Figure 8.1 Model of a biological membrane showing the different compositions of the interior and exterior sides. Glycolipids are seen on the outside, while phosphatidylserine and phosphatidylinositol predominate on the cytosolic side. See Fig. 8.2 for an impression of the membrane surface appearance.
Cholesterol is a major component of most mammalian biological membranes. Cholesterol forms complexes with phospholipids and its presence reduces the permeability of phospholipid membranes to water, cations, glycerol and glucose. The shape of the cholesterol molecule allows it to fit closely in bilayers with the hydrocarbon chains of unsaturated fatty acids (Fig. 8.1). Cholesterol condenses and rigidifies membranes without solidifying them. Its removal causes the membrane to lose its structural integrity and to become highly permeable. The flexibility of biological membranes is one of their important features, giving them their ability to re-form and to adapt to changed environments, a vital part of their function. The dynamic characteristics of biological membranes are due to their unique construction from small amphipathic molecules. On stretching (as with a soap film), the molecules at the surfaces become less concentrated in the bilayer but then are rapidly replenished from the bulk phase to maintain the original tension. That is, they are dynamic structures. Figure 8.2 shows a diagram of the ‘fluid mosaic’ model of a biological membrane.

Figure 8.2 Representation of the ‘fluid mosaic’ model of a biological membrane, showing embedded protein and protruding glycoproteins.
From Funhouse Films, with permission.
Although most of the data on permeation of non-electrolytes across biological membranes can be explained on the basis of the membrane behaving as a continuous hydrophobic phase, a fraction of the membrane may be composed of aqueous channels. These are effectively pores that offer a pathway parallel to the diffusion pathway through the lipid. In the absence of flow of water in one direction or the other, these pores play a minor part in the transfer of most drugs, although in the case of ions and charged drugs such as the quaternary ammonium compounds the pore pathway must be important. The fluid mosaic model of membranes allows the protein–lipid complexes to form either hydrophilic or hydrophobic ‘gates’ to allow transport of materials with different characteristics.
Overall, membrane permeability is controlled by the nature of the membrane, its degree of internal bonding and rigidity, its surface charge and the nature of the drug (solute) being transported. There are some similarities between solute transport in biological membranes and in synthetic membranes. As we have discussed in Chapter 7, the permeation of drugs and other molecules through hydrophobic membranes made of polydimethylsiloxane, for example, depends primarily on the solubility of the drug in the membrane. Drugs with little affinity for the membrane are unlikely to permeate, although in porous membranes, such as those of cellophane or collagen, even drugs with little affinity for the polymer may be transported through the pores.
Most biological membranes bear a surface negative charge, so one would imagine that this might influence permeation. Membranes with un-ionised surfaces (such as cellophane) or positively charged surfaces such as collagen have different permeability characteristics for ionic drugs. Crucially, anionic solutes permeate faster than cationic solutes. With amphoteric drugs such as sulfisomidine and sulfamethizole, a similar order of permeation may be observed depending on the pH of the medium, namely: un-ionised > anionic > cationic form. In acidic media the membrane is positively charged and cationic drugs will therefore be repelled from the surface.
In biological membranes, one might expect some preference for cationic drugs, other things being equal, but we have to remember that biological membranes are more complex and more dynamic than synthetic membranes and there are many confounding factors. One of these, which is outside the scope of this book, is the existence of efflux mechanisms centred on P-glycoprotein ‘pumps’. Some drugs are ejected from cells by the efflux pump, so that these drugs have a lower apparent absorption than predicted on physicochemical grounds. Some excipients (e.g. surfactants such as polysorbate 80) interact with these proteins, so both drug and excipients must be studied for their effect on the efflux pump activity.
In Chapter 4 we examined some relationships between the lipophilicity of drugs and their activity, controlled by their ability to pass across lipid barriers. Although of the same basic construction, biological membranes in different sites in the body serve different functions and thus one might expect them to have different compositions and physicochemical properties, as indeed they do. Tissues derived from the ectoderm (the epidermis, the epithelium of nose and mouth and the anus, and the tissues of the nervous system) have protective and sensory functions. Tissues evolved from the endoderm, such as the epithelium of the GI tract, have evolved mainly to allow absorption.
8.1.1 Lipophilicity and absorption
If one measures the absorption of a sufficiently wide range of substances in a series, one generally finds that there is an optimal point in the series for absorption. In other words, a plot of percentage absorption versus log P would be parabolic, with the optimum value designated as log Po (Fig. 8.3). By noting the values of optimal partition coefficient for different absorbing membranes and surfaces, one can deduce something about their nature. Some values are given in Table 8.1.
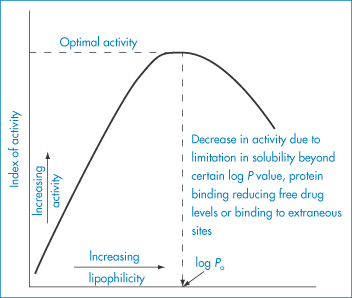
Figure 8.3 Parabolic nature of a typical activity–log P plot: the decrease in biological activity beyond the optimal log Po is probably due to the factors listed.
Table 8.1 Ideal lipophilic character of drugs (log Po) in different regions of the body
System | Solute or drug | Log Po (octanol/water) for maximal transport |
Buccal cavity (human) | Bases | 5.52 (undissociated) |
3.52 (dissociated) | ||
Acids | 4.19 (undissociated) | |
Epidermis (human) | Steroids | 3.34 |
Whole skin (rabbit) | Non-electrolytes | 2.55 |
Small intestine (rat) | Sulfonamides | 2.56–3.33 |
Stomach (rat) | Barbiturates | 2.01 |
Acids | 1.97 | |
Cornea (rabbit) | Steroids | 2.8 |
Biliary excretion | Sulfathiazoles | 0.60 |
Milk/plasma | Sulfonamides | 0.53 |
Prostatic/plasma ratio | Sulfonamides | 0.23 |
The parabolic nature of activity–log P plots is due to a combination of factors: with drugs with high log P values, protein binding, low solubility and binding to extraneous sites cause a lower measured activity than if it were possible to take the drug and place it at the receptor without it having to traverse the various lipid and aqueous hurdles that it has to overcome on its way to the site of action.
Molecular weight and drug absorption
The larger drug molecules are, the poorer will be their ability to traverse biological membranes. The change of permeability with increasing molecular size is shown in Fig. 8.4.
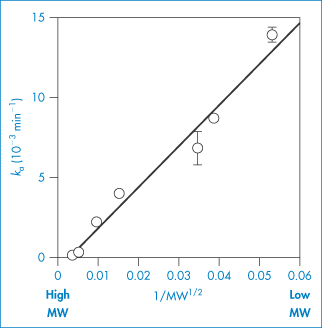
Figure 8.4 The relationship between the molecular weight of compounds and absorption rate constant. The membrane studied is the rat kidney surface, but similar plots hold for other sites.
Reproduced from Nishida K et al. Absorption characteristics of compounds with different molecular weights after application to the unilateral kidney surface in rats. Eur J Pharm Biopharm 2004;58:705–711. Copyright Elsevier 2004.
In a survey of over 2000 drugs, only 11% had molecular weights above 500 Da, and only 8% had molecular weights above 600 Da. Lipinski et al.1 devised the so-called ‘rule of 5’, which refers to the drug-like properties of molecules. It states that poor oral absorption is more likely when the drug molecule:
- has more than five hydrogen-bond donors (‒OH groups or ‒NH groups)
- has a molecular weight >500 Da
- has a log P > 5
- has more than 10 H-bond acceptors.
Note that all the numbers are factors of 5, hence the name of the rule. Compounds that are substrates for transporters are exceptions to the rule.
8.1.2 Permeability and the pH-partition hypothesis
If, in the first instance, the plasma membrane is considered to be a strip of lipoidal material, homogeneous in nature and with a defined thickness, one must assume that only lipid-soluble agents will pass across this barrier. As most drugs are weak electrolytes it is to be expected that the un-ionised form (U) of either acids or bases, the lipid-soluble species, will diffuse across the membrane, while the ionised forms (I) will be rejected. This is the basis of the pH-partition hypothesis, in which the pH dependence of drug absorption and solute transport across membranes is considered. The equations of Chapters 2 and 4 are relevant here.
For weakly acidic drugs such as acetylsalicylic acid (aspirin) and indometacin, the ratio of ionised to un-ionised species is given by the equation

For weak bases the equation takes the form

One can calculate from these equations (and equations 2.45 and 2.47) the relative amounts of absorbable and non-absorbable forms of a drug substance ([U] and [I], respectively), given the prevailing pH conditions in the lumen of the gut or the site of absorption. The profiles for un-ionised drug (%) versus pH for several drugs are given in Fig. 8.5. In very broad terms, one would expect acids to be absorbed from the stomach and bases from the intestine.
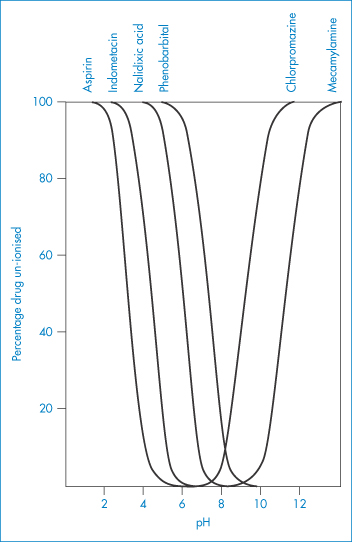
Figure 8.5 Plot of percentage drug un-ionised (that is, in its lipid-soluble form) as a function of solution pH, for the acidic drugs aspirin, indometacin, nalidixic acid and phenobarbital, and for the basic drugs chlorpromazine and mecamylamine.
A comparison of the intestinal absorption of several acids and bases at various pH values (Table 8.2) indicates the expected trend. Surprisingly, however, it will be seen that salicylic acid is absorbed from the rat intestine at pH 8, although with a pKa of 3.0 it is virtually completely ionised at this pH. There are two explanations: one is that absorption and ionisation are both dynamic processes and that the small amount of un-ionised drug absorbed is replenished; the second is that the bulk pH is not the actual pH at the membrane. Nevertheless, instilling solutions of different pH directly into the lumen of the rat stomach indicates the correct qualitative trends of absorption (62% absorption at pH 1 for salicylic acid and 13% at pH 8). Similarly, Box 8.1 shows that the absorption of acetylsalicylic acid is much greater than one would expect from a calculation of the pH dependency of its ionisation.
Table 8.2 Intestinal absorption of acids and bases in the rat at several pH values
Acid/base | pKa | Percentage absorption | |||
|
| pH 4 | pH 5 | pH 7 | pH 8 |
Acids | |||||
Salicylic acid | 3.0 | 64 | 35 | 30 | 10 |
Acetylsalicylic acid (aspirin) | 3.5 | 41 | 27 | – | – |
Benzoic acid | 4.2 | 62 | 36 | 35 | 5 |
Bases | |||||
Amidopyrine | 5.0 | 21 | 35 | 48 | 52 |
Quinine | 8.4 | 9 | 11 | 41 | 54 |
Box 8.1 Calculation of percentage ionisation |
Absorption values for acetylsalicylic acid at pH 4, 5, 7 and 8 are quoted in Table 8.2. The amount of drug in the un-ionised form at these pH values is obtained from equation (8.1). For example, at pH 4,
Percentage un-ionised
In the same way, we find 3.07% un-ionised at pH 5; 0.032% at pH 7; and less than 0.009% un-ionised at pH 9. Absorption is much greater than one would expect, being 41% at pH 4 and 27% at pH 5, although the trend is as predicted. |


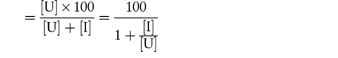
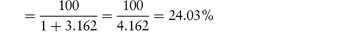
In attempts to explain away discrepancies between theoretical prediction and observed results we have already hinted that a local pH exists at the membrane surface that differs from the bulk pH (see below). This local pH is due to the attraction of hydrogen ions by the negative groups of membrane components so that, in the intestine, while the bulk pH is around 7 the surface pH is nearer 6. A ‘virtual’ surface pH can be calculated, which will allow results such as those in Table 8.2 to be explained. If we take as another example salicylic acid, again calculating the percentage of un-ionised species by equation (8.1), we obtain the following figures (taken from Table 8.2):
pH | 4 | 5 | 7 | 8 |
Percentage un-ionised | 9.09 | 0.99 | 0.0099 | 0.001 |
Percentage absorbed | 64 | 35 | 30 | 10 |
To have 30% of this compound in its un-ionised form when the bulk pH is 7 would require a surface pH of about 3.4, which is lower than anticipated. Again, however, we must remember that absorption and ionisation processes are dynamic processes. As the un-ionised species are absorbed, so the level of [U] in the bulk falls and, because of a shift in equilibrium, more of the un-ionised species appears in the bulk. In fact, if a pH of 5.3 is taken as the pH of the absorbing surface, the results in Table 8.2 become more explicable, as we discuss in the next section.
8.1.3 Problems with the quantitative application of the pH-partition hypothesis
There are several reasons why the pH-partition hypothesis cannot be applied quantitatively in practical situations. Some are discussed here.
Variability in pH conditions
The variation in the stomach pH in human subjects is remarkable, bearing in mind that each pH unit represents a 10-fold difference in hydrogen ion concentration. While the normally quoted range of stomach pH is 1–3, studies using pH-sensitive radiotelemetric capsules have shown a greater spread of values, ranging up to pH 7, as seen in Fig. 11.2 in Chapter 11. This means that the dissolution rate of many drugs will vary markedly in individuals – this is indeed one of the reasons for individual-to-individual variation in drug availability.
The scope for variation in the small intestine is less, although in some pathological states the pH of the duodenum may be quite low, owing to hypersecretion of acid in the stomach. Table 8.3 lists the normal pH of the blood and of different regions of the GI tract.
Table 8.3 pH of blood and contents of the human alimentary tract
Sample | pH |
Blood | 7.35–7.45 |
Buccal cavity | 6.2–7.2 |
Stomach | 1.0–3.0 |
Duodenum | 4.8–8.2 |
Jejunum and ileum | 7.5–8.0 |
Colon | 7.0–7.5 |
pH at membrane surfaces
Data on the relationship between absorption and pH of the intestinal contents on the one hand, and the percentage of drug in the un-ionised state on the other, can generally be rationalised if the apparent pH is reduced below the pH of the intestine. As described above, this pH ‘shift’ is due to the existence of a pH at the membrane surface lower than that of the bulk pH. One might expect that the hydrogen ion concentration at the surface would be greater (and hence pH lower) at the membrane surface as hydrogen ions would accumulate near anionic groups, leading to an effect that can be quantified by the equation
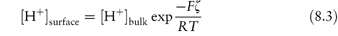
where F is the Faraday constant, R is the gas constant, T is absolute temperature and ζ is the zeta potential of the surface (see Chapter 6). Thus, in terms of pH,
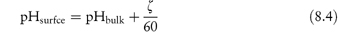
where ζ is expressed in millivolts.
The secretion of acidic and basic substances in many parts of the gut wall is also a complicating factor, as the local pH in the region of the microvilli of the small intestine will undoubtedly influence the absorption of weak electrolytes. A drug molecule in the bulk will diffuse towards the membrane surface and so meet different pH conditions from those in the bulk phase. Whether or not this influences the extent of absorption will depend on the pH changes and the pKa of the drug in question. The negative charge on the membrane will attract small cations towards the surface and small anions will be repelled; one might thus expect some selectivity in the absorption process. The existence of this ‘microclimate’ has been questioned, but experimental evidence for its existence has been obtained from the use of microelectrodes, which revealed the existence of a layer on the (rat) jejunum with a pH of 5.5 when the pH of the bathing buffer was 7.2. The existence of this more acid layer has also been demonstrated on the surface of the human intestine.
Other complications include convective water flow and unstirred layers, discussed below.
Convective water flow
The movement of water molecules into and out of the GI tract will affect the rate of passage of small molecules across the membrane. The reasons for water flow are the differences in osmotic pressure between blood and the contents of the lumen, and differences in hydrostatic pressure between lumen and perivascular tissue, resulting, for example, from muscular contractions. It can be appreciated that the absorption of water-soluble drugs will be increased if water flows from the lumen across the mucosa to the blood side, provided that drug and water are using the same route. Water movement is greatest within the jejunum.
Unstirred water layers
A layer of relatively unstirred water lies adjacent to all biological membranes. The boundary between the bulk water and this unstirred layer is indistinct but, nevertheless, it has a real thickness. During absorption, drug molecules must diffuse across this layer and then on through the lipid layer. The overall rate of transfer is the result of the resistance in both water layer and lipid layer. The flux, J, for a substance across the unstirred layer is given by the expression

where C1 and C2 are the concentrations of the substance in the bulk water phase and in the unstirred water layer, respectively, D is the diffusion coefficient and δ is the effective thickness of the unstirred layer. The flux of molecules that pass by passive diffusion through the lipid membrane can be written as

where Pc is the permeability coefficient. The rate of absorption must equal the rate of transport across the unstirred layer; that is,
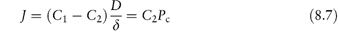
The rate of movement across the unstirred layer, as can be seen from the equations, is proportional to D/δ; the rate of absorption is proportional to Pc. Compounds with a large permeability coefficient may be able to penetrate across cell membranes much faster than they can be transported through the unstirred layer. Under these circumstances diffusion through the water layer becomes the rate-limiting step in the absorption process. Neglect of the unstirred layer causes errors in the interpretation of experimental flux data.
Effect of the properties of the drug
Drugs must be in their molecular form before diffusional absorption processes take place. We would expect bases to be more soluble than acids in the stomach, but it is impossible to generalise in this way. Although the basic form of a drug as its hydrochloride salt should be soluble to some extent in this medium, this is not always so. Indeed, the free bases of, for example, chlortetracycline, dimethylchlortetracycline and methacycline are more soluble than their corresponding hydrochlorides in the pH range of the stomach (see Chapter 4). It has been shown that mean plasma levels following administration of the free base and the hydrochloride of these tetracyclines reflect the differences in solubility, the bases giving higher levels. The reason is most likely that discussed in section 4.7.2, namely the influence of high ionic strength on the solubility of the drug substance (the common ion effect). As absorption of the tetracyclines takes place mainly from the duodenum, it is vital that they reach the intestine in a dissolved or readily soluble form, as their solubility is low at the pH conditions prevailing in the duodenum (pH 4–5).
The presence of buffer components in the formulation also creates a pH microenvironment around dissolving particles that may aid drug dissolution. If dissolution is the rate-limiting step in the absorption process, this will be significant in determining absorption. Bulk pH will then give little help in calculating the solution rate on the basis of a knowledge of saturation solubilities in bulk conditions.
Other complicating factors
The very high area of the surface of the small intestine also upsets the calculation of absorption based on considerations of theoretical absorption across identical areas of absorbing surface. The sheer complexity of the situation precludes mathematical precision, yet the pH-partition hypothesis is useful, especially in predicting what follows from a change in bulk pH – for example, on ingestion of antacids or drugs such as histamine receptor antagonists (H2 blockers) that reduce gastric acid secretion. The extent of the change in pH after administration of one of the first in the class, cimetidine, can be seen from Fig. 8.6, which shows a rise in resting pH from below 2 to near neutrality. The effect in turn depends on the formulation used. The fact that aspirin, an acid with a pKa of 3.5, is absorbed from the small intestine is due partly to the massive surface area available for absorption which allows significant absorption to occur, even though the percentage of absorbable species is very low, and partly to the dynamics of the process referred to earlier.
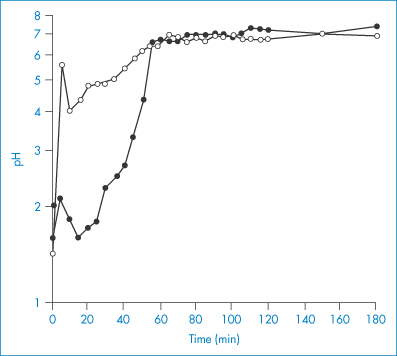
Figure 8.6 Intragastric pH after administration of (○) effervescent cimetidine and (●) standard cimetidine, plotted as median values from 13 patients with gastro-oesophageal reflux given 800 mg orally of the preparations.
Reproduced from Ström M et al. Intragastric pH rise with effervescent citrate-cimetidine. Lancet 1991;337:433.
A warning, however, about other complications: drugs that (1) are unstable in the GI tract (for example, erythromycin); (2) are metabolised on their passage through the gut wall; (3) are hydrolysed in the stomach to active forms (namely prodrugs); and (4) bind to mucin or form complexes with bile salts may not always be absorbed in the manner expected.
Ion pairing
The interaction of drugs in the charged form with other ions to form absorbable species with a high lipid solubility is a possible explanation for the ability of molecules such as quaternary ammonium compounds, ionised under all pH conditions, to be usefully absorbed. The origin of the ions that pair with drug ions is not clear, but there is evidence that ion-pair formation will aid absorption.
One could assume that small organic ions are absorbed through water-filled pores or channels in the membrane, but the effective diameter of such pores means that large drug ions would be excluded from this route. Although membranes are impermeable to large organic ions, nevertheless ion-pairing between a drug ion and an organic ion of opposite charge forming an absorbable neutral species is possible.
Two ionic species A+ and B− may exist in solution in several states:
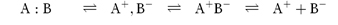
undissociated‘tight’‘loose’free ions
speciesion pairion pair
The formation of tight or loose ion pairs will depend on solvent–ion interactions: hydrophobic ions might be encouraged to form ion pairs by the mechanism of water-structure-enforced ion pairing, in which the water attempts to minimise the disturbance on its structuring, and achieves this end by reducing the polarity of the species in solution by ion-pair formation. Ion pairing in highly structured solvents, then, is due not to an electrostatic interaction but to a solvent-mediated effect. The significance of the phenomenon is that ion pairs have the property of being almost neutral species, so that the ion pair can partition into an oily phase when its parent ionic species cannot, a property that is important in drug absorption and drug extraction procedures, and that is put to use in chromatography.
The two reactions below are examples of (i) a quaternary amine pairing with a weak acid, and (ii) an alkyl sulfate with a weak base, both under pH conditions in which the solute is charged:
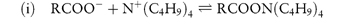
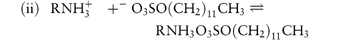
hydrophilicion-pair agent
solutehydrophobic pair
Figure 8.7 shows clearly the effect of chloride ion and other anions such as methane sulfonate on the apparent partition coefficient of chlorpromazine. The nature of the anion significantly affects the partitioning of the drug. Ion pairing in the GI tract obviously could influence absorption.
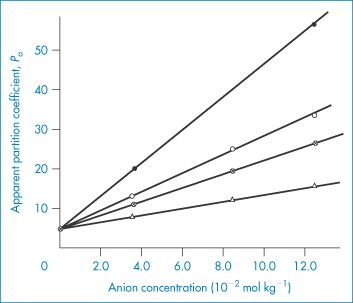
Figure 8.7 Apparent partition coefficients for chlorpromazine between n-octanol and aqueous buffers at pH 3.9, in the presence of various anions at 30°C: ○, chloride; ●, propanesulfonate;  , ethanesulfonate;
, ethanesulfonate;  , methanesulfonate.
, methanesulfonate.
Reproduced from Murthy LS, Zografi G. Oil-water partitioning of chlorpromazine and other phenothiazine derivatives using dodecane and n-octanol. J Pharm Sci 1970;59:1281. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
|
The pH of the stomach may be higher than the normally quoted range of 1–3. The pH at the membrane surface is lower than the bulk pH; there may also be secretion of acidic and basic substances in parts of the gut wall. The movement of water molecules, due to differences in osmotic pressure between blood and the contents of the lumen and differences in hydrostatic pressure between the lumen and the perivascular tissue, affects the rate of absorption of small molecules. A layer of unstirred water lies adjacent to the membrane, which is an additional barrier across which molecules must diffuse during their absorption. Although we would expect that bases would be more soluble than acids in the stomach, this may not always be the case because, for example, of the common ion effect. A variety of other factors may complicate the absorption process, including the complex nature and very high area of the surface of the small intestine and the properties of the drug, for example, its stability in the GI tract, its binding to mucin or complexation with bile salts and the formation of ion pairs. |

8.2 The oral route and oral absorption
8.2.1 Drug absorption from the gastrointestinal tract
The oral route is the most widely used and convenient route of drug administration for those drugs that can survive the acid of the stomach, that are resistant to enzymatic attack and that are absorbed across GI membranes. The primary function of the GI tract is the digestion and absorption of foods and other nutrients and it is not easy to separate these from that of drug delivery. Indeed, the natural processes in the gut frequently influence the absorption of drugs. This is not surprising when it is considered that, on average, approximately 500 g of solid and up to 2.5 litres of fluid are ingested each day. As well as this oral intake, litres of endogenous fluid are excreted each day into the intestine.
The pH of the gut contents and the presence of enzymes, foodstuffs, bile salts, fat and the microbial flora will all influence drug absorption. The complexity of the absorbing surfaces means that a simple physicochemical approach to drug absorption remains an approach to the problem and not the complete picture, as described above. Whatever the limitations of theory – and one should not expect simple theories to hold in the complex and dynamic circumstances that are involved in drug absorption – theory provides a starting point in rationalising the behaviour of drugs in the GI tract. While most drugs are absorbed by passive diffusion across the lipid membranes separating the gut contents from the rest of the body, certain molecules that resemble naturally occurring substances are actively transported by special mechanisms.
We first consider the physiological situation that might impinge on passive drug absorption. The delivery of macromolecules, including peptides and proteins, is treated in Chapter 13.
Particulate absorption from the gut
While the general rule is that only drugs in solution are absorbed from the GI tract, colloidal particles, some viruses, bacteria and prion proteins can gain entry in low amounts to the lymphatic system through absorption by the specialised cells (M cells) in the gut-associated lymphoid tissue (GALT) and also by enterocytes under certain conditions. In studies in rats, for example, model polystyrene particles above 300 nm in diameter were absorbed in minute amounts, with maximal absorption with particles around 50 nm. Comparisons of uptake between carboxylated microspheres and non-ionic systems, showed lower uptake of the former through the lymphoid tissue of the GI tract. Size is a key parameter: uptake increases with decreasing particle diameter. Adsorption of hydrophilic block copolymers on to polystyrene markedly reduces the uptake by intestinal GALT. Modification of the surface with specific ligands, such as by covalent attachment of tomato lectin molecules has indicated enhanced uptake following their binding to, and internalisation by, enterocytes. The ability to decrease and increase uptake is clear evidence of a phenomenon which has the potential for further control to allow it to be exploited fully for drug or vaccine delivery. The evidence to date with nanoparticles as carrier systems for labile drugs such as proteins by the oral route remains to be substantiated.
A lengthy discussion of this route of uptake is outside the scope of this book, but it is a route that is likely to be increasingly explored as a means of delivering proteins and perhaps genes in carrier nanoparticles, and for oral vaccination. It has yet to fulfil its promise.2 If the level of incorporation of the active ingredient is low and uptake is relatively low (a maximum perhaps of 5%) and if drug is released before uptake, this limits the absorption of the gut-labile active considerably. However, we should be aware that excipients such as titanium dioxide (rutile) have been found to be absorbed by the GALT, and we should be aware of the potential at least for other materials also to be taken up.
8.2.2 Structure of the gastrointestinal tract
We first consider the physiological situations and the structure and function of the GI tract that might impinge on passive drug absorption.
Figure 8.8 diagrammatically represents the GI tract and some of the factors involved in the process of drug absorption from this complex milieu. The stomach is not an organ that has evolved for absorption; the main site of absorption is the small intestine.
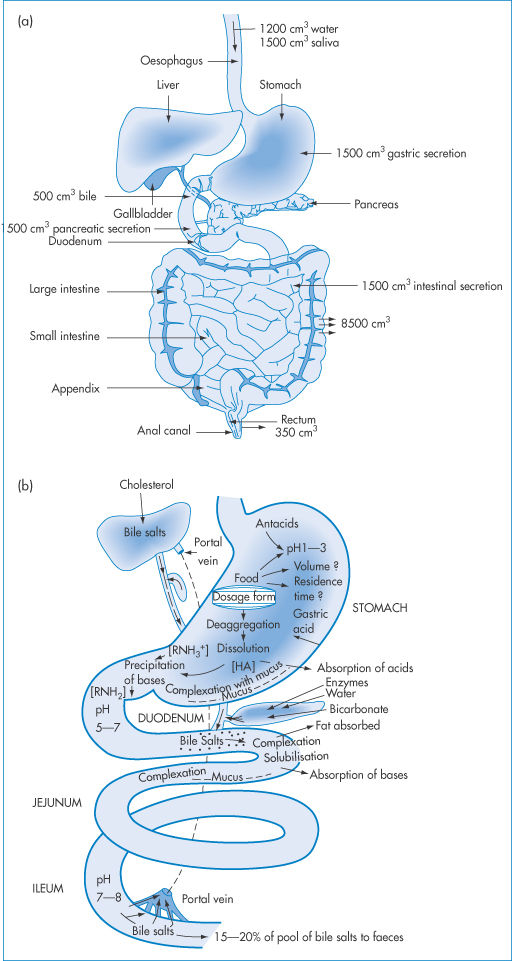
Figure 8.8 Representation of the processes occurring along the gastrointestinal tract, and of the factors that must be taken into account in considering drug absorption.
The stomach may be divided into its two main parts: (1) the body of the stomach, which is in effect a receptacle or hopper, which includes pepsin- and HCl-secreting areas; and (2) the pyloris (a churning chamber), the mucus-secreting area of the gastric mucosa. The stomach varies its luminal volume with the content of food and this is one reason why food intake can be so important in relation to drug absorption; the stomach of adults may contain a few millilitres or a litre or more of fluid. Hydrochloric acid is liberated from the parietal cells at a concentration of 0.58%, or 160 mmol dm–3. The gastric glands produce some 1000–1500 cm3 of gastric juice per day. It is in this somewhat chaotic environment that pharmaceutical dosage forms find themselves. Some of the events that result are shown in Fig. 8.8b.
The small intestine is divided anatomically into three sections: duodenum, jejunum and ileum. Histologically there is no clearly marked transition between these parts. All three are involved in the digestion and absorption of foodstuffs, absorbed material being removed by the blood and the lymph. The absorbing area is enlarged by surface folds in the intestinal lining that are macroscopically apparent: the surfaces of these folds possess villi and microvilli (Fig. 8.9). It has been calculated that, with a maximum of 3000 microvilli per cell in the epithelial brush border (so called because of its physical appearance), the number of microvilli in the small-intestine mucosa, even of the rat, is 2 × 108 per mm2. Although one would expect organic acids to be absorbed only from the stomach, where they will exist in the un-ionised lipid-soluble and membrane-diffusible form, the enormous surface area in the intestine allows significant absorption of acidic drugs from the intestine even though (as noted earlier) the fraction of un-ionised molecules is very small.
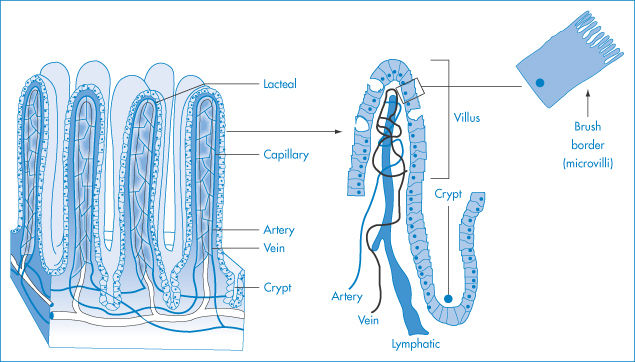
Figure 8.9 Representation of the epithelium of the small intestine at different levels of magnification. From left to right: the intestinal villi and microvilli that comprise the brush border.
Over the entire length of the large and small intestines and the stomach, the brush border has a uniform coating (3 nm thick) of mucopolysaccharides that consist of multibranched polymeric chains. This coating layer appears to act as a mechanical barrier to bacteria, cells or food particles, or as a filter. Whatever its function, the weakly acidic, sulfated mucopolysaccharides influence the charge on the cell membrane and complicate the explanations of absorption.
The goblet cells of the epithelium form mucus; secretions are stored in granule form in the apical cell region and are liquefied on contact with water to form mucus, which is composed of protein and carbohydrate and acts as a protective mechanism for the gut surface. Its role in the absorption of drugs is unclear.
The large intestine is concerned primarily with the absorption of water and the secretion of mucus to aid the intestinal contents to slide down the intestinal ‘tube’. Villi are therefore completely absent from the large intestine, but there are deep crypts distributed over its surface.
Differences in the absorptive areas and volumes of gut contents in experimental animals are important when comparing experimental results on drug absorption in various species and in humans.3 The all-important extrapolation to the human animal is complex. The human small intestine has a calculated active surface area of approximately 100 m2. No analogous calculations are available for the most commonly used laboratory animals, although the surface area of the small intestine of the rat is estimated to be 700 cm2.
Passive transport, carrier-mediated transport and specialised transport
In discussing the pH-partition hypothesis, it has been considered that drugs are absorbed by passive diffusion through epithelial cells – the enterocytes of the GI tract, for example. In fact, there is the possibility of some passage of drugs by way of the tight junctions (the paracellular route) and there are transcellular carrier-mediated uptake mechanisms as well as endocytosis. Figure 8.10 summarises these.
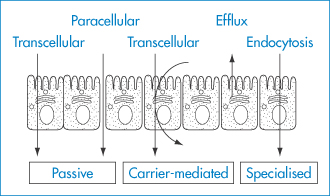
Figure 8.10 Gastrointestinal membrane transport. Transport through the enterocyte barrier can be divided into active, passive and specialised transport; and into the paracellular and transcellular routes. Efflux mechanisms can reduce absorption by these routes.
8.2.3 Bile salts and fat absorption pathways
Fat is absorbed by special mechanisms in the gut. The bile salts that are secreted into the jejunum are efficient emulsifiers and disperse fat globules, allowing the action of lipase at the much-increased globule surface. Medium-chain triglycerides are thought to be directly absorbed. Long-chain triglycerides are hydrolysed, and the monoglycerides and fatty acids produced form mixed micelles with the bile salts and are absorbed either directly in the micelle or, more probably, brought to the microvillus surface by the micelle and transferred directly to the mucosal cells, the bile salts remaining in the lumen. The bile salts are reabsorbed in the ileum and transported via the portal vein back into the bile salt pool.
There have been suggestions that lipid-soluble drugs may be absorbed by fat absorption pathways. Certainly, administration of drugs in an oily vehicle can significantly affect their absorption, increasing it in the case of griseofulvin and ciclosporin, but decreasing it in the case of vitamin D.
8.2.4 Gastric emptying, motility and volume of contents
The volume of the gastric contents will determine the concentration of a drug that finds itself in the stomach. The time the drug or dosage form resides in the stomach will determine many aspects of absorption. If the drug is absorbed lower down the gut, the residence time will determine the delay before absorption begins; if the drug is labile in acid conditions, longer residence times in the stomach will lead to greater stability; if the dosage form is non-disintegrating then retention in the stomach can influence the pattern of absorption. Gastro-retentive dosage forms are designed to achieve that control (see section 8.3.2).
The stomach empties liquids faster than solids. The rate of transfer of gastric contents to the small intestine is retarded by the activity of receptors sensitive to acid, fat, osmotic pressure and amino acids in the duodenum and the small intestine, and stimulated by material that has arrived from the stomach. Gastric emptying is a simple exponential or square-root function of the volume of a test meal – a pattern that holds for meals of variable viscosity. To explain the effect of a large range of substances on emptying, an osmoreceptor has been postulated that shrinks in hypertonic solutions and swells in hypotonic solutions.
Acids in test meals have been found to slow gastric emptying; acids with higher molecular weights (for example, citric acid) are less effective than those, such as HCl, with very low molecular weights. Natural triglycerides inhibit gastric motility, linseed and olive oils being effective. The formulation of a drug may thus influence drug absorption through an indirect physiological effect. The nature of the dose form – whether solid or liquid, whether acid or alkaline, whether aqueous or oily – may thus influence gastric emptying. The question of gastric emptying and transit down the GI tract has assumed further importance in relation to the design and performance of sustained-release preparations. The transit of pellets of different densities,4,5 for example, is shown in Fig. 8.11 and illustrates the influence of both density and food intake.
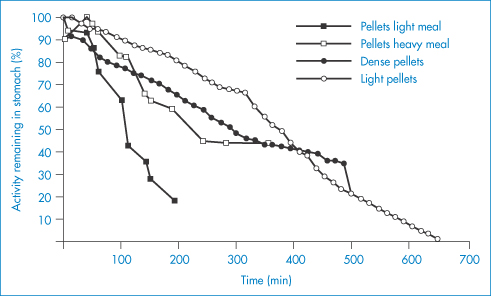
Figure 8.11 Gastric emptying of pellets of different density with various sizes of meal.
Reproduced with permission from Wilson CG, Washington N. Physiological Pharmaceutics. Chichester: Ellis Horwood; 1989.
Food, then, affects not only transit but also pH. The effect of a meal on the hydrogen ion concentration of the stomach contents is shown in Fig. 8.12; the effect of two antacids on gastric volume and pH is shown in Table 8.4. When considering the effect of an antacid, therefore, the effect of volume change and pH change and the effect on gastric emptying must all be considered. In designing delivery systems all these factors have ideally to be taken into account.
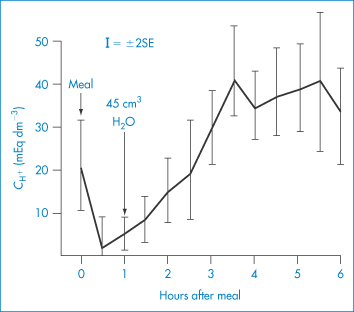
Figure 8.12 Hydrogen ion concentrations (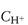 ) at intervals after a test meal (mean results are shown ±2 SEM); the zero samples were taken just before the meal was begun, and the 1-hour sample just before 45 cm3 of water.
) at intervals after a test meal (mean results are shown ±2 SEM); the zero samples were taken just before the meal was begun, and the 1-hour sample just before 45 cm3 of water.
Modified with permission from Fordtran J. Antacid pharmacology in duodenal ulcer. Effect of antacids on postcibal gastric acidity and peptic activity. N Engl J Med 1966;274:921.
Table 8.4 Effects of antacids on gastric volume and pH in the rata
| Water | Maalox | Amphojel |
Volume (cm3) | 0.29 ± 0.03 | 1.8 ± 0.3 | 4.0 ± 0.2 |
pH | 2.2 ± 0.2 | 7.4 ± 0.1 | 4.7 ± 0.2 |
aTen rats per group. Measurements on gastric contents made 20 minutes after the third hourly dose of 1 cm3 water or antacid by gastric intubation.
Reproduced from Hava M, Hurwitz A. The relaxing effect of aluminum and lanthanum on rat and human gastric smooth muscle in vitro. Eur J Pharmacol 1973;22:156. Copyright Elsevier 1973.
|
|

8.3 Oral dosage forms
A variety of modified-release oral dosage forms are discussed in other chapters of this book. Here we briefly touch on the conventional delivery systems used in the great majority of cases, namely tablets, capsules, suspensions and liquids, whose production and properties have also been discussed in earlier chapters. In the main, conventional – that is to say, fast-release – products are used as a means of administering large and small doses of drugs in convenient forms, to provide stable systems and reliable dosing. For very low-dose drugs (such as oral contraceptives) there is the problem even today of ensuring that the drug is dispersed evenly in the bulk powder or granules used for tableting, because of the phenomenon of particle segregation. On the other hand, it is possible by appropriate design of the dosage form to alter the pharmacokinetics that the drug would otherwise exhibit. Modified-, sustained-, delayed- or controlled-release forms (these descriptors are used interchangeably) can be used by the oral route to prolong the release and thus the action of drugs, so reducing the frequency of dosing with drugs having a short intrinsic half-life. One of the oldest forms of delayed-release product is the enteric-coated tablet or capsule. These have a polymer coat whose solubility is pH-dependent, this becoming porous or soluble in the lower intestine or colon. Other film coats and non-disintegrating matrix forms are dealt with in Chapter 7.
The preparation and formulation of tablets and capsules can be either straightforward or complex depending on the nature of the drug and the excipients. Particular problems arise with labile drugs that might be affected by moisture, low-dose drugs and dose forms containing more than one active ingredient, with the potential for the interaction between the drugs themselves or with the excipient materials, as discussed in Chapter 1. A cartoon of a typical coated tablet is shown in Fig. 8.13, which in a simple way illustrates the potential for problems, not least the uneven distribution of drug, the potential for drug–excipient interactions, drug–drug interactions even in the solid state, when more than one agent is incorporated into the dose form, and issues of overall stability, mostly discussed in earlier chapters.
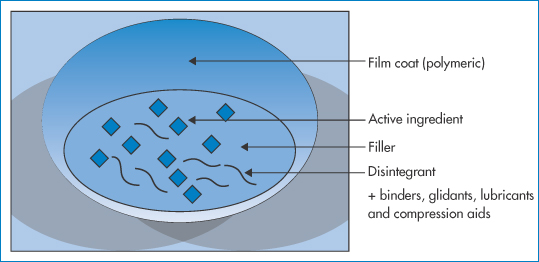
Figure 8.13 A cartoon of a conventional film-coated tablet, showing just some of the ingredients commonly found and the range of possibilities for interactions, such as drug–excipient, excipient–excipient, and changes in the polymer film coat.
|
Controlled-release formulations can moderate toxicity, prolong the activity of a drug and often have great benefits, as has been shown in clinical trials of prednisone in controlled release, reducing morning stiffness in the joints of patients with rheumatoid arthritis, when compared to conventional release forms, as shown in Fig. 8.14. |

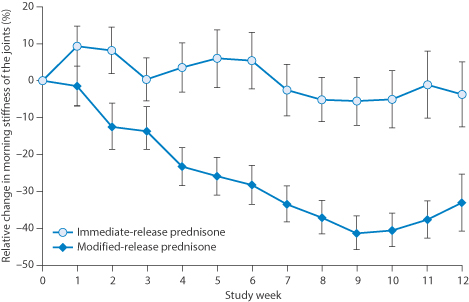
Figure 8.14 Index of stiffness relief in a 10-week clinical trial comparing immediate-release and modified-release oral prednisone tablets. The differences were found to be clinically relevant.
Reproduced from Buttgereit F et al. Efficacy of modified-release versus standard prednisone to reduce duration of morning stiffness of the joints in rheumatoid arthritis (CAPRA-1): a double-blind, randomised controlled trial. Lancet 2008;371:205–214.
8.3.1 The Biopharmaceutical Classification System (BCS)6
With the plethora of drugs now available and being tested, there was a need to assist drug developers to make decisions about the manner in which to deliver drugs by the oral route. Through the work of Gordon Amidon and colleagues, a valuable classification system was developed to relate drug intestinal absorption to the physical properties of drugs. Drugs were characterised into four types or classes, as shown in Table 8.5. The ideal drugs fall within class I. Different strategies must be adopted to formulate drugs in other classes to achieve efficient oral absorption.
Table 8.5 The four BCS drug classes
Class I: High solubility, high permeability | Class II: Low solubility, high permeability |
Class III: High solubility, low permeability | Class IV: Low solubility, low permeability |
There are, of course, drugs that are at the boundaries of these classifications: it is clear too that properties will vary widely within classes. The commonly seen grid is as shown in Fig. 8.15, where GI permeability is plotted not against drug solubility itself but against the dose/solubility ratio. Very poorly soluble drugs may be therapeutically effective orally if their dose is very low. High-dose drugs with a low solubility present the greatest pharmaceutical challenge. The figure also shows some of the stratagems to be adopted to achieve better absorption.
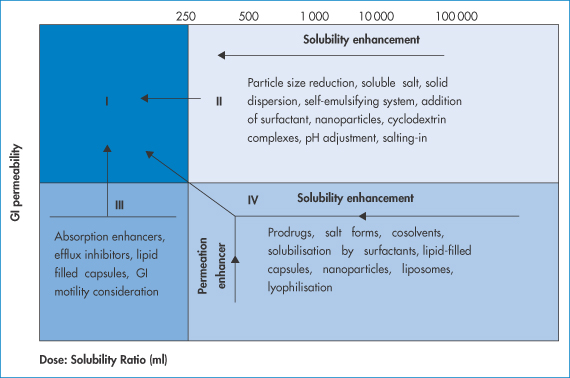
Figure 8.15 A BCS-type plot of the four classes of drug with suggested techniques to enhance absorption. Class II compounds benefit from particle size reduction, soluble salt formation and solubilisation by surfactants; class III drugs require absorption enhancers and efflux inhibitors, inter alia; class IV drugs require a range of techniques to attempt to enhance absorption, as shown.
Table 8.6 lists some well-known drugs and their classification.
Table 8.6 BCS classification of some well-known drugs
Class I | Class II | Class III | Class IV |
Chloroquine | Carbamazepine | Aciclovir | Coenzyme Q10 |
Diltiazem | Danazol | Atenolol | Ciclosporin A |
Metoprolol | Glibenclamide | Captopril | Ellagic acid |
Paracetamol | Ketoconazole | Cimetidine | Furosemide |
Propranolol | Nifedipine | Metformin | Ritonavir |
Theophylline | Phenytoin | Neomycin B | Saquinavir |
Verapamil | Troglitazone | Ranitidine | Taxol |
Of course, absorption is determined in part by the nature of the formulation used to deliver the drug. Formulation, as indicted in the legend to Fig. 8.15, can be used to enhance bioavailability, but there are also differences in the propensity of dose forms to display problems of bioavailability, especially with drugs which are themselves difficult to be absorbed. Table 8.7 shows those systems with high, medium and low potentials for problems, the order being self-evident from our understanding of the issues already discussed in terms of GI transit, pH, gastric emptying and other important factors, such as food intake.
Table 8.7 Relative potential of oral medicines to display bioavailability problems
High | Intermediate | Low |
Enteric-coated tablets | Suspensions | Solutions |
Sustained-release tablets | Chewable tablets |
|
Complex formulations | Capsules |
|
Slowly disintegrating tablets |
|
|
8.3.2 Variability in responses to oral medications
In this chapter so far we have discussed the difficulty in the strict application of physicochemical equations in spite of the fact that the physical chemistry is vitally important in understanding the processes occurring in drug absorption. One of the issues is the dynamic nature of absorption of drug and movement of dose forms down the GI tract. Smith and Rawlins’ 1973 book, Variability in Human Drug Response,7 succinctly explained the many reasons for the variation between individuals following the administration of identical medicines, which result from differences in:
- absorption
- distribution
- metabolism
- excretion
- tissue sensitivity.
These are in addition to problems with patient or carer compliance with medication regimes. Physiological, genetic and physicochemical variations can be crucial in determining the successful outcome of treatment. In cases where the effective dose and toxic doses are close then it is important that we understand better the processes involved. Here we are concerned mainly with absorption and how appropriate formulations can modulate differences. Figure 8.16 emphasises, in this case with ciclosporin, the importance of the product, which is a particular issue with drugs with a narrow therapeutic window. Chapter 16 returns to this topic when dealing with generic medicines and biosimilars.
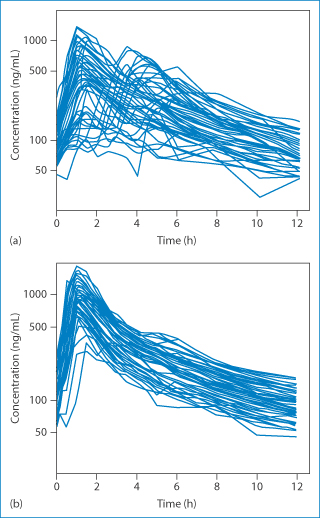
Figure 8.16 Plasma concentration of ciclosporin following oral administration of (a) Sandimmun and (b) Neoral, showing the improvement with the newer formulation, but the still large variation between individual liver transplant patients.
Reproduced from Trull AK et al. Absorption of cyclosporin from conventional and new microemulsion oral formulations in liver transplant recipients with external biliary diversion. Br J Clin Pharmacol 1995;39:627–631. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Ciclosporin is an immunosuppressant cyclic peptide that has an oral bioavailability of around 15% in its amorphous and unformulated form. This is in contrast to the majority of (non-cyclic) peptides, which have little or no oral availability. Ciclosporin, being poorly water-soluble, is difficult to formulate as an oral product. It has been available in several innovative forms: an emulsion-forming (Sandimmun) and a microemulsion-forming product (Neoral), as discussed in Chapter 6 (section 6.4.6). Neoral oral solution contains, in addition to ciclosporin, DL-α-tocopherol, ethanol, polyoxyethylated 40, hydrogenated castor oil, maize oil (corn oil mono-, di-, tri-glycerides) and propylene glycol. The oral concentrate forms a microemulsion. Neoral soft gelatin capsules also contain the above excipients and these also form microemulsions in vivo.
|
Figure 8.16 shows the variability of absorption after oral administration of Sandimmun and also with the improved microemulsion system, Neoral. The latter is less variable, that is, it improves the quality of effect but there is still variability. Reproducibility of effect is extremely important in transplantation patients to avoid underdosing and transplant rejection. In transplantation patients, initial treatment is by way of intravenous dosing and later maintenance through the oral route. |

It is logical to assume that the absorption of the drug would be improved by its formulation in a microemulsion form, where the lipid droplets are in the nanometre rather than micrometre size range. A simple surface area effect would operate – the smaller droplets, having a larger surface area per unit weight, present a greater surface from which the ciclosporin can diffuse directly into intestinal absorbing tissue. It has been suggested that, with the microemulsion, inhibition of metabolism in the gut wall leads to higher availability.
|
Several formulations of ciclosporin are available. The British National Formulary 68 (p. 619)8 warns: Patients should be stabilised on a particular brand of oral ciclosporin because switching between formulations without close monitoring may lead to clinically important changes in blood-ciclosporin concentration. Prescribing and dispensing of ciclosporin should be by brand name to avoid inadvertent switching. If it is necessary to switch a patient to a different brand of ciclosporin, the patient should be monitored closely for changes in blood-ciclosporin concentration, serum creatinine, blood pressure, and transplant function. |

Sandimmun is listed primarily as an intravenous injection but Sandimmun capsules and oral solution are available direct from Novartis for patients who cannot be transferred to a different oral preparation, of which there are Capimune and Deximune, as well as Neoral. It is not always straightforward to determine the ingredients of such formulations, which makes it difficult for pharmacists and physicians. The warning for the intravenous preparation – ‘observe patients for signs of anaphylaxis for at least 30 minutes after starting infusion and at frequent intervals thereafter’ – is due, as discussed in Chapter 12, to the presence of Cremophor EL, which can induce anaphylaxis in a minority of individuals.
As one of the sources of variation in absorption from the gut is the behaviour of the dosage form in the GI tract, the gastric emptying time, the time for transit in the intestine, the effect of food and concomitant therapies, not to mention gender and age all contribute to the inability to predict outcomes. Logarithmic plots of blood levels have to be read with caution.
Gastro-retentive products can go some way to make gastric emptying times less crucial. Drug is presented to the stomach and intestine as a solution. Four main categories of retentive system are shown in Fig. 8.17:
1. floating systems
2. bioadhesive systems
3. high-density systems
4. swellable systems.
These attempt to overcome the normal patterns of GI transit, particularly that of gastric emptying.
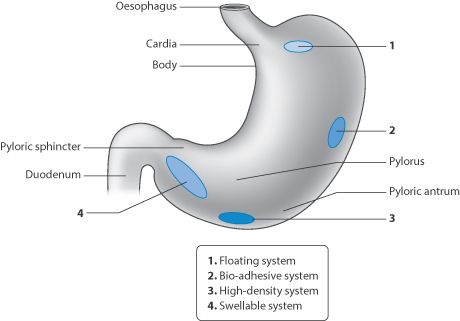
Figure 8.17 Representation of the four main types of gastro-retentive dose forms: floating systems, bioadhesive systems, high-density systems and swellable gel-like systems.
8.3.3 Targeting to sites in the gastrointestinal tract
Enteric-coated systems were the first to achieve targeting of delivery by preventing the dissolution of the dosage form in the stomach, until these units experience the higher pH of the intestine. Targeting to the colon is desirable for the treatment of ulcerative colitis and other pathologies of the gut. Some of the approaches adopted over the last few decades are represented in Fig. 8.18.
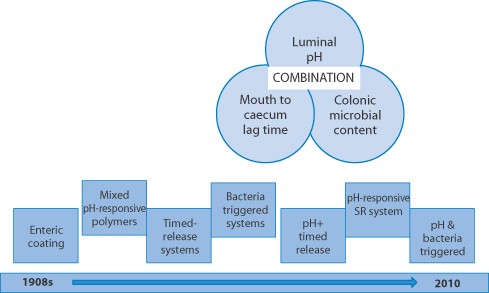
Figure 8.18 The evolution of the development of delivery strategies to target drugs to the colon using the differences in the pH of the gastrointestinal lumen in different regions, the transit times in the gut and the possibility of using the colonic microbiota to initiate release of drug or active drugs released in the vicinity of the colon. SR = sustained-release.
Adapted from Basit AW, McConnell EL. Drug delivery to the colon, Chapter 18. In Wilson CG, Crowley PJ (eds) Controlled Release in Oral Drug Delivery. New York: Springer-CRS; 2011, with kind permission from Springer Science and Business Media. See also: Florence AT. A short history of controlled release, pp. 1–26, in the same book.
Sulfasalazine represents one drug used in the management of inflammatory bowel diseases. Its activity is generally considered to lie in its metabolic breakdown product, 5-aminosalicylic acid, released in the colon (Fig. 8.19).
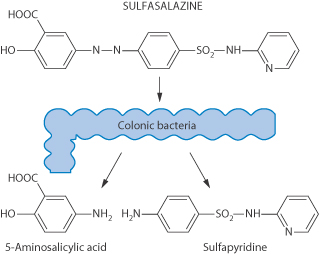
Figure 8.19 Sulfasalazine and the formation of its active metabolite as a result of the action of colonic bacteria.
|
|

8.4 Buccal and sublingual absorption
Mouthwashes, toothpastes and other preparations are introduced into the oral cavity for local prophylactic and therapeutic reasons. It is not known to what extent components of these formulations are absorbed and give rise to systemic effects. The absorption of drugs through the oral mucosa, however, provides a route for systemic administration that avoids exposure to the GI system. Drugs absorbed in this way bypass the liver and have direct access to the systemic circulation. The sublingual, buccal and gingival tissues are shown diagrammatically in Fig. 8.20. The oral mucosa functions primarily as a barrier, however, and it is not a highly permeable tissue, resembling skin more closely than the gut in this respect (Fig. 8.21) Table 8.8 lists some commercially available sublingual and buccal delivery systems. Mucosal adhesive systems have been studied for administration of buprenorphine through the gingiva.
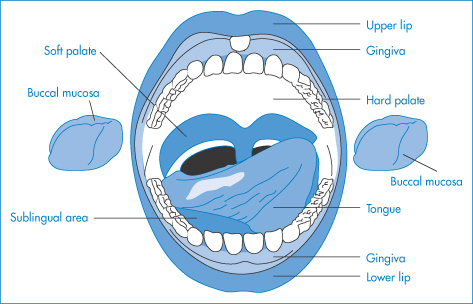
Figure 8.20 The anatomy of the oral cavity, including the buccal, sublingual and gingival cavities.
Reproduced from Kunth K et al. Hydrogel delivery systems for vaginal and oral applications. Formulations and biological considerations. Adv Drug Deliv Rev 1993;11:137. Copyright Elsevier 1993.
8.4.1 Mechanisms of oral mucosal absorption
The oral mucosa comprises:
- a mucus layer over the epithelium
- a keratinised layer in certain regions of the oral cavity
- an epithelial layer
- a basement membrane
- connective tissue
- a submucosal region.
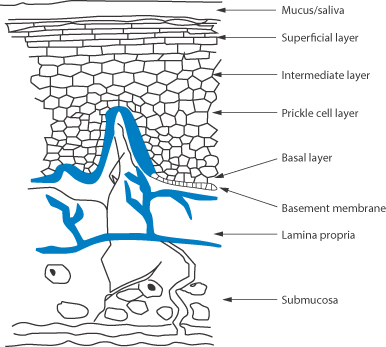
Figure 8.21 The structures of the oral mucosa.*
While cells of the oral epithelium and epidermis are capable of absorbing material by endocytosis, it does not seem likely that drugs or other solutes would be transported by this mechanism across the entire stratified epithelium. It is also unlikely that active transport processes are operative in the oral mucosa. There is considerable evidence that most substances are absorbed by simple diffusion. The linear relationship between percentage absorption through the buccal epithelium and log P of a homologous series is seen in Fig. 8.22. For example, the buccal absorption of some basic drugs increases (Fig. 8.23), and that of acidic drugs decreases, with increasing pH of their solutions.
Table 8.8 Commercially available drug-delivery systems for systemic delivery by the oral mucosal route
Mucosal site | Drug | Proprietary | Dosage |
Sublingual | Glyceryl trinitrate | Nitrostat | Tablets |
| Lenitral spray | Spray | |
| Susadrin | Bioadhesive | |
Isosorbide dinitrate | Risordan | Tablets | |
| Sorbitrate | Chewable | |
| Isocard spray | Spray | |
Erythritol trinitrate | Cardiwell | Tablets | |
Nifedipine | Adalat | Tablets | |
Buprenorphine | Temgesic; | Tablets | |
Apomorphine HCI | Apomorphine | Tablets
| |
Buccal | Prochlorperazine | Buccastem | Bioadhesive |
| Tementil | Solution | |
Phloroglucinol | Spasfon-Lyoc | Lyocs | |
Oxazepam | Seresta | Lyophilised | |
Lorazepam | Temesta | Lyophilised | |
Methyltestosterone | Metandren | Tablets | |
Nicotine | Nicorette | Chewing-gum |
Modified from Ponchel G. Formulation of oral mucosal drug delivery systems for the systemic delivery of bioactive materials. Adv Drug Deliv Rev 1994;13:75. Copyright Elsevier 1994.
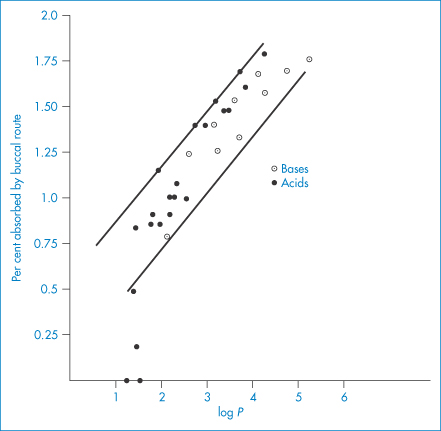
Figure 8.22 Relationship between the log P of solute and the percentage absorption through the buccal mucosa of human subjects for bases (○) and acids (●).
Plotted from data from Lien E et al. Buccal and percutaneous absorption. Drug Intell Clin Pharm 1971;5:38, with permission.
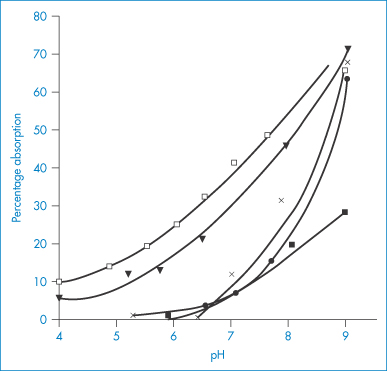
Figure 8.23 Buccal absorption of some basic drugs. The drugs were dissolved in buffered solutions of different pH and placed in the mouth of human subjects; absorption rates were determined from the decrease of drug concentration in expelled solutions: ◻, chlorphenamine (pKa = 8.99); ▼, methadone (pKa = 8.25); ×, amfetamine (pKa = 9.94); ●, pethidine (pKa = 8.72); ◼, nicotine (pKa = 8.02).
Reproduced with permission from Beckett AH, Triggs EJ. Buccal absorption of basic drugs and its application as an in vivo model of passive drug transfer through lipid membranes. J Pharm Pharmacol 1976;19:31S.
Experiments with some analgesics showed that the highly lipid-soluble etorphine was absorbed several times more rapidly from the buccal cavity than the less lipid-soluble morphine.9 Impressive absorption has been obtained with glyceryl trinitrate (which exerts its pharmacological action 1–2 minutes after sublingual administration), methacholine chloride, isoprenaline, some steroids such as desoxycortisone acetate10 and 17β-estradiol (at one-quarter of its normal oral dose), morphine, captopril and nifedipine.
It has been claimed that an oil/water partition coefficient in the range 40–2000 (log P of 1.6–3.3) is optimal for drugs to be used by the sublingual route. Drugs with a log P greater than 3.3 are so oil-soluble that it is difficult for sufficiently high levels of drug to be soluble in the aqueous salivary fluids. Drugs less lipophilic than those with a log P of 1 would not be absorbed to any great extent and would thus require large doses by this route. A comparison of the sublingual/subcutaneous dose ratio serves as an index of the usefulness of the route.11 For example, cocaine with a P of 28 requires a sublingual/subcutaneous dosage ratio of 2 to obtain equal effects; atropine with a P of 7 requires 8 times the subcutaneous dose; and for codeine (P 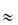 2) over 15 times the subcutaneous dose must be given sublingually.
2) over 15 times the subcutaneous dose must be given sublingually.
An oromucosal spray: Sativex
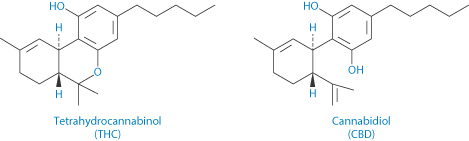
Structures of tetrahydrocannabinol (THC) (I) and cannabidiol (II), active components of Sativex oromucosal spray
The only cannabis product for medicinal use licensed in the UK and increasingly in other countries is Sativex, containing tetrahydrocannabinol (THC, I) and cannabidiol (II), mainly for the relief of pain in multiple sclerosis. The spray is a solution of the cannabis extracts in a vehicle consisting of propylene glycol and ethanol flavoured with peppermint oil. Table 8.9 shows the Cmax and tmax of the THC compared to inhaled extract and the very different results from smoking cannabis (note that the doses are different!).
Table 8.9 Pharmacokinetic parameters for Sativex, vaporised tetrahydrocannabinol (THC) extract and smoked cannabis
| Cmax THC (ng mL–1) | tmax THC (min) | AUC (0-t) THC ng/mL/min |
Sativex (providing 21.6 mg THC) | 5.40 | 60 | 1362 |
Inhaled vaporised THC extract (providing 8 mg THC) | 118.6 | 17.0 | 5987.9 |
Smoked cannabisa (providing 33.8 mg THC) | 162.2 | 9.0 | No data |
aHuestis et al. J Analyt Toxicol 1992;16:276–282.
Orodispersible fllms
Orodispersible films show promise because of their versatility for personalised medication. They can be fabricated on a small scale. Casting solutions containing hypromellose, carbomer and glycerol are used for example. Poorly soluble drugs such as diazepam can be prepared in film form using ethanol as cosolvent. The water-soluble active pharmaceutical ingredients as well as ethanol influence the viscosity of the casting solution, the mechanical properties and disintegration times of the orodispersible films. Chapter 15 discusses an in vitro testing procedure for the determining the mechanical strength of such films.
Nicotine gum
Nicotine in a gum vehicle, when chewed, is absorbed through the buccal mucosa; nicotine levels obtained are lower than those obtained by smoking cigarettes and the high peak levels are not seen. Buccal glyceryl trinitrate has been found to be an effective drug. The buccal route has the advantages of the sublingual route – the buccal mucosa is similar to sublingual mucosal tissue – but a sustained-release tablet can be held in the cheek pouch for several hours if necessary. Mucosal adhesive formulations can have obvious advantages.
Orally disintegrating tablets
Tablets which are designed to disintegrate rapidly in the mouth frequently do so within 30 seconds of placing the dose on the tongue. Some are made by lyophilisation procedures from solutions or suspensions of the active ingredient. This mode of administration again avoids the passage of solid-dose forms down the GI tract and hence can minimise some of the variables we have discussed.
|
|

Summary
We have considered the basic mechanisms of absorption that apply to all routes of administration, dealing with the factors that depend on the drug itself, its chemical and physicochemical characteristics, the nature of the medium and the subtle differences in biological barriers that drugs experience in vivo. While the equations that relate to many of the processes are given, it is emphasised that the dynamic processes of drug absorption – dynamic in two senses, in that the drug is moving down the GI tract at the same time as it is being absorbed – deny us a physicochemical ‘theory of everything’. Nonetheless, estimates are valuable in understanding what is going on.
The emphasis on the oral route of delivery and sublingual and buccal delivery provided details of the underlying processes. The following chapter considers parenteral (non-oral) routes of drug administration. In all of these, the actual processes of absorption of drug once it reaches the absorbing surface are the same as those discussed in the present chapter. Sometimes the route to that surface, such as after inhalation, is very tortuous, as we discuss in the next chapter, and sometimes, as with topical preparations, there is a more direct contact between the formulation and the barrier to be overcome. In all, there are at least four important factors: the drug, the route and the formulation (both its physical form and the excipients it contains). These four factors feature as strongly in the discussions in the next chapter.
References
1. Lipinski CA et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;4:63–26.
2. Florence AT. Nanoparticle uptake by the oral route: fulfilling its potential? Drug Discov Today: Technology 2005;2:75–81.
3. Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos 1995;16:351–380.
4. Davis SS et al. Transit of pharmaceutical dosage forms through the small intestine. Gut 1986;27:886–892.
5. Davis SS et al. The effect of food on the gastrointestinal transit of pellets and an osmotic device (Osmet). Int J Pharm 1984;21:331–340.
6. Amidon GL et al. A theoretical basis for a biopharmaceutical classification system: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 1995;12:413–420.
7. Smith SE, Rawlins MD. Variability in Human Drug Response. London: Butterworths, 1973.
8. British National Formulary 68. London: British Medical Association and the Royal Pharmaceutical Society, September 2014–March 2015.
9. Dobbs EH et al. Uptake of etorphine and dihydromorphine after sublingual and intramuscular administration. Eur J Pharmacol 1969;7:328.
10. Gibaldi M, Kanig JL. Absorption of drugs through the oral mucosa. J Oral Ther Pharmacol 1965;31:440–450.
11. Speirs CF. Oral absorption and secretion of drugs. Br J Clin Pharm 1977;4:97–100
Further reading
Freire AC et al. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. Int J Pharm 2011;415:15–28.
Pinto JF. Site-specific drug delivery systems within the gastro-intestinal tract: from the mouth to the colon. Int J Pharm 2010;395:44–52.
Sattar M et al. Oral transmucosal drug delivery: current status and future prospects, Int J Pharm 2014;471:498–506.
Varum FJO et al. Oral modified release formulations in motion. The relationship between gastro-intestinal transit and drug absorption. Int J Pharm 2010;395:26–36.
Wilson CG, Crowley PJ (eds) Controlled Release in Oral Drug Delivery. New York: Springer-CRS, 2011.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


