances that have leached from the containers. The aluminium tube is still the most widely used container for topical creams and ointments; the tubes are lacquered to prevent direct interaction between product and aluminium. Epoxy resins are used as the protective layer in most cases, particularly bisphenyl A diglycidyl ether (BADGE)-based resins. Leaching of BADGE and its congeners depends to an extent on the formulation and the mechanical stresses applied to the tubes.30
Adverse effects due to these leachables have not been assessed but they are a potential cause, for example, of contact dermatitis.
Figure 12.10 shows the time scale of the leaching process from poly(vinyl chloride) bags into intravenous etoposide formulations.
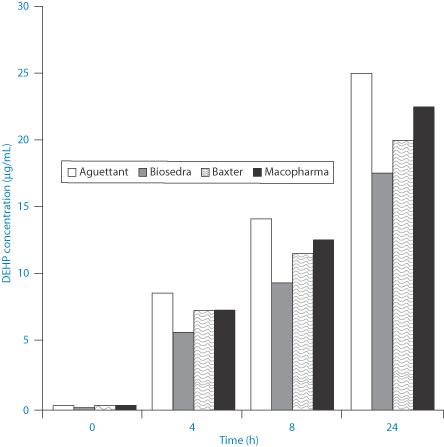
Figure 12.10 Levels of di(2-ethylhexylphthalate) (DEHP) (μg mL–1) leached from bags of intravenous etoposide from different manufacturers, as shown.
Reproduced with permission from J Clin Hosp Pharm 2002;27:139–142.
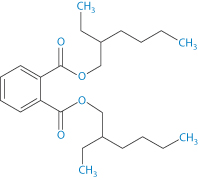
Structure VIII Di(2-ethylhexylphthalate) (DEHP)
Di(2-ethylhexylphthalate) (DEHP) (VIII) has a log P (octanol/water) of 7.5 and an aqueous solubility of 3 μg L–1. Hence lipids and surfactants in formulations will encourage the transfer from the bags to the contents.
12.11.5 Delivery devices and materials
There is an increasing number of devices and biomaterials that are used to deliver drugs, genes and vaccines. Issues can arise with the materials used in such devices, which include syringes and giving sets, or with the manner in which the device as a whole behaves (system plus active material). There might also be technical failure of a pump or reservoir, or conditions in the device that destabilise the drug, especially if it is a protein.
12.11.6 Insulin pumps
One example of problems that arise through system–drug interactions is the phenomenon of protein aggregation in insulin pumps,31 as aggregation is accompanied by a significant loss of biological activity. Various problems have been encountered with insulin pumps: obstruction in the infusion set or leakage at the infusion set connection or from the infusion site. Insulin aggregation (when insulin fibrils form) can also occur owing to agitation of the pump during wear and to temperature fluctuations. The stability is influenced by the type of insulin, the solvent and concentration.32 Metal-ion contamination has also been implicated and the use of ethylenediaminetetraacetic acid has been recommended to sequester ions in formulations.33 Insulin aggregates will at times block delivery channels.
Figure 12.11 illustrates the different deposition patterns of insulin in solution following administration by pen injector, a jet injector and an external insulin pump. The diffusion of insulin from sites of administration is important for its activity; retention of insulin and its degradation at the site of deposition might reduce biological activity. Pharmacokinetic differences between these modes of administration might also be found.
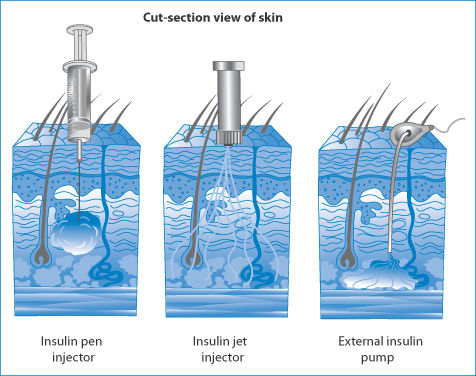
Figure 12.11 Different distribution patterns after administration of insulin by injector pen, jet injector and external insulin pump.
Reproduced from http://adam.about.com/care/diabetes.
In one study of bovine zinc-insulin, aggregation occurred only when there was both agitation and hydrophobic surfaces.34
Other proteins also degrade physically in pumps: interleukin-2 can lose 90% of its activity over a 24-hour period of infusion. Adsorption, which is a precursor of aggregate formation in some insulin systems, does not occur with interleukin-2, but irreversible structural changes can occur.35
Adsorption of proteins on to solid surfaces is a well-known phenomenon. Insulin adsorbs to glass, so that in low concentrations drug can be lost. Figure 12.12 shows the rate at which AspB25 insulin adsorbs on to a silanated silica wafer surface.36 At the higher concentrations adsorption is rapid, as one might expect from diffusion-controlled kinetics. One technique to avoid loss of insulin by adsorption is to add small amounts of albumin to the infusion as the albumin adsorbs first and inhibits further adsorption of the active.
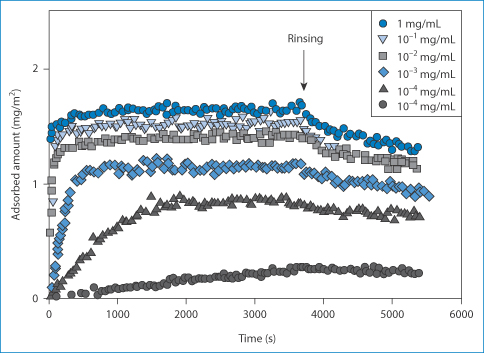
Figure 12.12 Adsorption on to silanised silica surfaces of AspB25 insulin, showing the influence of concentration.
Reproduced from Mollman SH et al. Adsorption of human insulin and AspB28 insulin on a PTFE-like surface. J Colloid Interface Sci 2005;286:28–35. Copyright Elsevier 2005.
12.11.7 Drug-eluting stents
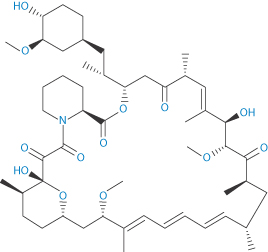
Drug-eluting stents have made an impact in the treatment of restenosis – the proliferation of smooth-muscle cells in response to pressure from foreign objects, such as during balloon angioplasty. So, while the stent opens the artery flow, it simultaneously diminishes the stenosis. Hence, such stents have been considered a breakthrough, acting to reduce the need for repeat revascularisation after bare-metal stenting.37 The first drug-eluting stents to appear were those with non-biodegradable (or ‘durable’) polymer coats as the drug reservoir. There has been debate about whether the so-called ‘durable’ and biodegradable coated stents are equivalent therapeutically, as there have been concerns that the former may induce inflammation. Biodegradable polymers on stents were suggested to be safer because, when degraded, the bare-metal stent is left, reducing any inflammation caused by the polymer.** Of course, some adverse effects might be due to the drug used. Among those incorporated are paclitaxel, and the antiproliferative macrolide lactams sirolimus (IX), everolimus and zotarolimus (X).
Sirolimus is formulated in a controlled-release polymer coating following coronary intervention. Several large clinical studies have demonstrated lower restenosis rates in patients treated with sirolimus-eluting stents when compared to bare-metal stents, resulting in fewer repeat procedures. A sirolimus-eluting coronary stent has been marketed by Cordis, a division of Johnson & Johnson, under the trade name Cypher. Figure 12.13 shows the proximity of the polymer drug layer on a drug-eluting stent to the tissues being treated, hence the importance of both polymer and drug in beneficial and adverse effects.
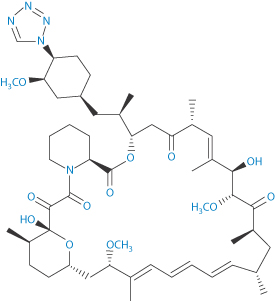
Structure IX Sirolimus Structure X Zotarolimus
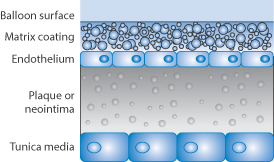
Figure 12.13 Diagram showing the proximity of the polymer drug layer on the stent surface to the endothelial cells, the plaque or neointima.
Resistance of the stent to withdrawal has been ascribed to the ‘stickiness’ of the stent, perhaps due to friction between the stent delivery balloon and the drug–polymer coating on the stent. The foregoing emphasises the complex nature of the development of such devices and the need for postmarketing surveillance.
12.11.8 Catheters and infection
Intravascular catheter-related infections can be an important source of blood stream infections in patients who are critically ill.38 It is said that more than 250 000 vascular catheter-related bacterial or fungal infections occur each year in the USA alone, with mortalities in critically ill patients of up to 25%. Bacteria can adhere to and form biofilms on catheter surfaces (see Fig. 12.14). Preventive stratagems include cutaneous antisepsis, use of sterile barriers during insertion, application of chlorhexidine-impregnated sponges and the use of antimicrobial, antibiotic-coated or silver-impregnated catheters, which depend generally on antimicrobial polymer coats, both on the external and internal surfaces of the catheters. Antimicrobial polymers are discussed in Chapter 7 and these bear many similarities to polymer therapeutic systems.
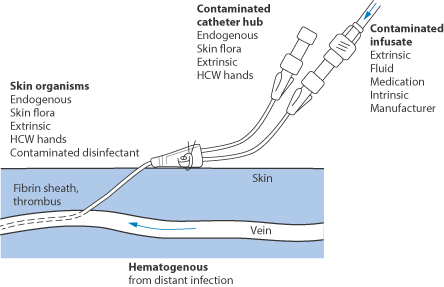
Figure 12.14 Points of entry of bacteria to an indwelling catheter. HCW = health care worker.
Urinary catheters also suffer from bacterial film formation. There are several possible points of entry of bacteria in such systems, as seen in Fig. 12.15. This is clearly an area of great importance, but some of the issues are outside the scope of this book. What is clear is that antibiotic-coated catheters are drug delivery devices and that the bacterial biofilms that form on catheters are the result of bacterial adhesion processes.
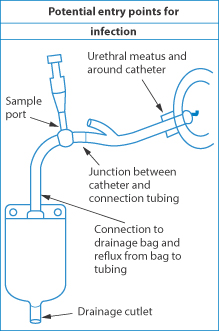
Figure 12.15 Diagram of the arrangement of a urinary catheter, with sources of infection indicated by arrows.
Bacterial adhesion
Bacteria adhere to solid surfaces and in so doing cause many problems. Bacterial hydrophobicity can be correlated with the adhesion of bacteria to experimental surfaces such as negatively charged polystyrene. Different bacteria have different hydrophobic properties. Contact angles of water with bacterial surfaces correlate with their hydrophobicity: the higher the contact angle, the higher the hydrophobicity and the greater the potential for adsorption.39 Different strains of bacteria, e.g. of Pseudomonas, behave differently, as can be seen in Table 12.7. However, it is generally unlikely that only one parameter correlates with bacterial adsorption on to a wide range of surfaces, which are not always smooth, as in experimental systems. Nonetheless, knowing the surface properties of bacteria and of the catheter material can be valuable in understanding problems.
Table 12.7 Contact angles of water on different Pseudomonas spp.
Strain number and name | Contact angle (°) |
Pseudomonas fluorescens | 21.2 |
Pseudomonas aeruginosa | 25.7 |
Pseudomonas putida | 38.5 |
Pseudomonas sp. strain 26-3 | 20.1 |
Pseudomonas sp. strain 52 | 19.0 |
Pseudomonas sp. strain 80 | 29.5 |
Values for other organisms can range up to 60° for an Arthrobacter sp. strain 177 and 70° for a Corynebacter sp. strain 125.
Cutaneous antisepsis is clearly demanded using povidone-iodine, for example, and by maximising sterile barriers. Several other approaches are in use38:
- chlorhexidine-impregnated sponges (placed over site of insertion)
- antibacterial catheter locks
- antimicrobial catheters:
antiseptic catheters
antibiotic-coated catheters
silver-impregnated catheters.
Biofilms
Biofilms built by accumulation of bacteria are complex structures (Fig. 12.16), which makes access of antimicrobial and antibiotic agents difficult (similar to the difficulties encountered with the hindered delivery of drugs to tumours, discussed in Chapter 14). As with diffusion of solutes into any complex structure, like porous media, the architecture of the structure matters. One of the most important features in the control of access of antibiotics and other solutes is their diffusion into the biofilm, as Stewart40 emphasised, pointing out that such diffusion can be summarised as follows.
- Diffusion is the predominant solute transport process within cell clusters.
- The time scale for diffusive equilibration of a non-reacting solute will range from a fraction of a second to tens of minutes in most biofilm systems.
- Diffusion limitation readily leads to gradients in the concentration of reacting solutes and hence to gradients in physiology.
- Water channels can carry solutes into or out of the depths of a biofilm, but they do not guarantee access to the interior of cell clusters.
Studies of the relative diffusion coefficient of small solutes such as sucrose shows that the ratio Dbiofilm/Do, (where Dbiofilm and Do are the diffusion coefficients in the biofilm and in water respectively) falls to around 0.2. The poor penetration and thus the action of several antibiotics are evidenced in the clinical point below.
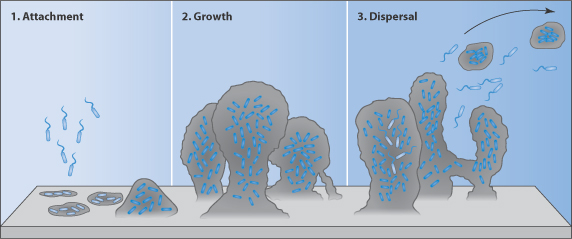
Figure 12.16 The biofilm life cycle. (1) Free-floating bacteria encounter a submerged surface and within minutes can become attached. They produce extracellular polymeric substances (EPS) and colonise surfaces. (2) EPS production allows the emerging biofilm community to develop a complex, three-dimensional structure. Biofilm communities can develop within hours. (3) Biofilms can propagate through detachment of small or large clumps of cells, or by a type of ‘seeding dispersal’ that releases individual cells.
Reproduced from: http://www.hypertextbookshop.com/biofilmbook/v004/r003/index.html; used with permission from Montana State University Center for Biofilm Engineering.
|
‘Biofilm-related infections remain a scourge’, states Siala and colleagues.41 In an in vitro model of biofilms using Staphylococcus aureus reference strains, delafloxacin and daptomycin were found to be the most active among the antibiotics from eight different pharmacological classes. At clinically meaningful concentrations, delafloxacin, daptomycin and vancomycin caused a ≥25% reduction in viability of the biofilms studied, respectively. The antibiotic penetration within the biofilms ranged from 0.6 to 52% for delafloxacin, 0.2 to 10% for daptomycin and 0.2 to 1% for vancomycin; for delafloxacin, this was inversely related to the polysaccharide proportion in the matrix. Six biofilms were acidic, explaining the high potency of delafloxacin (which has a lower minimum inhibitory concentration at acidic pH). |

12.11.9 Transdermal patches as devices
Transdermal patches can be considered to be devices. Problems that have arisen with these include adverse reactions to the adhesive used to adhere the patch to the skin. Allergic contact dermatitis from hydroxymethylcellulose has been reported with an estradiol patch.42 Mishaps related to use of transdermal patches include many involving patches falling off under a variety of conditions, including subsequent adhesion to another person! Other events include swelling and itching at the application site; too strong adhesion, leading to pain on removal; excessive adhesion of the plastic backing to the adhesive layer, leading to tearing of the patch; inflexible patches that do not flex with the skin; and cases of drug crystals being seen on patches.43 Cases of burning have been reported when medicated patches containing even small amounts of aluminium are worn during magnetic resonance imaging, as a result of overheating in the area of the patch.44
12.12 Crystallisation
Figure 12.17 summarises some of the situations in which the solid state is important clinically. Highlighted are crystalluria, gout, the precipitation of drugs before or after injection, inhalation therapy and understanding the potential toxic effects of particulates.
A case of crystalluria45 illustrates one of the propositions put forward in this book. The case concerned a 60-year-old man infected with HIV whose medications included efavirenz, emtricitabine, tenofovir and pravastatin sodium: a heady cocktail. Two hours after he had received aciclovir, his urine became cloudy and white in the proximal part of a Foley catheter. Microscopic analysis showed birefringent needle-like crystals ‘consistent with the precipitation of acyclovir [aciclovir]’, as shown in Fig. 12.18. |

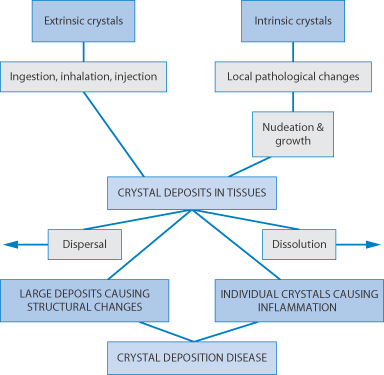
Figure 12.17 Potential clinical issues with crystal forms of drugs.
Additional treatment with intravenous aciclovir did not result in urinary crystallisation of the drug. Aciclovir (pKa values: 2.27 and 9.25) has a solubility in water at 25°C of >100 mg mL–1. At physiological pH, aciclovir sodium is un-ionised and has a minimum solubility in water (at 37°C) of 2.5 mg mL–1 (Fig. 12.18b).46 The concentration of aciclovir in human urine after oral administration of 200 mg reaches 7.5 μg mL–1,47 clearly not exceeding its aqueous solubility. As urine is concentrated as it passes along nephrons, urine drug concentrations increase. Determination of saturation of drugs in urine is more predictive of problems.
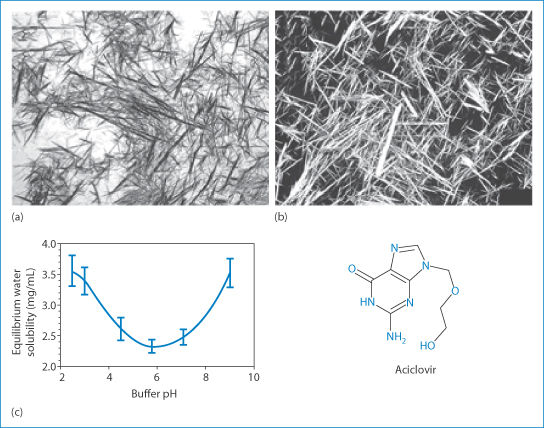
Figure 12.18 (a, b) Photomicrographs of aciclovir crystals harvested from the urine of the patient in question (a), and a pure sample (b). (c) The solubility–pH relationship for aciclovir is shown along with its chemical structure.
(a and b) Reproduced from reference 45.
(c) Reproduced from reference 46.
This case illustrates that a knowledge of the solution properties of drugs and the pH at which drugs might precipitate or become saturated in body fluids and compartments is essential if we are to make contributions to patient care using our unique knowledge. Knowledge of the solution properties of drugs is of course applied directly in the formulation and delivery of intravenous mixtures of drugs or when drugs are added to infusion fluids.
Some time ago an editorial in the Lancet48 discussed the topic of crystals in joints. It pointed out that it is not only in gout that crystals (of monosodium urate monohydrate, or of calcium pyrophosphate in pseudogout) appear but that calcium hydroxyapatite deposits cause apatite deposition disease. The discussion indicated that synovial fluid may contain pieces of cartilage, strands of fibrin, cholesterol crystals and, in some patients, steroid crystals remaining after intra-articular injection. Biological systems are of course complex. Urine and blood are more complex than water, so simple theories of solution properties cannot be applied directly, especially when we introduce formulations into already multicomponent environments. Nevertheless, theory and equations give clues as to what might be happening in vivo. Without them we only guess.
12.13 Abnormal bioavailability and adverse events
Classical examples where the bioavailability has changed after a period when patients have been stabilised and physicians accustomed to responses from a particular dose form or brand include that of digoxin (Lanoxin). In the 1970s a change in the manufacturing process led to a marked difference in bioavailability through an increase in particle size, resulting from a change in the processing of the drug. Another example was when a change in the main excipient from calcium carbonate to lactose in a brand of phenytoin sodium led to a marked increase in bioavailability and overdosing. The calcium was clearly forming a poorly soluble calcium salt of the drug, decreasing its intrinsic solubility and its rate of dissolution. Its removal led to faster absorption and toxic effects in titrated patients. Other examples of formulation-related effects are discussed in Chapters 8 and 9.
12.13.1 Testing for adverse effects
How can pharmacists become more involved in searching for the causes of adverse effects and reactions related to dosage forms and devices? This depends on:
- a thorough knowledge of the nature of the ingredients and their purpose in the medication concerned
- understanding the nature of the drug substance and its likely interactions with excipients
- understanding the structure of systems such as controlled-release preparations
- carrying out straightforward tests to investigate potential problems
- improving the scoring of severity and adopting more quantitative approaches to this.
One example derives from work between the University of Strathclyde and the Contact Dermatitis Unit at Belvidere Hospital, Glasgow, some years ago, where modes of quantitative measurement of skin reactions to substances applied to the skin were explored. The aim was to replace more subjective (–, +, ++, +++) clinical scoring of effects. Contact dermatitis is a (skin) reaction resulting from exposure to allergens (allergic contact dermatitis) or irritants (irritant contact dermatitis). Phototoxic dermatitis occurs when the allergen or irritant is activated by sunlight. Contact dermatitis can occur from contact with jewellery but also from drugs and devices (e.g. transdermal patch adhesives).
Contact dermatitis testing
Suspected contact dermatitis is usually tested for by application of a series of patches containing putative causative agents. The formulation of these materials has often been fairly crude (for example, by dispersing nickel sulfate in a paraffin base) and this might affect the outcome. Poor attention to pharmaceutical principles of release, poor choice of vehicle and lack of consideration of particle size all contribute to imprecision.49 Proprietary patch formulations are available. Test results are often evaluated against a control or controls by clinical scoring of reactions from severe (+++), through (++), (+), 0 and equivocal (<+). Skin reflectance measurements were evaluated as a measure of skin haemoglobin content and correlated well with clinical scoring.50 The following is an example of a study conducted some time ago.49
A group of 43 patients with a clinical history of nickel allergy who exhibited an equivocal or no allergic reaction to a patch test at 48 hours were further challenged using several different formulations of nickel sulfate. This experimental test battery comprised aqueous, dimethyl sulfoxide and propylene glycol solutions of nickel sulfate, and nickel sulfate incorporated into Cetomacrogol cream and yellow soft paraffin (Paraffin Molle Flavum: PMF). Although some of these vehicles were irritant, a formulation-dependent test response was observed, such that in terms of the number of responses per unit weight of nickel sulfate applied to the skin, the vehicles could be ranked: dimethyl sulfoxide > propylene glycol > aqueous solution > Cetomacrogol cream > PMF preparations. This ranking could be correlated with the relative ease with which nickel sulfate could be dialysed from each vehicle in vitro. This study demonstrates that, for nickel sulfate, the vehicle can influence the outcome of patch testing, apparently by modifying the quantity of nickel released into the skin for elicitation of the allergic response. (Cetomacrogol is a non-ionic surfactant with a C16 hydrocarbon chain and an average 24-unit polyoxyethylene oxide hydrophilic chain.)
Release of nickel sulfate from the test preparations varied considerably, as shown in Fig. 12.19. The correlation between skin blood flow (plotted in Fig. 12.20 as the haemoglobin index) and clinical scoring is good. The response is often a weal which reflects increased blood flow and, of course, heat output. Infrared thermography has also been used.51 This is a convenient non-invasive technique that employs an infrared camera and can be used to discriminate between irritant and allergic responses and to quantify the latter.
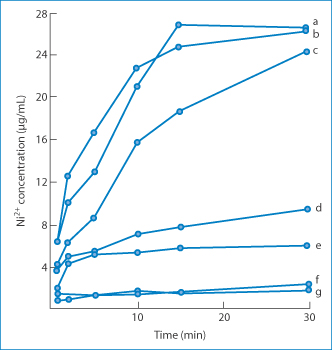
Figure 12.19 In vitro release of nickel sulfate from various vehicles at 37°C using a dialysis technique. Curve a, an aqueous solution of nickel sulfate (NiSO4); curve b, propylene glycol solution; curve c, dimethyl sulfoxide (DMSO) solution; curve d, Cetomacrogol cream; curve e, 2.5% NiSO4 in yellow soft paraffin (proprietary formulation); curve f, yellow soft paraffin; curve g, 5% NiSO4 in yellow soft paraffin (proprietary formulation).
Reproduced from Mendelow AY et al. Patch testing for nickel allergy. The influence of the vehicle on the response rate to topical nickel sulphate. Contact Dermatitis 1985;13:29–33. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
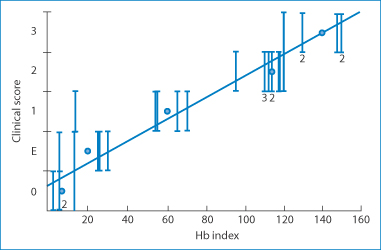
Figure 12.20 Correlation between clinical scores of patient responses to a variety of proprietary test patches and the haemoglobin (Hb) index derived from a skin reflectance technique. The individual test patches contained separately nickel, chromium, colophony fragrances, thiomersal, benzoic acid, parabens, neomycin and a range of other materials implicated in contact dermatitis.
Reproduced from Mendelow AY et al. Skin reflectance measurements of patch test responses. Contact Dermatitis 1986;15:73–78. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Figure 12.21 shows some results after application of the contact dermatitis test patches, both the appearance of the reactions to different formulations and the results when assessed using the infrared camera. The infrared thermograms, which detect the increased blood flow and heat profiles, show not only the intensity but also the spread of the reaction on the skin.
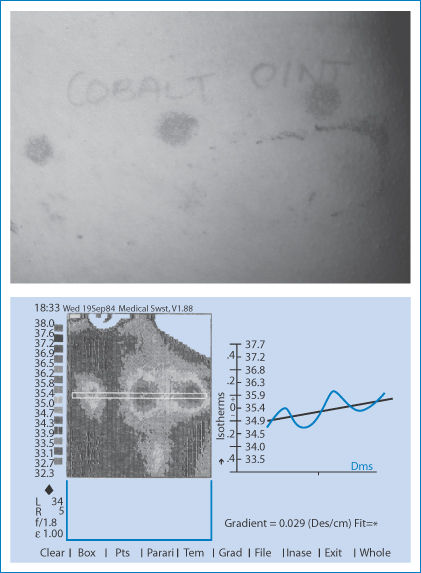
Figure 12.21 (a) Typical appearance of positive reactions to contact with patch tests on the back of a subject. (b) An infrared thermogram of the back of a (different) patient who has been assessed for contact dermatitis to nickel with three formulations of nickel sulfate. The image shows not only differences in intensity but also difference in the spread of the response. These thermograms are more informative than physicians’ scoring systems.
The simple in vitro tests for the disintegration behaviour of emopronium bromide tablets discussed earlier in this chapter illustrate clearly that investigation does not necessarily involve expensive equipment. Dissolution testing can also show differences or confirm patients’ concerns about generic products, or perhaps detect counterfeit medicines emanating from internet suppliers.
Mechanisms of toxicity of nanosystems are discussed in Chapter 14. Undoubtedly, new adverse effects will arise from the use of nanosytems. Much has still to be learned about the potential downsides of nanomedicines and assurances on the safety of the materials from which they are made are not always sufficient to instill confidence in the complete safety of the nanoparticles. Because of the vast array of possible systems, it is impossible to generalise. Each system will have to be examined and tested on its merits.
The last section deals with light-induced reactions, either following the administration of a drug such as a fluoroquinolone or through photodynamic therapy (PDT). This section has a specific focus on light but it illustrates the multifaceted nature of reactions to drugs and formulations.
12.14 Photochemical reactions and photoinduced reactions
First, it is essential to define a variety of light-induced effects. These include:
- photoallergy: an acquired immunological reactivity dependent on antibody- or cell-mediated hypersensitivity
- photosensitivity: a broad term used to describe an adverse reaction to light after drug administration, which may be photoallergic or phototoxic in nature
- phototoxicity: the conversion of an otherwise non-toxic chemical or drug to one that is toxic to tissues after absorption of electromagnetic radiation
- photodynamic effects: photoinduced damage requiring the presence of light, photosensitiser and molecular oxygen
- PDT: therapy in which photoactive drugs inactive in the unexcited state are administered and activated at particular sites in the body.
Many drugs decompose in vitro after exposure to light, but the consequences depend on the nature of the breakdown products (Fig. 12.22). Some derivatives of nifedipine have a very short photochemical half-life, sometimes of the order of a few minutes, while others decompose only after several weeks’ exposure. A drug may not decompose after exposure to light, but may be the source of free radicals or of phototoxic metabolites in vivo. Adverse reactions occur when the drug or metabolites are exposed to light and the absorption spectrum of the drug coincides with the wavelength of light to which it is exposed (the wavelengths of ultraviolet-A (UV-A) are 320–400 nm; of UV-B 280–320 nm; and of UV-C 200–290 nm). To behave as a photoallergen, a drug or chemical must be able to absorb light energy present in sunlight and on absorption of the light generate a chemical species capable of binding to proteins in the skin, either directly or after metabolism.52
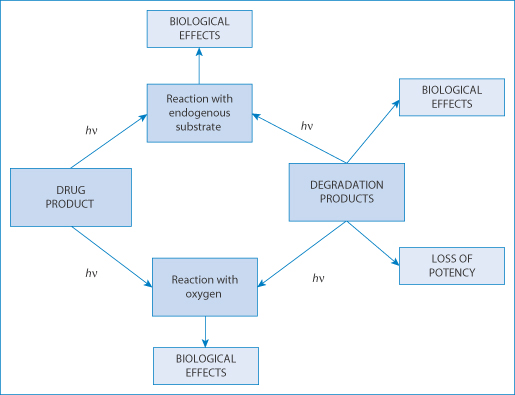
Figure 12.22 The consequences for a light-sensitive drug of interaction with light energy, causing biological effects and loss of potency depending on the compounds involved.
Redrawn with permission from Tønneson HH (ed.). Photostability of Drugs and Drug Solutions. London: Taylor and Francis; 1996.
12.14.1 How do photosensitisers work?
In PDT, drugs that can be activated by light are administered intravenously. The drug remains inactive until exposed to light with a wavelength that can penetrate the skin but is not completely attenuated by the blood. Most photosensitive drugs respond best to blue or green light in vitro, but these wavelengths can pass through only a thin layer of human skin. Red blood cells absorb blue and green light, making it impossible for most photosensitisers to work in deep or bloody places. Texaphyrins, on the other hand, respond best to a specific red light that passes through blood. Light energy is directed to the required site through a fibreoptic device. When activated, the drug – usually a porphyrin derivative – creates oxygen radicals that destroy tissue in its vicinity. Principal side-effects of drugs like Photofrin have a skin sensitivity to light for up to 6 weeks as the drug is available systemically and partitions into lipid layers.
Other drugs cause light sensitivity; these are those that deposit in the skin and either interact with light or degrade to form coloured complexes. Coloration is not always a sign of pathological change.
Temoporfin (XI: Foscan) and porfimer sodium (XII: Photofrin) are used in PDT of various tumours. Activated by laser light they produce a cytotoxic effect in the tissues in which they accumulate. Temoporfin is licensed in the UK for the therapy of advanced head and neck cancers, while porfimer sodium is used in the PDT of non-small-cell lung cancer and oesophageal cancer.
|
The cautions given in the British National Formulary for porfimer are to ‘avoid exposure of the skin to direct sunlight or bright indoor light for at least 30 days’, and for temoporfin ‘for at least 15 days’, but to avoid prolonged exposure of the injection site to direct sunlight for 6 months after administration. |

Structures XI and XII Temoporfin and the oligomer (n = 0–6) porfimer sodium
12.14.2 Chemical photosensitivity
In chemical photosensitivity, patients develop redness, inflammation and sometimes brown or blue discoloration in areas of skin that have been exposed to sunlight for a brief period. This reaction occurs after ingestion of drugs, such as tetracycline, or the application of compounds topically in consumer products such as perfumes or aftershaves. These substances (Table 12.8) may make some skin types more sensitive to the effects of UV light. Some develop hives with itching, which indicates a type of drug allergy triggered by sunlight.
Table 12.8 Some substances that sensitise skin to sunlight
Type | Examples |
Anxiolytics | Alprazolam, chlordiazepoxide |
Antibiotics | Quinolones, sulfonamides, tetracyclines, trimethoprim |
Antidepressants | Tricyclics |
Antifungals (oral) | Griseofulvin |
Antihypertensives | Sulfonylureas |
Antimalarials | Chloroquine, quinine |
Antipsychotics | Phenothiazines |
Diuretics | Furosemide, thiazides |
Chemotherapeutics | Dacarbazine, fluorouracil, methotrexate, vinblastine |
Antiacne drugs (oral) | Isotretinoin |
Cardiovascular drugs | Amiodarone, quinidine |
Skin preparations | Chlorhexidine, hexachlorophene, coal tar, fragrances, sunscreens |
Problems have been reported during a clinical trial with Foscan. This contains temoporfin as its active ingredient. In the trial it was alleged that a high proportion of patients suffered burns.53 The manufacturer of the product complained that the results were at odds with more extensive trials of the product.54 It subsequently emerged that in the trials the formulation of the product was different from that of the marketed product. It was claimed that a new solvent had been added so that the drug would be more soluble and less painful to administer. Here was the scenario: trial data are disputed, there is some confusion or inadequate reporting of the formulation used (there are many instances of this, including the problems that arise with the different formulations of amphotericin) and an underlying effect of the active substance, a porphyrin. Later, two of the physicians involved in the trial55 admitted that there may have been a connection between the leakage and the effect of the solvent as the active agent had spread from the point of administration. The adverse events were not due to extravasation injury itself as they occurred only after photoactivation.56 This is a good example of the interlocking effects of drug, formulation and the nature of clinical trials, where the influence of the formulation was underestimated or neglected. |

12.15 Conclusions
Detecting the causes of adverse events is not an easy task, especially when drugs or drug products are novel. It is essential that one looks for analogies, reliable reports on closely chemically and pharmacologically related drugs and similar formulations. This requires a breadth of knowledge of similar events recorded in the literature or from experience. The few examples noted in this chapter may assist in asking the right questions. The concluding illustration (Fig. 12.23) summarises how one might approach the issues. A fuller record of adverse events and the involvement of delivery systems can be read: the older literature should not be discarded as there may be clues there which might have relevance to new medicines and treatments.
Potential mechanisms should be proposed and the following questions asked:
- Is the event due to the drug?
- Is the event linked to the use of another medication?
- Is it an excipient effect?
- Is it a vehicle or matrix effect?
- Is it a result of the physical nature of the dosage form?
- Is it the result of concomitant pathologies?
- Is it unrelated to the medication?
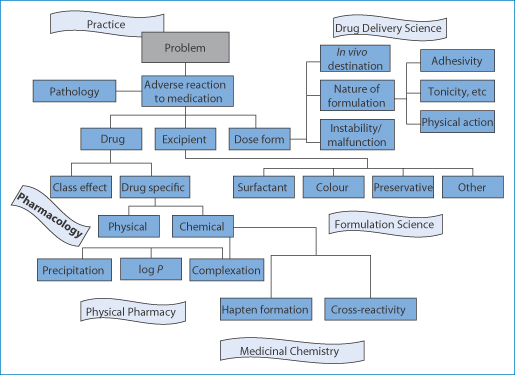
Figure 12.23 Charting the causes of adverse reactions to medications. The many causes are shown in relation to drug, excipient, nature of formulation, the dosage form or device and whether there are physical or chemical causes.
The determination or, better, the prediction of potential adverse reactions is an important task. It requires constant attention to the literature, a good knowledge of the chemistry of offending drugs, the influence of the formulation, determining the mechanisms if possible, and certainly looking for trends and patterns. It is a fascinating and vital subject. Nearly all the topics raised here can be the subject of chapters in themselves.
References
1. Uchegbu IF, Florence AT. Adverse drug events related to dosage forms and delivery systems. Drug Saf 1996;14:39–67.
2. Balbani APS et al. Pharmaceutical excipients and the information on drug labels. Rev Bras Otorinolaringol 2006;72:400–406.
3. Millar JS. Pitfalls of ‘inert’ ingredients. Br J Gen Pract 2001;51:570.
4. Cubitt GT. Pitfalls of ‘inert’ ingredients. Br J Gen Pract 2001;51:756.
5. Pifferi G, Restani P. The safety of pharmaceutical excipients. Il Farmaco 2003;58:541–550.
6. Seidenari S et al. Cross-sensitisations between azo dyes and para-amino compound: a study of 236 azo-dye-sensitive subjects. Contact Dermatitis 1997;36:91–96.
7. ten Tije AJ et al. Pharmacological effects of formulation vehicles: implications in cancer chemotherapy. Clin Pharmacokinet 2003;42:665–685.
8. Phillips DM, Zacharopoulous VR. Nonoxynol-9 enhances rectal infection by herpes simplex virus in mice. Contraception 1998;57:341–348.
9. Smith CM et al. Pluronic F-68 reduces the endothelial adherence and improves the rheology of liganded sickle erythrocytes. Blood 1987;69:1631–1636.
10. Orringer EP et al. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease. JAMA 2001;286:2099–2106.
11. Lee R. Pipe dream or paradigm shift? University of Chicago Magazine 2006;98(3). Available online at: magazine.uchicago.edu/0602/features (accessed 24 March 2015).
12. Medicine.net.com. Pleurisy. Available online at: http://www.medicinenet.com/pleurisy/article.htm (accessed 20 September 2009).
13. Aelony Y. Talc pleurodesis and acute respiratory distress syndrome. Lancet 2007;369: 1494–1496.
14. Marchi E et al. Talc for pleurodesis. Hero or villain? Chest 2003;124:416–417.
15. Ferrar J et al. Influence of particle size on extrapleural talc dissemination after talc slurry pleurodesis. Chest 2002;1221018–1027.
16. Bogman K et al. The role of surfactants in the reversal of active transport mediated by multidrug resistance proteins. J Pharm Sci 2003;92:1250–1261.
17. Hu Z et al. A novel emulsifier, Labrasol enhances gastrointestinal absorption of gentamicin. Life Sci 2001;69:2899–2810.
18. Singh MK et al. Another reason to dislike medication. Lancet 2008;371:1388.
19. Levy DJ. An aspirin tablet and a gastric ulcer. N Engl J Med 2000;343:863.
20. Florence AT, Salole EG (eds). Formulation Factors in Adverse Reactions. London: Wright; 1990.
21. Holly FJ. Formation and rupture of the tear film. Exp Eye Res 1973;15:515–525.
22. Njobuenwu DO. Spreading of trisiloxanes on thin water film: dry spot profile. Leonardo J Sci 2007;6:165–178.
23. Lawrenson JG, Murphy PJ. The neonatal tear film. Contact Lens Anterior Eye 2003;26:197–202.
24. McMonnies CW. Incomplete blinking: exposure keratopathy, lid wiper epitheliopathy, dry eye, refractive surgery and dry contact lenses. Contact Lens Anterior Eye 2007;30:37–51.
25. Firestone BA et al. Solubility characteristics of three fluoroquinolone ophthalmic solutions in an in vitro tear model. Int J Pharm 1998;164:119–128.
26. WHO Drug Information 2008;22:84–85.
27. Beyer T et al. Quality assessment of unfractionated heparin using 1H nuclear magnetic resonance spectroscopy. J Pharm Biomed Anal 2008;48:13–19.
28. Matsuno Y-K et al. Electrophoresis studies on the contaminating glycosaminoglycan in commercially available hyaluronic acid products. Electrophoresis 2008;29:3628–3635.
29. Jouyban A, Kenndler E. Impurity analysis of pharmaceuticals using capillary electromigration methods. Electrophoresis 2008;9:3531–3551.
30. Haverkamp JB et al. Contamination of semi-solid dosage forms by leachables from aluminium tubes. Eur J Pharm Biopharm 2008;70:921–928.
31. Guilhem I et al. Technical risks with subcutaneous insulin infusion. Diabetes Metab 2006;32:279–284.
32. Hirsh IB et al. Catheter obstruction with continuous subcutaneous insulin infusion. Effect of insulin concentration. Diabetes Care 1992;15:593–594.
33. James DE et al. Insulin precipitation in artificial infusion devices. Diabetologia 1981;21:554–557.
34. Sluzky V et al. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc Natl Acad Sci USA 1991;88:9377–9381.
35. Tzannis ST et al. Irreversible inactivation of interleukin 2 in a pump-based delivery environment. Proc Natl Acad Sci USA 1996;93:5460–5465.
36. Mollman SH et al. Adsorption of human insulin and AspB28 insulin on a PTFE-like surface. J Colloid Interface Sci 2005;286:28–35.
37. Waksman R, Maluenda G. Polymer-drug eluting stents: is the future biodegradable? Lancet 2013; 378:1900–1902.
38. Raad I et al. Intravascular catheter-related infections: advances diagnosis, prevention and management. Lancet Infect 2007;7:645–657.
39. van Loosdrecht MCM et al. The role of the bacterial cell wall hydrophobicity in adhesion. Appl Environ Microbiol 1987;53:1893–1897.
40. Stewart PS. Diffusion in biofilms. J Bacteriol 2003;185:1485–1491.
41. Siala W et al. Antimicrob Ag Chemother 2014;58:6385–6397.
42. Schwartz BK, Glendinning WE. Contact Dermatitis 2006;18:106–107.
43. Wokovich AM et al. Transdermal drug delivery systems (TDDS) adhesion as a critical safety, efficacy and quality attribute. Eur J Pharm Biopharm 2006;64:1–8.
44. Lowry F. Medicated patch can cause burns during MRI, FDA warns. Medscape Medical News 2009. Available online at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm149537.htm (accessed 14 May 2009).
45. Mason WJ, Nickols HH. Crystalluria from acyclovir use. N Engl J Med 2008;358:e14.
46. Shojaei AH et al. Transbuccal delivery of acyclovir: I. In vitro determination of routes of buccal transport. Pharm Res 1998;15:1181–1188.
47. Testerecí H et al. The determination of acyclovir in sheep serum, human serum, saliva and urine by HPLC. East J Med 1998;3:62–66.
48. Editorial. Crystals in joints. Lancet 1980;1:1006–1007.
49. Mendelow AY et al. Patch testing for nickel allergy. The influence of the vehicle on the response rate to topical nickel sulphate. Contact Dermatitis 1985;13:29–33.
50. Mendelow AY et al. Skin reflectance measurements of patch test responses. Contact Dermatitis 1986;15:73–78.
51. Baillie AJ et al. Thermographic assessment of patch-test responses. Br J Dermatol 1990;122:351–360.
52. Barratt MD. Structure–activity relationships and prediction of the phototoxicity and phototoxic potential of new drugs. ATLA 2004;32:511–524.
53. Hettiaratchy S et al. Burns after photodynamic therapy. Br Med J 2000;320:1245.
54. (a) Bryce R. Burns after photodynamic therapy. Br Med J 2000;320:1731. (b) Dow R. Br Med J 2000;321:53.
55. Täubel J, Besa C. Burns after photodynamic therapy. Br Med J 2000;320:1732.
56. Hettiaratchy S, Clarke J. Burns after photodynamic therapy. Br Med J 2000;320:1731.
Further reading
Gonçalo M. Phototoxic and photoallergic reactions. Chapter 18. In Johansen JD et al. (eds) Contact Dermatitis. Berlin: Springer-Verlag; 2010.
Hatashi N et al. New findings on the structure-photosensitivity relationship and photostability of fluoroquinolones with various substituents at position 1. Antimicrob Agn Chemother 2004;48:799-803.
McCann MT, Gilmore BF, Gorman SP. Staphylococcus epidermidis device-related infections: pathogenesis and clinical management. J Pharm Pharmacol 2008;60:1551–1571.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


