Figure 11.1 Solubility curves for total parenteral nutrition solutions containing amino acid (15 g dm–3) and 10% dextrose at pH 5.5. Dotted line: Aminosyn. Solid line: TrophAmine. Dashed lines: relative calcium-to-phosphate ratios.
Modified with permission from Fitzgerald KA, MacKay MW. Calcium and phosphate solubility in neonatal parenteral nutrient solutions containing TrophAmine. Am J Hosp Pharm 1986;43:88–93.
11.1.2 In vivo pH effects
The sensitivity of the properties of most drugs to changes in the pH of their environment means that the hydrogen-ion concentration will be an important determinant of solubility, crystallisation and partitioning. Gastric pH is 1–3 in normal subjects, but the measured range of pH values in the human stomach is wide, as can be seen in Fig. 11.2. Remembering that pH is a logarithmic scale, the order of the change in the gut and its effect on aqueous and lipid solubility in particular can be appreciated. Changes in the acid–base balance therefore have a marked influence on the absorption and thus on the activity of drugs.
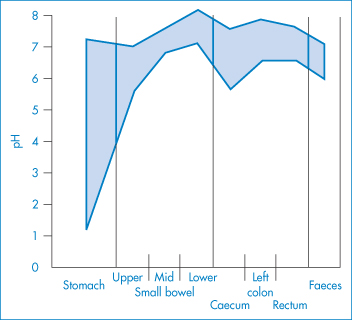
Figure 11.2 pH profile in the gut as measured by radiotelemetric capsule; the shaded area represents extremes of values observed in 9 subjects.
Reproduced with permission from Watson BW et al. pH profile of gut as measured by radiotelemetry capsule. Br Med J 1972;2:104.
Ingestion of antacids, food and weak electrolytes will all change the pH of the stomach. Weakly acidic drugs, being un-ionised in the stomach, will be absorbed from the stomach by passive diffusion. One might expect, therefore, that concomitant antacid therapy would delay or partially prevent absorption of certain acidic drugs. The main mechanism would be an increase in pH of the stomach, increasing ionisation of the drug and reducing absorption. A problem in generalisations of this kind is that the acid-neutralising capacity of antacids is very variable, as the results quoted in Table 11.2 show. Some acidic drugs, listed in Table 11.3, are also known to be absorbed in the intestine, in which case the co-administration of an antacid is not necessarily prohibited, as its effects may be transitory.
Table 11.2 Amounts of various antacids required to neutralise 50 mEq HCl
Antacid | Neutralising capacity of 1 g or 1 cm3 | Dose required to neutralise 50 mEq | Weight of tablet (g) | No. of tablets | |
cm3 0.1 mol dm–3 HCl | mEq HCl | ||||
Powders | |||||
NaHCO3 | 115 | 11.5 | 4.4 g |
|
|
MgO | 85 | 8.5 | 5.9 g |
|
|
CaCO3 | 110 | 11.0 | 4.5 g |
|
|
Magnesium trisilicate | 10 | 1.0 | 50 g |
|
|
MgCO3 | 8 | 0.8 | 63 g |
|
|
Suspensions |
|
|
|
|
|
Al(OH)3 gel | 0.7 | 0.07 | 715 cm3 |
|
|
Milk of Magnesia | 27.7 | 2.8 | 17.8 cm3 |
|
|
Titralac | 24 | 2.4 | 20.6 cm3 |
|
|
Aludrox | 1.7 | 0.17 | 294 cm3 |
|
|
Oxaine | 2.4 | 0.24 | 208 cm3 |
|
|
Mucaine | 1.7 | 0.17 | 294 cm3 |
|
|
Kolantyl gel | 3.4 | 0.34 | 147 cm3 |
|
|
Tablets | |||||
Gastrogel | 5.0 | 0.5 | 100 g | 1.08 | 93 |
Gastrobrom | 15.0 | 1.5 | 33.3 g | 1.48 | 23 |
Glyzinal | 2.5 | 0.25 | 200 g | 0.72 | 278 |
Actal | 7.7 | 0.77 | 65 g | 0.60 | 109 |
Amphotab | 2.5 | 0.25 | 200 g | 1.04 | 192 |
Gelusil | 2.5 | 0.25 | 200 g | 1.36 | 147 |
Nulacin | 10.0 | 1.0 | 50 g | 3.12 | 17 |
Kolantyl wafer | 5.0 | 0.50 | 100 g | 1.64 | 61 |
Titralac | 42.5 | 4.25 | 11.8 g | 0.65 | 18 |
Almacarb | 3.0 | 0.3 | 167 g | 1.28 | 130 |
Dijex | 4.6 | 0.46 | 109 g | 1.65 | 66 |
Reproduced from Piper DW, Fenton BH. Antacid therapy of peptic ulcer. II. An evaluation of antacids in vitro. Gut 1964;5:585, copyright 1964, with permission from BMJ Publishing Group Ltd.
Note that proprietary preparations available in different countries may not have the same formulation.
Table 11.3 Drugs whose absorption may be affected by antacid administration
Drug whose activity would be reduced | Drug whose activity would |
Tetracyclines | Theophylline |
Nalidixic acid | Chloroquine |
Nitrofurantoin | Mecamylamine |
Benzylpenicillin | Amfetamine |
Sulfonamides | Levodopa |
|
The effects of antacids are not always clear-cut, as pointed out at the beginning of this chapter. There are contradictory reports of the effect of antacids on the absorption of levodopa, as one example. Levodopa is metabolised within the gastrointestinal tract and more rapidly degraded in the stomach than in the intestine, so the rate at which the drug is emptied from the stomach can affect its availability. It has been suggested that, when an antacid is administered prior to the drug, serum levodopa concentration is increased because it is transferred to the intestine more rapidly. |

The use of ranitidine, nizatidine, famotidine and other H2-antagonists has given rise to the possibility of a drug interaction involving the subsequent increase in gastric pH, as these drugs inhibit gastric acid secretion. A few subjects have transient achlorhydria after oral cimetidine, so increased absorption of acid-labile drugs is a predictable side-effect, as breakdown is reduced.
Sodium bicarbonate (sodium hydrogen carbonate) is one of the most effective antacids in terms of neutralising capacity. It can greatly depress the absorption of tetracycline – the mean amount of drug appearing in the urine of patients receiving only drug was 114 mg at 48 hours and was 53 mg for those also given sodium bicarbonate. Chelation (see section 11.5) is not possible with the monovalent Na+ ion as it is with the multivalent components of other antacids, nor does adsorption occur. If the drug is dissolved prior to administration, the antacid does not affect the excretion of the antibiotic, suggesting that in normal dosage forms it is the rate of dissolution of the drug that is affected by the antacid, as explained below in the case of tetracycline.
The aqueous solubility of tetracycline at pH 1–3 is 100-fold greater than at pH 5–6. Consequently, its rate of solution, dc /dt, at this lower pH is greatly reduced as, according to equation (11.1),

where cs is the solubility. A 2 g dose of NaHCO3 will increase the intragastric pH above 4 for a period of 20–30 minutes, sufficient time for 20–50% of the undissolved tetracycline particles to be emptied into the duodenum, where the pH (at 5–6) is even less favourable for solution to occur. The fraction of drug absorbed is therefore decreased.
Effects of antacids other than directly on pH
The effect of antacids on gastric-emptying rate is a factor that makes difficult a direct physicochemical analysis of the problem. The difficulty in predicting the effect of antacids is clearly shown by studies with naproxen, a weakly acidic non-steroidal anti-inflammatory with a pKa of 4.2. Several textbooks of drug interactions state that antacids decrease the absorption of acidic drugs such as nalidixic acid, nitrofurantoin and benxylpenicillin (as indicated in Table 11.3), but antacids both increase and decrease the absorption of naproxen. Magnesium carbonate, magnesium oxide and aluminium hydroxide decrease absorption and, as these are insoluble agents, adsorption effects that reduce the quantity of free drug in solution are suspected (Fig. 11.3). On the other hand intake of Maalox, which contains magnesium and aluminium hydroxides, slightly increased the area under the curve. The extent to which Al3+ and Mg2+ ions chelate with nalidixic acid is not clear, but the structure of the drug suggests it has chelating potential (see section 11.5).
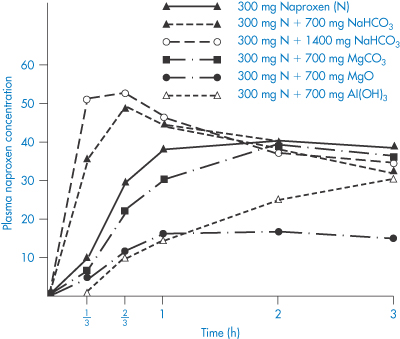
Figure 11.3 Mean plasma concentrations (μg cm–3) of naproxen in 14 male volunteers with and without intake of sodium bicarbonate, magnesium oxide or aluminium hydroxide.
Modified with permission from Segre EJ et al. Letter: Effects of antacids on naproxen absorption. N Engl J Med 1974;291:582.
|
Even though gastric emptying tends to become more rapid as the gastric pH is raised, antacid preparations containing aluminium or calcium are prone to retard emptying, and magnesium preparations to promote it. The caution about the co-administration of antacids containing divalent or trivalent metals and tetracyclines should be extended to antacids containing sodium bicarbonate or any substance capable of increasing intragastric pH. |

Similar principles can be applied to intestinal absorption, although here the pH gradients between the contents of the intestinal lumen and capillary blood are smaller. Sudden changes in the acid–base balance may, none the less, change the concentration of drugs able to enter cells, providing that the pH change does not alter binding of the drug to protein, or drug excretion, which of course it invariably does. Accurate prediction of the effect of a change in the acid–base balance on the activity of any drug requires a knowledge of its site of action and the potential effect of pH changes on its excretion and biotransformation, and requires knowledge of the extent of pH changes throughout the body.
|
Ingestion of some antacids over a period of 24 hours will increase urinary pH and hence affect renal resorption and handling of some drugs. For example, administration of sodium bicarbonate with aspirin reduces blood salicylate levels by about 50%, probably owing to its increased excretion in the urine. Although high doses of alkalising agents that raise the pH of the urine will increase the renal excretion of free salicylate and result in lowering of plasma salicylate levels, in some commercial buffered aspirin tablets (such as Bufferin) there is insufficient antacid to cause a change in the pH of the gastric fluids. The small amount of antacid is sufficient, however, to aid the dissolution of the acetylsalicylic acid and this leads to more favourable absorption rates. |

The importance of urinary pH
Change in the pH of urine will change the rate of urinary excretion of many drugs (as represented in Fig. 11.4). When a drug is in its un-ionised form it will more readily diffuse from the urine to the blood. In an acidic urine, acidic drugs will diffuse back into the blood from the urine. Acidic compounds such as nitrofurantoin are excreted faster when the urinary pH is alkaline. Amfetamine, imipramine and amitriptyline are excreted more rapidly in acidic urine. The control of urinary pH in studies of pharmacokinetics is thus vital. It is difficult, however, to find compounds to use by the oral route for deliberate adjustment of urinary pH. Sodium bicarbonate and ammonium chloride may be used but are unpalatable. IV administration of acidifying salt solutions presents one approach, especially for the forced diuresis of basic drugs in cases of poisoning.
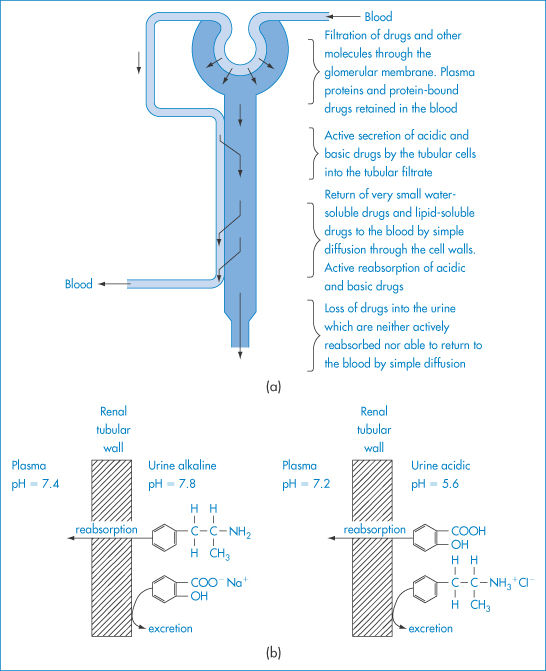
Figure 11.4 (a) A highly simplified diagram of a kidney tubule to illustrate the filtration and secretion of drugs from the blood into the tubular filtrate, and their subsequent reabsorption or loss in the urine. (b) Schematic representation of the influence of urinary pH on the passive reabsorption of a weak acid and a weak base from the urine in the renal tubules; at a high pH the passive reabsorption of the weak base and the excretion of the weak acid are enhanced, while at a low pH value the reabsorption of the weak acid and the excretion of the weak base are enhanced.
|
Urinary pH can be important in determining drug toxicity more directly. A preparation containing the urinary antiseptic methenamine mandelate and sulfamethizole caused turbidity in the urine of 9 out of 32 patients. The turbidity was higher in acidic urine, and was caused by precipitation of an amorphous sulfonamide derivative containing 63% sulfamethizole. The efficacy of both agents is reduced by precipitation and the danger of renal blockade is, of course, increased.2 |

Precipitation of drugs in vivo
Pain on injection may be the result of precipitation of a drug at the site of injection brought about either by solvent dilution or by alteration in pH. Precipitation of drugs from formulations used intravenously can, of course, lead to thromboembolism. The kinetics of precipitation under realistic conditions must be appreciated, since a sufficiently slow rate of infusion may obviate problems from this source as the drug precipitates and then redissolves. A simple but important equation3 yields the flow rate (Q) of blood or normal saline required to maintain a drug in solution during its addition to an IV fluid:

where R is the rate of injection of drug in mg min−1 and Sm is the drug’s apparent maximum solubility in the system (mg cm−3). Using diazepam and normal saline as an example, if R is 5 mg min−1, Sm is approximately 0.3 mg cm−3, so Q would have to exceed 17 cm3 min−1 to prevent observable precipitation. As this is a high rate of infusion, it is evident that the administration of diazepam through the tubing of an IV drip is likely to result in precipitate formation.
|
Too rapid injection of the diazepam preparation directly into the venous supply might result in precipitation; slow venous blood flow would contribute to the effect. This would perhaps explain a finding that thrombophlebitis occurs less frequently when smaller veins are avoided and when injection of diazepam is followed by rigorous flushing of the infusion system with normal saline.4 |

11.2 Dilution of mixed-solvent systems
In several cases the special nature of a formulation will preclude dilution by an aqueous infusion fluid. Injectable products containing phenytoin, digoxin and diazepam may come into this category if they are formulated in a non-aqueous but water-miscible solvent (such as an alcohol–water mixture) or as a solubilised (e.g. micellar) preparation. Addition of the formulation to water may result in precipitation of the drug, depending on the final concentration of the drug and solvent. It has been suggested that precipitation of the relatively insoluble diazepam may account for the high (3.5%) incidence of thrombophlebitis that occurs when diazepam is given intravenously.
Other additives in formulations may give rise to subtle problems that are not immediately obvious. One diazepam injection contains 40% propylene glycol and 10% ethanol; it is buffered with sodium benzoate and benzoic acid and preserved with benzyl alcohol. Addition of this formulation to normal saline results in the formation of a precipitate; a precipitate also forms on addition of the diazepam solution to human plasma.
A graphical technique has been described to predict whether a solubilised drug system will become supersaturated and thus have the potential to precipitate. When a drug dissolved in a cosolvent system is diluted with water, both drug and cosolvent are diluted. The logarithm of the solubility of a drug in a cosolvent system generally increases linearly with the percentage of cosolvent present (Fig. 11.5a). On dilution, the drug concentration falls linearly with a fall in the percentage of cosolvent. The aim of the graphical method is to plot dilution curves and solubility curves on the same graph. This is achieved in Fig. 11.5b, where the dilution curves have been plotted semilogarithmically for three systems containing initially 1, 2 and 3 mg cm−3 of drug substance (plots I, II and III, respectively). With solution III, dilution below about 30% cosolvent causes the system to be supersaturated; with solution II, below 20% cosolvent the solubility line and the dilution line touch. Only solutions containing 1 mg cm−3 can be diluted without precipitation.
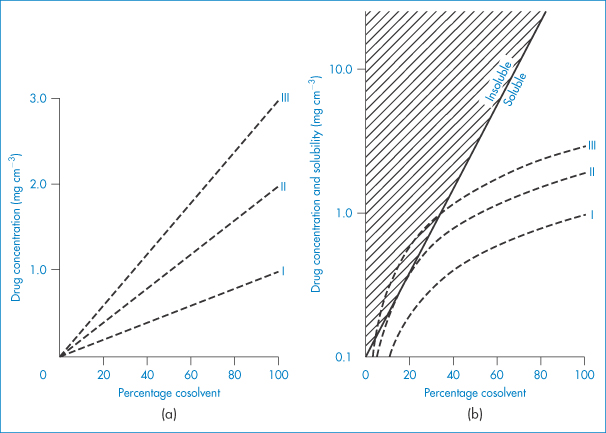
Figure 11.5 (a) Dilution profiles for three solutions (I, II and III) in pure cosolvent containing 1, 2 and 3 mg cm−3 drug, respectively. (b) Curves from (a) plotted on semilog scale along with typical solubility line.
Reproduced with permission from Yalkowsky SH, Valvani S. Precipitation of solubilized drugs due to injection or dilution. Drug Intell Clin Pharm 1977;11:417.
11.3 Cation–anion interactions
The interaction between a large organic anion and an organic cation may result in the formation of a relatively insoluble precipitate. Complexation, precipitation or phase separation can occur in these circumstances, the product being affected by changes in ionic strength, temperature and pH. Examples of cation–anion interactions include those between procainamide and phenytoin sodium, procaine and thiopental sodium, and hydroxyzine hydrochloride and benzylpenicillin (Scheme 11.1). The nature of many of these interactions has not been studied in detail. In the absence of this it is necessary to predict possible incompatibilities from a knowledge of the physical properties of the drug and other components in the formulation. Sometimes, as when chlorpromazine and morphine injections are mixed, the incompatibility is not due to an interaction between the two drugs but to drug–bactericide interaction; the chlorocresol contained in the morphine injection precipitates with the chlorpromazine, possibly by an anion–cation interaction. Nitrofurantoin sodium must be diluted prior to use with 5% dextrose or with sterile water for injection; alkyl p-hydroxybenzoates (parabens), phenol or cresol, all of which tend to precipitate the nitrofurantoin, must be absent.
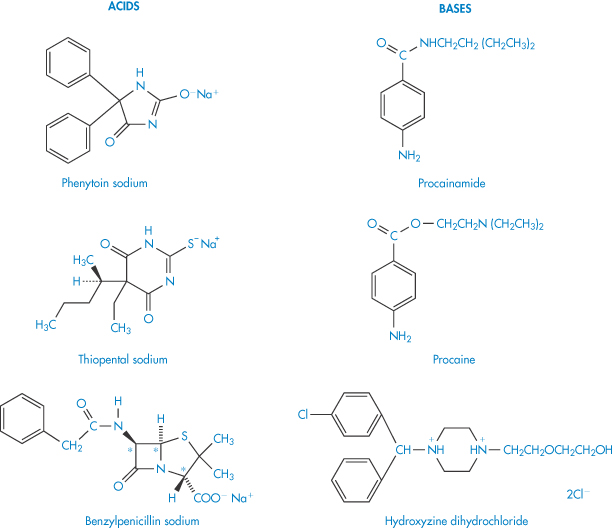
Scheme 11.1
Phase diagrams such as that shown in Fig. 11.6 are useful in determining regions of incompatibility and compatibility in cation–anion mixtures because admixture is not always contraindicated. The example shown here is for mixtures of disodium cromoglicate (DSCG, sodium cromoglicate) which is a di-anionic drug with a cationic surfactant, tetradecyldimethylammonium bromide (C14BDAC). As can be seen, the interaction is strongly concentration-dependent. In some regions, below the line AB, the two ions coexist. Ion pairs (see below) form in the shaded region below AB. Above this solubility product line, turbidity occurs in the hatched region. On increasing the concentration of surfactant, the complex is solubilised so that the interaction is masked.
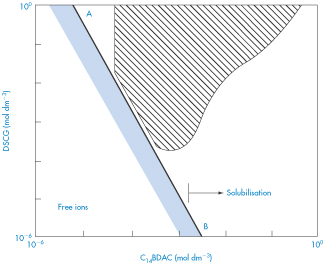
Figure 11.6 Phase diagram for mixtures of disodium cromoglicate (DSCG, sodium cromoglicate) and tetradecyldimethylammonium bromide (C14BDAC).
Reproduced with permission from Tomlinson E. Pharm Int 1980;1:156–158.
The incompatibility of organic iodide contrast media and antihistaminic drugs (added to reduce anaphylactic reactions) is due to the acidity of the antihistamine solutions, resulting in the precipitation of the organic iodide.5 One of the antihistamines, promethazine, reacted strongly with all the contrast media, probably because its solution had the lowest pH of the drugs studied. It is not only with IV fluids that such interactions may occur. Examples have been quoted of the inadvisable mixture of syrups; for example, immediate precipitation in a prescribed mixture of a syrup containing cloxacillin sodium and a syrup containing the bases codeine and promethazine. Precipitation was followed by a 20% loss in antibiotic activity in 5 hours and 99% loss in 5 days. The double-decomposition reactions involved are likely to be those shown in Fig. 11.7.
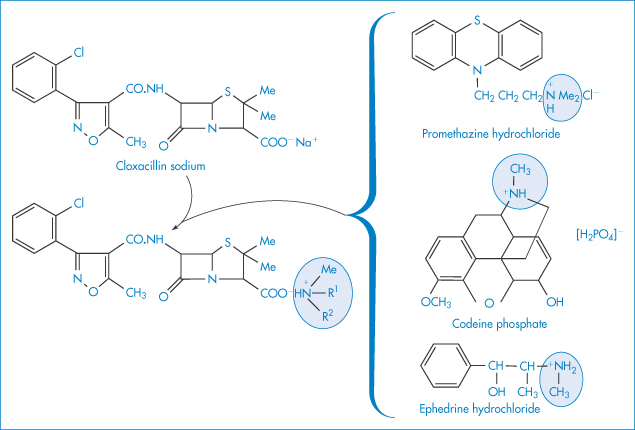
Figure 11.7 The interaction of cloxacillin sodium with promethazine hydrochloride, codeine phosphate and ephedrine hydrochloride.
Complexes that form are not always fully active. A well-known example is the complex between neomycin sulfate and sodium lauryl sulfate that will form when Aqueous Cream BP is used as a vehicle for neomycin sulfate. Aqueous cream comprises 30% emulsifying ointment, which itself is a mixture of emulsifying wax that contains 10% of sodium lauryl sulfate or a similar anionic surfactant.
Interactions are also not always visible. The formation of visible precipitates depends to a large extent on the insolubility of the two combining species in the particular mixture and the size to which the precipitated particles grow.
Interactions between drugs and ionic macromolecules are another potential source of problems. Heparin sodium and erythromycin lactobionate are contraindicated in admixture, as are heparin sodium and chlorpromazine hydrochloride or gentamicin sulfate. Tables of incompatibilities abound with such examples. Interference with the sulfate groups reduces the anticoagulant activity of heparin.
Ion-pair formation
Ion-pair formation may be responsible for the absorption of highly charged drugs such as the quaternary ammonium salts and sulfonic acid derivatives, the absorption of which is not explained by the pH-partition hypothesis. Ion pairs may be defined as neutral species formed by electrostatic attraction between oppositely charged ions in solution, which are often sufficiently lipophilic to dissolve in non-aqueous solvents. The formation of an ion pair (Fig. 11.8) results in the ‘burying’ of charges and alteration to the physical properties of the drug. Interactions between charged drug species and appropriate lipophilic ions of opposite charge may constitute a drug interaction and may occur in vitro or in vivo.
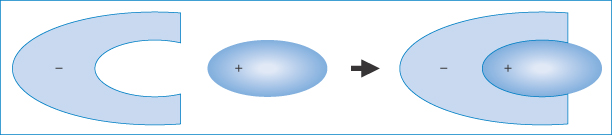
Figure 11.8 Representation of an organic ion-pair – the anion is shown here interacting with a cationic molecule of complementary shape (purely schematic), thus masking the exposure of the charge to the aqueous environment.
11.4 Polyions and drug solutions
The extensive clinical use of polyionic solutions for IV therapy means that drugs are frequently added to systems of a complex ionic nature. Reduction in the solubility of both weak electrolytes and non-electrolytes can occur through salting out, a phenomenon discussed in section 4.2.3.
Most physicochemically based drug interactions can take place in the body, or outside it, or during concomitant drug administration, so it is probably not profitable to consider them separately. Some interactions, such as complexation, which are probably more important in vivo than in vitro, are discussed below.
|
Sodium sulfadiazine and sulfafurazole diolamine in therapeutic doses (1 mg) added to 5% dextrose and 5% dextrose and saline solution have been found to be compatible, yet when added to commercial polyionic solutions (such as Abbott Ionosol B, Baxter electrolyte No. 2), both rapidly form heavy precipitates. pH and temperature are two vital parameters, but the pH effect is not simply a solubility-related phenomenon. Polyionic solutions of a lower initial pH (4.4–4.6) cause crystallisation of sulfafurazole at room temperature within 2.5 hours, the pH values of the admixtures being 5.65 and 5.75, respectively. Other solutions with slightly higher initial pH levels (6.1–6.6) formed crystals only after preliminary cooling to 20°C at pH values from 4.25 to 4.90. If the temperature remains constant, the intensity of precipitation varies with the composition and initial pH of the solution used as a vehicle.6 |

11.5 Chelation
The term chelation (derived from the Greek chele, meaning lobster’s claw) relates to the interaction between a metal atom or ion and another species, known as the ligand, by which a heteroatomic ring is formed. Chelation changes the physical and chemical characteristics of both the metal ion and the ligand. It is simplest to consider the ligand as the electron-pair donor and the metal the electron-pair acceptor, the donation establishing a coordinate bond. Many chelating agents act in the form of anions that coordinate to a metal ion. For chelation to occur there must be at least two donor atoms capable of binding to the same metal ion, and ring formation must be sterically possible. For example, ethylenediamine (1,2-diaminoethane, NH2CH2CH2NH2) has two donor nitrogens and acts as a bidentate (two-toothed) ligand. When a drug forms a metal chelate, the solubility and absorption of both drug and metal ion may be affected, and drug chelation can lead to either increased or decreased absorption. Tetracyclines have similar chelating groups in their structure. Therapeutic chelators are used in syndromes where there is metal ion overload. Deferiprone (I) chelates iron. Ethylenediaminetetraacetic acid (EDTA) (II) as the monocalcium disodium salt is used in the treatment of lead poisoning.
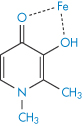
Structure I Deferiprone, shown as chelate with iron
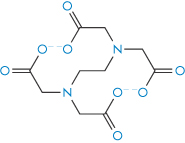
Structure II Ethylenediaminetetraacetic acid (EDTA)
Tetracyclines
Probably the most widely quoted example of complex formation leading to decreased drug absorption is that of tetracycline chelation with metal ions. Polyvalent cations such as Fe2+ and Mg2+, and anions such as trichloracetate or phosphate, interfere with absorption in both model and real systems. Figure 11.9 shows the effect of a dose of 40 mg ferrous ion on serum levels following 300 mg of tetracycline. As can be seen from this figure, the nature of the iron salt ingested is important. Ferrous sulfate has the greatest inhibitory effect on tetracycline absorption, perhaps because it dissolves in water more quickly than organic iron compounds. The ability of the various iron compounds to liberate ferrous or ferric ions in the upper part of the gastrointestinal tract before tetracycline is absorbed would seem to be an essential step in the interaction. The order of activity of the different iron salts in the chelation process in vivo turns out to be the same as the order of the intestinal absorption of these iron compounds. All the active tetracyclines form stable chelates with Ca2+, Mg2+ and Al3+. The antibacterial action of the tetracyclines depends on their metal-binding activity, as their main site of action is on the ribosomes, which are rich in magnesium.
The tetracyclines have an avidity for divalent metals similar to that of glycine (III) but they have a greater affinity for the trivalent metals, with which they form 3 : 1 drug–metal chelates. Therapeutically active tetracyclines form 2 : 1 complexes with cupric, nickel and zinc ions while inactive analogues form only 1 : 1 complexes.
The structures of some tetracyclines are reproduced in Table 11.5, together with their pKa values; from these data it should be possible to determine something of the relative affinities of the tetracyclines for metal ions. One piece of evidence relating to the site of chelation is that isochlortetracycline, which lacks the C-11, C-12 enolic system, does not chelate with Ca2+ ions.
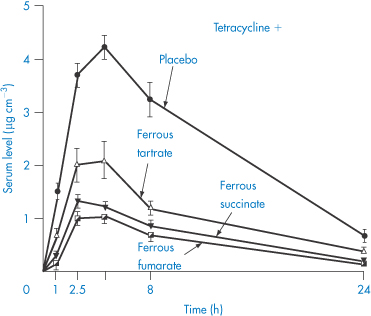
Figure 11.9 The effect of simultaneous ingestion of various iron salts (dose equivalent to 40 mg elemental iron) and tetracycline hydrochloride (500 mg) on serum levels of tetracycline (mean levels in 6 patients).
Reproduced from Neuvonen PJ, Turakka H. Inhibitory effect of various iron salts on the absorption of tetracycline in man. Eur J Clin Pharmacol 1974;7:357, with kind permission from Springer Science and Business Media.
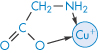
Structure III The 1:1 copper-glycine chelate
|
The complexing of tetracyclines with calcium poses a problem in paediatric medicine. Discoloration of teeth results from the formation of a coloured complex with the calcium in the teeth; the deposition of drug in the bones of growing babies can lead to problems in bone formation.7 Table 11.4 shows that there is no correlation between the binding capacity of a tetracycline with iron and that with calcium, suggesting different modes of complexation. The in vitro data are simpler to interpret: the serum levels are the result of two processes, the chelation of the tetracycline and the partitioning of the chelate. Clinical studies have shown that the absorption of doxycycline is not significantly affected by milk in conditions where the absorption of tetracycline itself is reduced. |

Table 11.4 Relative calcium-binding capacities of tetracycline derivatives and decreases in serum levels after 200 mg ferrous sulfate
Tetracycline | Calcium bindinga (%) | Decrease in serum concentration (%) after 200 mg ferrous sulfate |
Demeclocycline | 74.5 | n.s. b |
Chlortetracycline | 52.7 | n.s. b |
Tetracycline | 39.5 | 40–50 |
Methacycline | 39.5 | 80–85 |
Oxytetracycline | 36.0 | 50–60 |
Doxycycline | 19–22 | 80–90 |
a Percentage of dissolved antibiotic (5 mg/50 cm3 H2O) bound to calcium phosphate after overnight shaking. Reproduced from Osol A, Pratt R. The United States Dispensatory, 27th edn. Philadelphia, PA: Lippincott, p. 1155ff.
b n.s. = not studied.
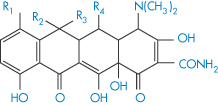
Table 11.5 Structures and pKa values for some tetracyclines
| |||||||
| R1 | R2 | R3 | R4 | pKa1 | pKa2 | pKa3 |
Chlortetracycline | Cl | Me | OH | H | 3.27 | 7.43 | 9.33 |
Oxytetracycline | H | Me | OH | OH | 3.5 | 7.6 | 9.2 |
Tetracycline | H | Me | OH | H | 3.33 | 7.7 | 9.5 |
Demeclocycline | Cl | H | OH | H | 3.3 | 7.16 | 9.25 |
Doxycycline | H | Me | H | OH | 3.4 | 7.7 | 9.7 |
The decreased aqueous solubility of chelates suggests increased lipophilicity but, in the case of the tetracycline chelates, precipitation would decrease the biological activity of the drug as it would be less available for transport across membranes; the larger molecular volume of the chelate would also prevent easy absorption in the intact form.
Complex formation of a coumarin derivative occurs between magnesium ions present in antacid formulations. In this case the formation of a more absorbable species is indicated, since plasma levels of bishydroxycoumarin are elevated in the presence of magnesium hydroxide but are unaffected by aluminium hydroxide; neither antacid influenced warfarin absorption.8 A magnesium–bishydroxycoumarin chelate having the structure (IV) has been isolated. Adsorption may counteract a chelate-mediated increase in absorption, so the adsorptive properties of aluminium hydroxide may be responsible for the lack of effect of this antacid on the absorption of the drug. Results are shown in Fig. 11.10. It has been suggested that the more rapid and complete absorption of warfarin makes it less susceptible to interactions of this type.8
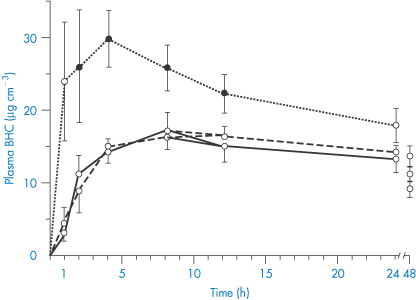
Figure 11.10 Plasma levels of bishydroxycoumarin (BHC), also known as dicoumarol, in 6 subjects after a 300 mg oral dose with water (solid line), magnesium hydroxide (dotted line) or aluminium hydroxide (dashed line). Closed data points represent a significant difference from control.
Reproduced with permission from Ambre JJ, Fischer LF. Effect of coadministration of aluminium and magnesium hydroxides on absorption of anticoagulants in man. Clin Pharm Ther 1973;14:231–237.
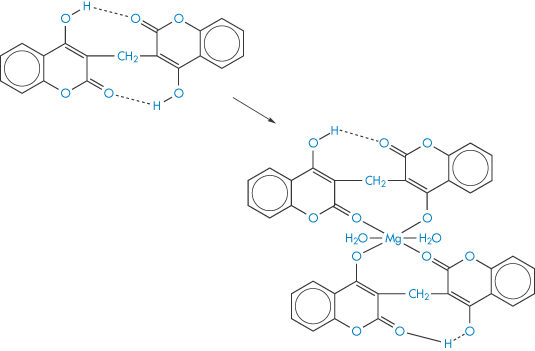
Structure IV Suggested form of the bishydroxycoumarin-Mg chelate
Chelation of ciprofloxacin (V) by aluminium hydroxide and calcium carbonate reduces bioavailability, as seen in Fig. 11.11. Other quinolones (VI–IX) undoubtedly suffer the same fate.
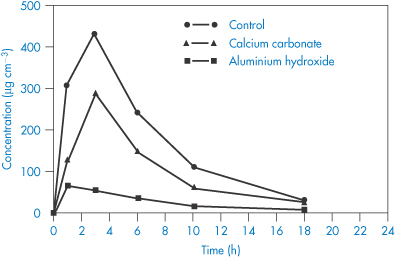
Figure 11.11 Concentrations of ciprofloxacin in the urine following administration of 50 mg to 12 healthy volunteers in a three-way randomised crossover design by RW Frost et al. (Antimicrob Agents Chemother 1992;36:830), who write: ‘The precise conditions required for ciprofloxacin to form chelate complexes with other cations have not been fully elucidated, but the carboxyl group seems to be the most likely site for chelation. Thus, the gastric pH would need to be elevated sufficiently to ionise the carboxyl group. Therefore, the lack of interaction with dairy products might be attributable to a gastric pH that was not elevated sufficiently to ionise the carboxyl group. Alternatively, it could be due to a decrease in the availability of calcium to chelate ciprofloxacin because of a lipid barrier.’
Following treatment of acrodermatitis enteropathica with diiodohydroxyquinolone (X), the absorption and retention of dietary zinc and other trace metals have been found to be greater than in subjects not receiving the drug. Chelation of zinc and other trace metals by the drug could have resulted in the increased absorption from the intestinal lumen.
Desferrioxamine (as the mesylate) is used as a drug to sequester iron in iron poisoning or chronic iron overload; penicillamine is similarly used to aid the elimination of copper in Wilson disease. Chelation is also used for the safe delivery of toxic ions such as gadolinium (Gd3+), which is used as a magnetic resonance imaging enhancing agent. One such preparation is gadobenic acid (XI), which is a gadolinium–benzyloxypropionic tetraacetate (BOPTA) (XIa) octodentate chelate.
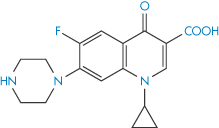
Structure V Ciprofloxacin
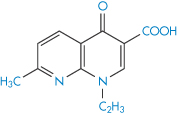
Structure VI Nalidixic acid
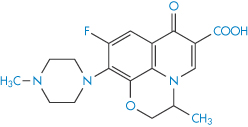
Structure VII Ofloxacin
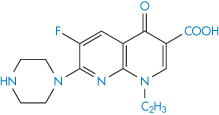
Structure VIII Enoxacin
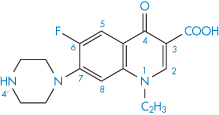
Structure IX Norfloxacin
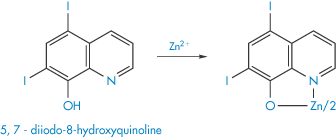
Structure X Suggested form of the chelate of zinc-diiodohydroxyquinolone
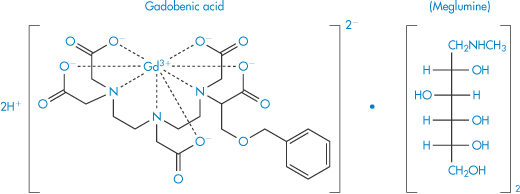
Structure XI Gadobenic acid as the meglumine salt
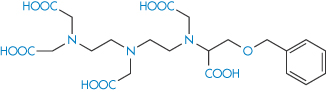
Structure XIa Benzyloxypropionic tetraacetate (BOPTA)
11.6 Other complexes
Molecular complexes of many types may be observed in systems containing two or more drug molecules. Generally, association follows from attractive interactions (hydrophobic, electrostatic or charge transfer interactions) between two molecules. In the charge transfer system one component is usually an aromatic compound; the second may be a saturated moiety containing an electron lone pair (donor atom) or a weakly acidic hydrogen (acceptor atom). The interaction therefore takes place between electron-rich donors and electron-poor acceptors. Interactions between aromatic rings in which there is a parallel overlap of π-systems fall into this category, as in the following example involving nicotinamide (acceptor) and the indole moiety of tryptophan (donor) (XIIScheme 11).
The imidazole moiety (XIII) is involved in many interactions; it is regarded as aromatic and the molecule is planar. Not surprisingly, caffeine (XIV) and theophylline (XV) are frequently implicated in interactions with aromatic species. Caffeine increases the solubility of not only ergotamine but also that of benzoic acid. The marked difference in the solubilising properties of caffeine and dimethyluracil (XVI) towards substances such as benzoic acid suggests that the imidazole ring of the xanthine nucleus is the portion involved in the interaction. In the main, 1 : 1 complexes are formed but 2 : 1 drug–caffeine complexes may also be found. Hydrophobic interactions are also implicated in the interaction, since caffeine has a greater solubilising capacity than theophylline, which, as can be seen, lacks the NCH3 group in the imidazole nucleus.
11.6.1 Interaction of drugs with cyclodextrins
Compounds that have interior cavities large enough to incarcerate organic ‘guest’ molecules have been named molecular container compounds.
Over the last decade there has been growing interest in the interaction of drugs with one group of container compounds, the β-cyclodextrins (cyclic D-glucose oligomers) and their now myriad derivatives. The cyclodextrin molecules have cyclic structures with internal diameters of 0.6–1 nm (see section 4.6). The interior cavity of the cyclodextrin ring is hydrophobic in nature and binds a hydrophobic portion of the ‘guest’ molecule, usually forming a 1 : 1 complex. The possible structure of β-cyclodextrin complexes of aspirin is shown in Fig. 11.12.
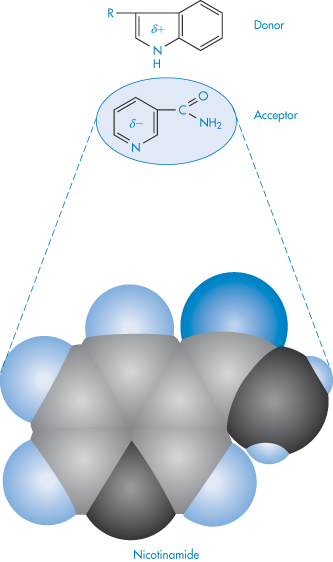
Structure XII Example of donor–acceptor interaction between tryptophan (donor) and nicotinamide
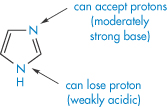
Structure XIII Imidazole
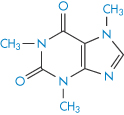
Structure XIV Caffeine
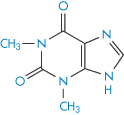
Structure XV Theophylline
Structure XVI Dimethyluracil
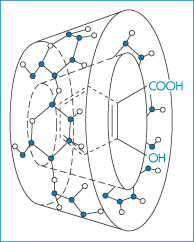
Figure 11.12 Schematic drawing of an aspirin–β-cyclodextrin complex.
Reproduced from Loftsson T, Masson M. Cyclodextrins in topical drug formulations: theory and practice. Int J Pharm 2001;225:15–30. Copyright Elsevier 2001.
Inclusion of some drugs (including peptides) into cyclodextrins increases their solubility, chemical stability and absorption. The solubilising effect of β-cyclodextrin on flufenamic acid suggests that a 1 : 1 complex is formed in aqueous solution. A relationship between the oil/water partition coefficient of a drug and its tendency to form inclusion complexes has generally been found.9 This indicates that the hydrophobic cavity of the cyclodextrin ‘eye’ is more attractive to lipophilic guest molecules. The formation constants for mefenamic acid were, however, lower than expected from the Me or Cl ortho substituent. α-Cyclodextrin showed no appreciable interaction with the anti-inflammatory acids, mefenamic acid and meclofenamic acid, suggesting that its cavity is not large enough to contain the drug molecules.10
The altered environment of the drug molecules leads to changes in stability. The cyclodextrins catalyse a number of chemical reactions, such as hydrolysis, oxidation and decarboxylation. Although interactions between drugs and cyclodextrins have been mostly the result of deliberate attempts to modify the behaviour or properties of the drug, they are included here to illustrate an additional mode of interaction with other components that may well occur inside the body as well as in the laboratory.
Sugammadex: a beneficial interaction with anaesthetics
Three of the widely used general anaesthetics, the neuromuscular blocking agents cis-atracurium, vecuronium and rocuronium, have durations of action of around 37–81 minutes, 36–137 minutes and 33–119 minutes respectively, according to one survey. Such anaesthetics may require reversal depending on the surgical procedures being undertaken. Atracurium was designed to have a short duration of action, but vecuronium and rocuronium have significantly longer actions. These latter are distinguished from atracurium in having their –N-onium groups separated by a steroidal backbone, as can be seen from their structures, and they interact in the blood with sugammadex when it is administered IV. This complex renders them inactive, thus reversing their action. This is a good example of a beneficial interaction. Sugammadex is a γ-cyclodextrin derivative which effectively acts as an antagonist11 (Fig. 11.13) and is therefore a modified excipient used therapeutically. The complex is excreted mainly in the urine.
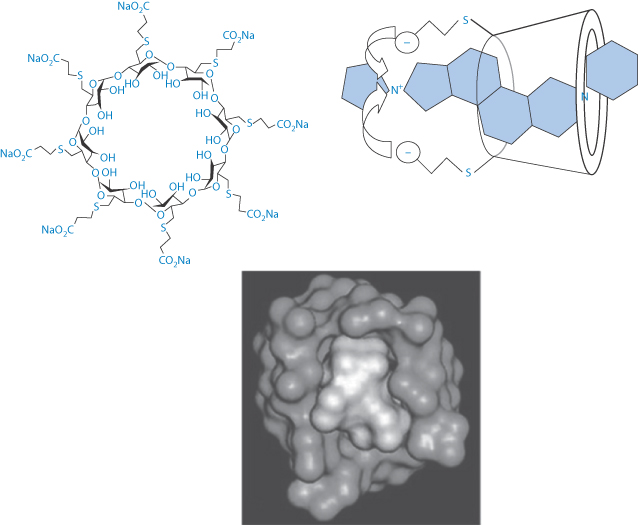
Figure 11.13 (Top left) Sugammadex structure with a cavity of 1.1 nm; (top right) a schematic view of the interaction of sugammadex with the steroidal neuromuscular-blocking agent, rocuronium and (below) a space-filling model of the sugammadex with rocuronium, this being reproduced with permission from Raft J et al. 51o Congrès National d’Anésthesie et de Réanimation. Elsevier-Masson; 2009.
According to Chemistry Beta (chemistry.stackexchange.com): ‘Sugammadex binds rocuronium really tightly. The reason for this is straightforward – lipophilic interactions with the inner cavity of the cyclodextrin, which is the perfect size … The negatively charged carboxylate groups lock into place with the protonated amine’.
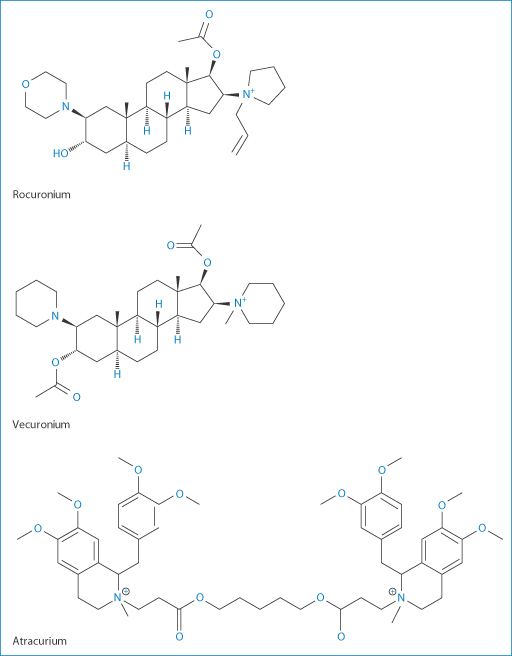
Figure 11.14 Neuromuscular blocking agents: rocuronium, vecuronium and atracurium.
Clearly sugammadex has affinities with other anaesthetic molecules of similar dimensions (Fig. 11.14) such as vecuronium, but the affinity for vecuronium is 2.5 times lower. It does not interact with atracurium which, as can be seen from its structure, has bulky benzylisoquinolinium groups that cannot be accommodated in the cavity of this particular cyclodextrin.
|
Levels of atracurium will not be changed by administration of sugammadex, but vecuronium levels are reduced. Although its affinity for sugammadex is weaker, it is itself seven times more potent than rocuronium, so that fewer molecules are present. Vecuronium blockage can thus be successfully achieved. The possibilities of forming complexes with other steroidal molecules such as aldosterone and glucocorticoids have been found to be insignificant. |

11.6.2 Ion-exchange interactions
Ion-exchange resins are now being used medicinally and as systems for modified release of drugs (Fig. 11.15). Colestyramine and colestipol are insoluble quaternary ammonium anion-exchange resins that, when administered orally, bind bile acids and increase their elimination because the high-molecular-weight complex is not absorbed. As bile acids are converted in vivo into cholesterol, colestyramine is used as a hypocholesteraemic agent. When given to patients receiving other drugs as well, the resin would conceivably bind anionic compounds and reduce their absorption. Phenylbutazone (XVII), warfarin (XVIII), chlorothiazide (XIX) and hydrochlorothiazide are bound strongly to the resin in vitro.
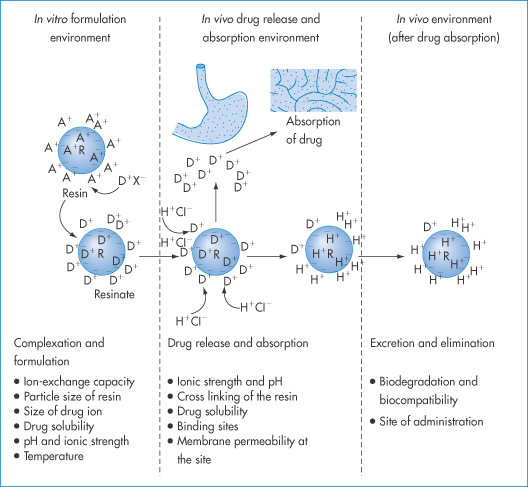
Figure 11.15 The basis of the ion-exchange process in drug delivery with factors affecting the therapeutic efficacy of the system at each stage. R and a blue circle represent resin; a minus sign depicts the integral ion of the resin, and A+ is the counterion. D is the drug ion, X– is the ion associated with drug cation and H+Cl− is hydrochloric acid. Ions inside and outside the resin indicate ions adsorbed at the surface, as well as in the interior, of the resin structure.
Reproduced from Anand V et al. Drug Discov Today 2001;6:905–915.
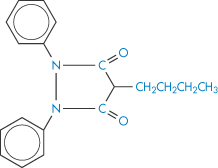
Structure XVII Phenylbutazone
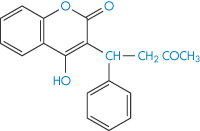
Structure XVIII Warfarin
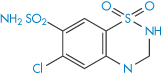
Structure XIX Chlorothiazide
Colestyramine had no effect in animal experiments on the blood levels of these drugs. However, 95% of both warfarin and phenylbutazone were bound and peak blood levels of the latter were halved, although the same total amount was eventually absorbed. A single dose of resin did not significantly reduce the pharmacological effect of warfarin. Nevertheless, it is prudent to administer drugs orally a short time before colestyramine to preclude delay in the action of the drug through adsorption and slow leaching. Table 11.6 shows the effect of very high resin levels on the serum concentration of sodium fusidate. Since the binding is pH-dependent (Table 11.7), an ion-exchange resin can bind any drug with an appropriate charge under pH conditions in which both species are ionised. Decreased drug absorption caused by colestyramine or colestipol in the main has been reported with thyroxine, aspirin, phenprocoumon, warfarin, chlorothiazide, cardiac glycosides and ferrous sulfate as a consequence of binding. Absorption of vitamin K and vitamin B12 is decreased indirectly by competition of the resin for binding sites on intrinsic factor molecules.12,13
Table 11.6 The effect of colestyramine administration on sodium fusidate levels
Resin dose (mg kg–1) | Mean serum concentration (μg cm–3) |
0 | 3.7 |
72 | 2.5 |
215 | 1.9 |
356 | 0.8 |
Reproduced from Johns WH, Bates TR. Drug-cholestyramine interactions. II. Influence of cholestyramine on GI absorption of sodium fusidate. J Pharm Sci 1972;61:735. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Table 11.7 Binding of acetylsalicylic acid (aspirin) in vitro to colestyramine
| Percentage binding | |||
| pH 2 | pH 3 | pH 5 | pH 7.5 |
Buffer | 5 | – | 6 | 6 |
Water | 51 | 95 | 98 | 97 |
Reproduced from Halin K-J et al. Effect of cholestyramine on the gastrointestinal absorption of phenprocoumon and acetylosalicylic acid in man. Eur J Clin Pharmacol 1972;4:142, with kind permission from Springer Science and Business Media.
The influence of buffer ions on the binding of the drug in Table 11.7 indicates one of the problems of making in vitro measurements and attempting to assess the biological significance of the results. Binding of cefalexin, clindamycin (XX) and the components of co-trimoxazole to colestyramine has been measured; in vivo absorption of these substances in the presence of the resin is delayed and somewhat reduced, but the changed pattern of absorption is unlikely to affect therapeutic efficacy, except perhaps with cefalexin, where it is more pronounced.
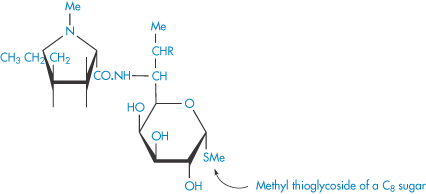
Structure XX Lincomycin (R = OH) and clindamycin (R = Cl)
Colestilan
A quite recent introduction is the product colestilan, a crosslinked copolymer of 2-methylimidazole and epichlorohydrin (XXI) which acts as an anion exchanger resin with an affinity for phosphate. It binds the ions in the gut, removing them from the enterohepatic circulation. Colestilan is not absorbed from the gut. It is used in the treatment of hyperphosphataemia in adult patients with chronic kidney disease receiving haemodialysis or peritoneal dialysis.
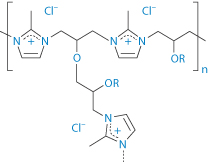
Structure XXI Colestilan
11.7 Adsorption of drugs
The process of adsorption and its medical and pharmaceutical applications have been dealt with in some detail in Chapter 5. The use of adsorbents to remove noxious substances from the lumen of the gut is considered there. Adsorbents generally are non-specific so will adsorb nutrients, drugs and enzymes when given orally. It is not uncommon for adsorbents to be administered in combination with various drugs and it becomes a matter of practical importance to determine the extent to which adsorbents will interfere with the absorption of the drug substance from the gastrointestinal tract.
Several consequences of adsorption are possible. If the drug remains adsorbed until the preparation reaches the general area of the absorption site, the concentration of the drug presented to the absorbing surfaces will be much reduced. The driving force for absorption would then be reduced, resulting in a slower rate of absorption. During the course of absorption of the drug, it is probable that the adsorbate will dissociate in an attempt to re-establish equilibrium with drug in its immediate environment, particularly if there is competition for absorption sites from other substances in the gastrointestinal tract. As a consequence, the concentration of free drug in solution would be maintained at a low level and the absorption rate would be reduced. Alternatively, the release of drug from the adsorbent might be complete before reaching the absorption site, possibly hastened by the presence of electrolytes in the gastrointestinal tract, in which case absorption rates would be virtually identical to those in the absence of adsorbent. Figure 11.16 shows the reduced amounts of promazine excreted in the urine when the promazine was administered with activated charcoal – evidence of a reduced absorption rate of this drug in this situation.
A further example is the delayed absorption of lincomycin (XX) when administered with kaolin and pectinic acid (Kaopectate) (Fig. 11.17). The dramatic effect of several antacids on the adsorption of digoxin from elixirs is shown in Fig. 11.18. In view of the relatively low doses of these glycosides, these adsorption effects are likely to impair digoxin bioavailability.
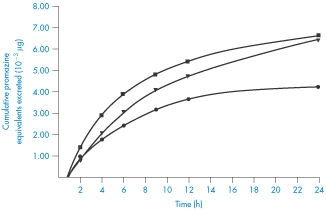
Figure 11.16 Cumulative amounts of promazine equivalents excreted in the urine following administration of various dosage forms to humans: ◼, promazine in simple aqueous solution; ▼, promazine plus activated attapulgite; ●, promazine plus activated charcoal.
Reproduced from Sorby DL. Effect of adsorbents on drug absorption. I. Modification of promazine absorption by activated attapulgite and activated charcoal. J Pharm Sci 1965;54:677. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
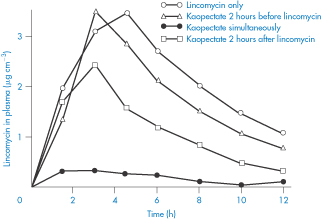
Figure 11.17 The influence of Kaopectate (kaolin and pectinic acid) on the absorption of lincomycin in humans after oral application; note the decrease in plasma levels caused by the simultaneous administration of the absorbent.
Reproduced with permission from Wagner JG. Design and data analysis of biopharmaceutical studies in man. Can J Pharm Sci 1966;1:55.
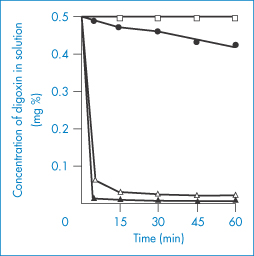
Figure 11.18 Adsorption of digoxin from Lanoxin paediatric elixir at 37 ± 0.1°C by: ●, Aluminium Hydroxide Gel BP (10% v/v); 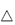 , Magnesium Trisilicate Mixture BPC (10% v/v); ▲, Gelusil suspension (10% v/v); ◻, Lanoxin elixir diluted 1 : 10 with water.
, Magnesium Trisilicate Mixture BPC (10% v/v); ▲, Gelusil suspension (10% v/v); ◻, Lanoxin elixir diluted 1 : 10 with water.
Reproduced from Khalil SAH. The uptake of digoxin and digitoxin by some antacids. J Pharm Pharmacol 1974;26:961. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
If the possibility of adsorption on to formulation ingredients is not kept in mind, erroneous conclusions may be drawn from bioavailability studies, as was the case in the suspected in vivo interaction between p-aminosalicylic acid (PAS) and rifampicin. It was thought that PAS impaired the intestinal absorption of rifampicin, reducing its serum levels to about half those occurring when it was administered by itself (Fig. 11.19). It was later found that bentonite, a naturally occurring mineral (montmorillonite), consisting chiefly of hydrated aluminium silicate, present in the PAS granules was adsorbing the antibiotic and delaying its absorption14 (Fig. 11.19). It can readily be seen how the initial erroneous conclusion was drawn.
Figure 11.19 Plasma concentrations of rifampicin (RMP) after oral administration in solution (10 mg kg–1) given alone (▲), with p-aminosalicyclic acid (PAS) granules (●) or with Na-PAS tablets (○); mean values (n = 6 patients) are indicated.
Reproduced from Boman G et al. Mechanism of the inhibitory effect of PAS [p-aminosalicylic acid] granules on the absorption of rifampicin. Adsorption of rifampicin by an excipient, bentonite. Eur J Clin Pharm 1975;8:293–299, with kind permission from Springer Science and Business Media.
Loss of activity of preservatives can arise from adsorption on to solids commonly used as medicaments. Benzoic acid, for example, can be adsorbed to the extent of 94% by sulfadimidine. Adsorption can be suppressed, however, with hydrophilic polymers. In solid-dosage forms, talc, a commonly used tablet lubricant, has been reported to adsorb cyanocobalamin and consequently to interfere with intestinal absorption of this vitamin.
11.7.1 Protein and peptide adsorption
The pharmaceutical properties of peptides and proteins are discussed in Chapter 13. Adsorption can be a problem because of the amphipathic nature of many peptides, and becomes pharmaceutically important when they are present in low concentrations in solution. The adsorption of peptides on to glass has been ascribed to bonding between their amino groups and the silanol groups of the glass. A decapeptide derivative of LHRH, the natural luteinising hormone-releasing hormone, has two basic amino groups positively charged at low pH, providing an opportunity for binding to silanol groups.15 Siliconisation of the glass did not inhibit adsorption completely, suggesting that ionic binding was not the only mechanism of interaction. Phosphate buffer at 0.1 mol dm−3 concentration and acetate ions at 0.16 mol dm−3 concentration (both at pH 5) were most effective in preventing adsorption.
11.7.2 Protein aggregation and interactions
As Bee and co-authors discuss,16 ‘proteins can aggregate during processing, shipping, storage and delivery to the patient. Formulation conditions are chosen to maximise protein stability over its shelf-life. However, even thermodynamically and colloidally stable proteins may aggregate after interaction with the container-closure system, container leachates, or particulate contaminants because the stability with respect to surfaces may involve different physical processes than those that determine stability in bulk solution’. We can classify at least some of these adverse events as incompatibilities and interactions. The following are listed by Bee et al.16:
- nucleation of the aggregation of recombinant human platelet-activating factor acetylhydrolase has been attributed to the presence of silica nanoparticles from glass containers
- Teflon as the cause aggregation of insulin17
- silicone rubber delivery tubing decreasing the activity of interleukin-2 by 97%18
- silicone oil syringe lubricant inducing aggregation of several model proteins19
- leachates from tungsten causing protein precipitation
- stainless-steel particles shed from a filler pump in the laboratory environment caused aggregation of a monoclonal antibody
- Teflon containers fostering aggregation of an IgG2 during freeze thawing.
Protein aggregation in ambulatory pumps can be serious: not only can aggregation reduce the biological activity of macromolecules, but the aggregates can cause blockage of needles. There are several mechanisms of protein aggregation, but the two most relevant to the present chapter are nucleation-controlled aggregation and surface-induced aggregation.20 In the first, the native monomer interacts with a nucleus and adsorbs on to the nucleus, often with partial unfolding; this can lead to growth and visible precipitation. In the latter mechanism, the native monomer adsorbs on to the surface of containers or closures and partly unfolds; this can trigger growth through aggregation.
11.8 Drug interactions with plastics
In Chapter 7 we discussed the adsorption of insulin on to glass and plastic materials used in syringes and giving sets. The plastic tubes and connections used in IV containers and giving sets can adsorb or absorb a number of drugs (Fig. 11.20), leading to significant losses in some cases. Drugs that show a significant loss when exposed to plastic, in particular poly(vinyl chloride) (PVC), include insulin, glyceryl trinitrate, diazepam, chlormethiazole, vitamin A acetate, isosorbide dinitrate and a miscellaneous group of drugs such as phenothiazines, hydralazine hydrochloride and thiopental sodium.
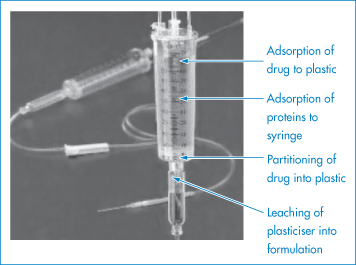
Figure 11.20 The possible modes of interaction between a drug formulation and the plastic of a giving set.
A theoretical treatment to account for the loss of glyceryl trinitrate from solution to plastic containers has been developed21 (Derivation Box 11A).
Some idea of the rate and extent of disappearance of warfarin sodium from PVC infusion bags can be gained from Fig. 11.21. The marked effect of pH is seen. Losses can obviously be significant. When medazepam is present at an initial concentration of 40 μg cm−3 in 100 cm3 normal saline stored in similar bags, a 76% loss of the drug occurs in 8 hours at 22°C (Table 11.8).22
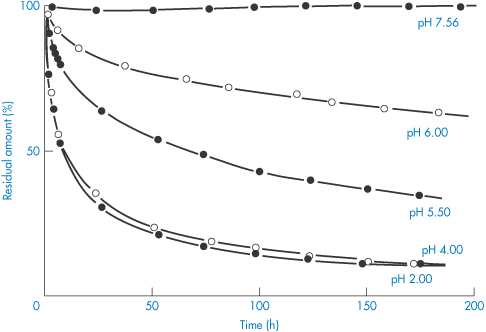
Figure 11.21 Disappearance of warfarin sodium from aqueous buffered solutions stored in 100 cm−3 poly(vinyl) chloride infusion bags at room temperature.
Reproduced with permission from Illum L, Bundgaard H. Sorption of drugs by plastic infusion bags. Int J Pharm 1982;10:339–351. Copyright Elsevier 1982.
Table 11.8 Loss of drugs from normal saline solutions in poly(vinyl chloride) (PVC) bags stored at 22°C for 8 hours
Drug | Initial solute conc. | % loss | |
PVC | PVC | ||
Diazepam | 40 | 60 | 31 |
| 120 | 58 | 31 |
Medazepam | 40 | 76 | – |
Oxazepam | 40 | 22 | 12 |
Nitrazepam | 40 | 15 | 10 |
Warfarin sodiuma | 20 | 49 | – |
Warfarin sodium | 20 | 29 | – |
Glyceryl trinitrate | 200 | 54 | – |
Thiopental sodiumb | 30 | 25 | – |
Pentobarbital sodium | 30 | 0 | 0 |
Hydrocortisone acetate | 20 | 0 | 0 |
Reproduced from Illum L, Bundgaard H. Sorption of drugs by plastic infusion bags. Int J Pharm 1982;10:339–351. Copyright Elsevier 1982.
a At pH 2 and pH 4.
b At pH 4.0 and pH 7.2.
Preservatives such as the methyl and propyl parabens present in formulations can be sorbed into rubber and plastic membranes and closures, thus leading to decreased levels of preservative and, in the extreme, loss of preservative activity.
11.9 Protein binding
This is a topic that requires a chapter to itself for full coverage. Here we have space only for a treatment of mechanisms of protein binding that might enable readers to recognise molecules likely to be protein-bound. High levels of protein binding alter the biological properties of the drug as free drug concentrations are reduced. The bound drug assumes the diffusional and other transport characteristics of the protein molecule. In cases where drug is highly protein-bound (around 90%), small changes in binding lead to drastic changes in the levels of free drug in the body. If a drug is 95% bound, 5% is free. Reduction of binding to 90% by, for example, displacement by a second drug doubles the level of free drug. Such changes are not evident when binding is of a low order.
Most drugs bind to a limited number of sites on the albumin molecule. Binding to plasma albumin is generally easily reversible, so that drug molecules bound to albumin will be released as the level of free drug in the blood declines. Drugs bound to albumin (or other proteins) are attached to a unit too large to be transported across membranes. They are thus prevented from reacting with receptors or from entering the sites of drug metabolism or drug elimination until they dissociate from the protein.
Plasma albumin consists of a single polypeptide chain of molecular weight 67 000 ± 2000 Da. Human albumin contains between 569 and 613 amino acid residues. It is a globular molecule with a diameter of 5.6 nm (calculated assuming the molecule to be an anhydrous sphere). With an isoelectric point of 4.9, albumin has a net negative charge at pH 7.4, but it is amphoteric and capable of binding acidic and basic drugs. Subtle structural changes can occur on the binding of small molecules. Fatty-acid binding produces a volume increase and a decrease in the axial ratio. This is due primarily to non-polar interaction between the hydrocarbon tail of the fatty-acid molecule and the binding site, and reflects the adaptability of the albumin molecule. When a hydrophobic chain penetrates into the interior of the globular albumin molecule, the helices of the protein separate, producing a small change in the tertiary structure of the protein.
What are the binding sites? Organic anions are thought to bind to a site containing the amino acid sequence Lys-Ala-Try-Ala-Val-Ala-Arg. The five non-polar side-chains of these residues form a hydrophobic ‘pool’. A cationic group is present at each end. It has been proposed that the ε-amino group of lysine is the point of attachment for anionic drugs. Albumin is, however, capable of binding molecules that are not ionised at all, such as cortisone or chloramphenicol. Acidic drugs such as phenylbutazone, indometacin and the salicylates may establish primary contact with the albumin by electrostatic interactions, but the bond can be strengthened by hydrophobic interactions. The dual interaction would therefore limit the number of possible binding sites for anionic organic drugs. In spite of electrostatic interactions, a number of drugs bind to albumin according to their degree of lipophilicity. Evidence for the importance of the hydrophobic interaction has been obtained from studies of the binding of phenol derivatives, which depends mainly on the hydrophobic character of the substituent, the phenolic hydroxyl group playing no significant role in the binding process. Similarly, penicillin derivatives and phenothiazines bind in a manner that depends on the hydrophobic characteristics of side-chains. It has been concluded that binding to albumin is a process comparable to partitioning of drug molecules from a water phase to a non-polar phase. The hydrophobic sites are not necessarily ‘preformed’. Fatty acids and warfarin are both capable of inducing conformational changes that result in the formation of hydrophobic ‘pools’ in the protein.
Plasma proteins other than albumin may also be involved in binding; examples of such interactions are shown in Fig. 11.22. Blood plasma normally contains on average about 6.72 g of protein per 100 cm3, the protein comprising 4.0 g of albumin, 2.3 g of globulins and 0.24 g of fibrinogen. Although albumin is the main binding protein, dicoumarol is also bound to β- and γ-globulins, and certain steroid hormones are specifically and preferentially bound to particular globulin fractions.
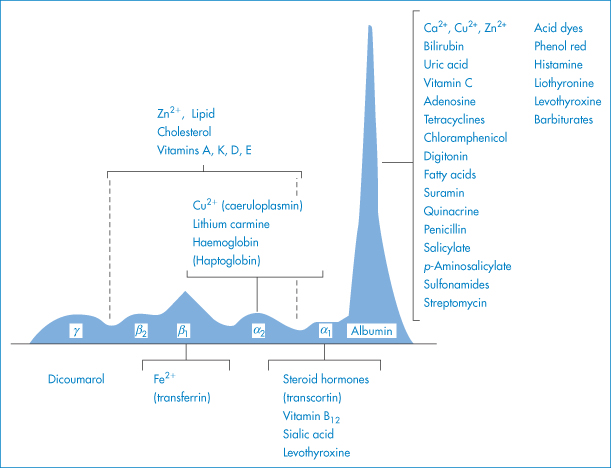
Figure 11.22 Interactions of drugs and chemicals with plasma proteins; plasma proteins are depicted according to their relative amounts.
Modified from Putnam FW. In: Neurath H (ed.) The Proteins, 2nd edn, vol. 3. New York: Academic Press; 1965.
If protein binding is assumed to be an adsorption process obeying the law of mass action, then the following equations can be derived (Derivation Box 11B) for the number of moles of drug, r, bound to the protein in a system,

or

where K is the equilibrium constant, Df is the molar concentration of unbound drug and n is the number of binding sites per protein molecule.
Protein-binding results are often quoted as the fraction of drug bound, β. This fraction varies generally with the concentration of both drug and protein, as shown in equation (11.5), which relates β with n, K and concentration:
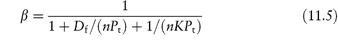
where Pt is the total molar concentration of protein.
11.9.1 Thermodynamics of protein binding
Estimation of the thermodynamic parameters of binding allows interpretation of the mechanisms of interaction. Table 11.9 gives the thermodynamic parameters of binding of a number of agents to bovine serum albumin. The negative value of ΔG implies that binding is spontaneous. ΔH is negative, signifying an exothermic process and a reduction in the strength of the association as temperature increases. ΔS is positive, most likely signifying loss of structured water on binding. A diagrammatic representation of this last process is shown in Fig. 11.23. This diagram shows the change in the ordered water in the hydrophobic cavity of the albumin and around the non-polar portion of the drug. The loss of the structured water gives rise to the positive entropy change, which contributes to a negative free-energy change.
Table 11.9 Thermodynamic parameters of binding of drugs to bovine serum albumin
Drug | Percentage bound | ΔG | ΔH | ΔS |
|
| (kJ mol–1) at 22 °C | (kJ mol–1) | (J mol–1 K–1) |
Aminopyrine (aminophenazone) | 24.7 | –17.7 | –5.19 | +42.2 |
Antipyrine (phenazone) | 21.4 | –17.1 | –4.98 | +41.0 |
4-Aminoantipyrine | 22.7 | –17.4 | –5.27 | +41.0 |
Phenylbutazone | 84.6 | –28.9 | –6.1 | +77.4 |
Oxyphenylbutazone | 80.3 | –27.4 | –5.90 | +73.2 |
Reproduced with permission from Ozeki S, Tejima K. Drug interaction II. Binding of some pyrazolidine derivates to bovine serum albumin. Chem Pharm Bull 1974;22:1297.
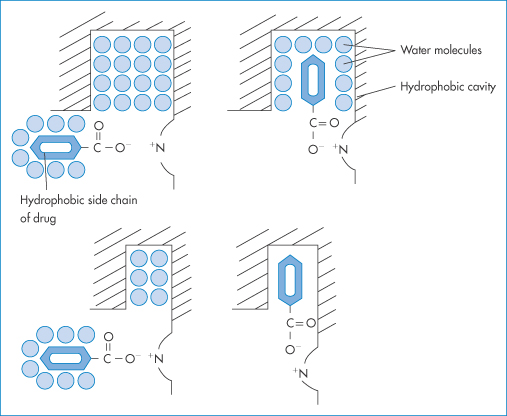
Figure 11.23 Electrostatic contact and hydrophobic binding of a molecular model with anionic and hydrophobic properties.
Reproduced with permission from Hasseblatt A. Interactions of drugs at plasma protein binding sites. Proc Eur Soc Study Drug Toxic 1972;13:89.
11.9.2 Lipophilicity and protein binding
Given the data in Table 11.9 it is not surprising to find that the extent of protein binding of many drugs is a linear function of their partition coefficient P (or log P). A linear equation of the form log(percentage bound/percentage free) = 0.5 log P − 0.665 has been found to be applicable to serum binding of penicillins. Although there may be an electrostatic component to the interaction, the binding increases with the degree of lipophilicity, suggesting, as is often the case, that more than one binding interaction is in force.
|
Binding to protein outside of the plasma may determine the characteristics of drug action or transport. Muscle protein may bind drugs such as digoxin and so act as a depot. Differences in the bioavailability of two antibiotics following intramuscular administration have been ascribed to differences in protein binding. Dicloxacillin, 95% bound to protein, is absorbed more slowly from muscle than ampicillin, which is bound only to the extent of 20%. |

Drug binding often changes with drug concentration and with protein concentration. On increasing the drug/protein ratio, saturation of some sites can occur and there may be a decrease in binding (as indicated by total percentage). Hence the importance of determining binding at realistic albumin concentrations (about 40 g dm−3). There may be more than one binding site for a drug.
In determining the pharmacological importance of protein binding, several factors have to be considered. If drug molecules not bound to plasma protein are freely distributed throughout the body, on leaving the blood they enter a volume 13 times as large as the plasma volume. Entry into the cerebrospinal fluid (CSF) depends on the concentration of free, diffusible drug in the plasma. Sulfanilamide enters CSF faster than sulfamethoxypyridazine because less is bound to serum albumin. Such binding factors may override the intrinsic lipophilicities, which in some cases may suggest a different order of penetration from that observed.
When binding occurs with high affinity and the total amount of drug in the body is low, drug will be present almost exclusively in the plasma. Drugs with lower association constants (K  106 or 107) will be distributed more in the body water spaces. When the number of available binding sites is reduced by a second drug, it will appear as if there has been an increase in overall drug concentration.
106 or 107) will be distributed more in the body water spaces. When the number of available binding sites is reduced by a second drug, it will appear as if there has been an increase in overall drug concentration.
Although drugs are predominantly bound to albumin, the amount taken up by erythrocytes must not be neglected. More directly, the effect of protein binding on antibiotic action is worth considering.
|
Penicillins and cephalosporins bind reversibly to albumin. Only the free antibiotic has antibacterial activity. Oxacillin in serum at a concentration of 100 μg cm−3 exhibits an antibacterial effect similar to that of 10 μg cm−3 of the drug in water. A high degree of serum protein binding may nullify the apparent advantage of higher serum levels of some agents (Table 11.10). An increase in the lipophilicity of penicillins results in decreased activity, although one normally expects that this should enhance penetration of bacterial cell walls and also absorption. The hydrophobic binding of penicillins to serum proteins reduces their potency in vivo by decreasing their effective concentration. |

Table 11.10 Protein binding and other characteristics of some penicillins and cephalosporins
Antibiotic | Log Pa | Serum protein binding (%) | Serum concentration (μg cm–3) during continuous infusion | Peak serum levels after 500 mg kg–1 | ||
|
|
| Total | Free | Total | Free |
Dicloxacillin | 3.24 | 96–98 | 25 | 1 | 15 | 0.3 |
Cloxacillin | 2.49 | 94–96 | 15 | 0.9 | – | – |
Oxacillin | 2.38 | 92–94 | 10 | 0.8 | 15 | 0.8 |
Nafcillin | – | 89–90 | 9 | 0.9 | 6 | 0.6 |
Benzylpenicillin | 1.72 | 59–65 | 16 | 5.6 | 4.5 | 1.6 |
Methicillin | 1.06 | 37–60 |
|
| 16 | 10 |
Ampicillin | – | 20–29 | 29 | 23.2 | 12 | 9 |
Carbenicillin | – | 50 | 73 | 36.5 | – | – |
Ceftriaxone | – | 95 | – | – | – | – |
Cefazolinb | – | 89 | – | – | – | – |
Cefditoren | – | 88 | – | – | – | – |
Cefalotin | 0.5 | 65 | 18 | 6.3 | 7.6 | 2.6 |
Cefaloridine | – | 20 | 24.7 | 19.7 | 18.5 | 14.8 |
Cefalexin | 0.7 | 15 | 27 | 23 | – | – |
Cefradineb | –1.2 | 14 | – | – | – | – |
a Log P values from Ryrfeldt A. J Pharm Pharmacol 1971;23:463 and Jack DB. Handbook of Chemical Pharmacokinetic Data. London: Macmillan; 1992.
b Additional values from Singhri SM et al. Chemotherapy 1978;24:121.
Reproduced with permission from Lien EJ et al. Diffusion of drugs into prostatic fluid and milk. Drug Intell Clin Pharm 1974;8:470 (originally published in Annals of Pharmacotherapy.
Comparisons between antibiotics are best made with activity–time plots and not serum concentration profiles, as the free levels often differ from the total antibiotic levels (see, for example, values for cloxacillin and benzylpenicillin in Table 11.10).
The second important consequence of protein binding is related to the fact that only free drug is able to cross the pores of the capillary endothelium. At equilibrium, levels of free drug on both sides of the capillary wall are equal. Albumin levels in most sites are considerably less than those in serum, so there is little bound drug in extravascular regions. There is, however, no correlation between the degree of protein binding and peak serum levels of the penicillins and cephalosporins of Table 11.10; other factors such as rates of elimination also determine the peak levels. Protein binding will affect transport into other tissues, however, as the gradient that determines movement is the gradient caused by free drug. Both ampicillin (50 mg kg−1 every 2 hours) and oxacillin (50 mg kg−1 h−1) produced similar peak levels in the serum when given as repeated IV boluses. Levels of free drug are markedly different, however, as oxacillin is 75% bound and ampicillin is 17.5% bound in rabbit serum.
11.9.3 Penetration of specialised sites
Muscle, bone and synovial and interstitial fluid are readily accessible to intravascular antibiotics by way of aqueous pores in the capillary supply. The CSF, brain, eye, intestines and prostate gland lack such pores, and entry into these areas is by way of a lipoidal barrier. There is evidence that extensive protein binding may prevent the access of antibiotics into the eye and the inflamed meninges.
The level of free drug in serum is important in determining the amount of drug that reaches tissue spaces.23 The total concentration of a drug in tissue fluid can be predicted from the serum concentration, the extent of serum protein binding and the protein binding in the tissue fluid. Equation (11.6) has been shown to be highly predictive under equilibrium conditions23:

where Ct is the tissue fluid drug concentration, Cs is the serum drug concentration and fs and ft are the free fractions of drug in serum and tissue fluid, respectively. If protein binding in the serum and tissue fluid is identical then, provided the same proteins are present in both ‘phases’, the tissue fluid drug concentration will equal the serum concentration; this is true for both high and low protein-containing extravascular fluid.
|
In the treatment of bacterial prostatitis; most currently available antibiotics do not readily pass across the prostatic epithelium. The pH of the prostatic fluid is lower than that of plasma, being around 6.6; milk has a pH of 6.8. In passage of drugs both into prostatic fluid and into milk, the degree of dissociation of the drug appears to be the most important factor determining the degree of penetration. The partition coefficient plays a secondary role. |

When the prostatic fluid/plasma ratios of sulfonamides were studied it was found that the results could be predicted more closely from the ratio of undissociated/dissociated drug rather than from log P values. It is probable that this is because lipophilic drugs will be highly protein-bound and thus unable to penetrate the boundary membranes.
11.10 Reflections on interactions
The information in this chapter demonstrates the multitude of ways in which drugs can interact with other drugs, with excipients, with the body, with containers. Lists of course can be consulted, but lists (such as the one which closes this chapter) are a starting point to understanding. The subject is undoubtedly complex and to make predictions from the body of knowledge that there is to cases involving new drugs or new formulations and dose regimes requires a high degree of understanding of the possibilities. We have covered many of these in this chapter, and address others in the following chapter on adverse events. But the following is a case history as it were of a search of recent medico-scientific literature on one group of drugs, which begins with the observation of the effects of a group of anticancer drugs and the effect on their absorption of the ingestion of acid-reducing agents, antacids, proton pump inhibitors or buffered medications. It points out that sometimes other factors are involved, not least the bioavailability of the product in question and whether or not it is optimised in a formulation (see the case of vemurafenib below), the dose/solubility ratio and the situation when over-the-counter (OTC) medicines are themselves involved in the picture (see the first clinical point below).
|
The availability of OTC medicines as combination formulations of an antacid and an H2-receptor antagonist can be a concern because these will increase the risk of drug–drug interactions by two mechanisms: a gastric pH-dependent mechanism by the drug itself and a cation-mediated chelation mechanism by antacids. *From Ogawa R, Echizen H. Clinically significant drug interactions with antacids. Drugs 2011;71:1839–1864. |

11.10.1 A narrative of anticancer drugs and proton pump inhibitors (PPI)
*
|
Many cancer patients frequently take acid-reducing medicines to alleviate symptoms of gastro-oesophageal reflux, thus potentially affecting the absorption of anticancer drugs and possibly jeopardising therapy. Clinical data suggest that the interactions are most relevant for anticancer drugs whose solubility varies over the pH range 1–4, the latter value being the pH of the stomach when the patient has used acid-reducing agents. Figure 11.24 shows the effect on the maximum absorbable dose of such drugs whose solubility is affected by an acid-reducing agent. |

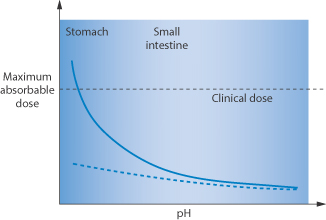
Figure 11.24 Graph showing (solid line) the maximum absorbable dose of drug as a function of pH, the clinical dose and (dashed line) the potential influence of acid-reducing agents in the stomach/small intestine.
Protein kinase inhibitors such as dasatinib, erlotinib, gefitinib (Fig. 11.25) and lapatinib inter alia are used in the treatment of non-small-cell lung cancer with activating epidermal growth factor receptor mutations. Their use comes with a long list of potential side-effects, many of which will be determined by their pharmacokinetics. They have a pH-dependent solubility. Vemurafenib differs in that it has a low solubility over all pH ranges, and has a high dose regimen up to 960 mg. Thus the effect of acid-reducing agents on a drug such as dasatinib will differ from that on vemurafenib (hence the danger of assuming that drugs in a pharmacological class behave identically).
Table 11.11 shows recorded effects of famotidine or omeprazole on dasatinib.
Table 11.11 Effect of acid-reducing agents (ARA) on the area under the curve (AUC) and peak plasma concentration (Cmax) of dasatinib
Drug | ARA | Mean change AUC | Cmax |
Dasatinibb | Famotidine 40 mg 10 h ptda |
|
|
| Famotidine 40 mg 2 h after dose | NSEd | NSE |
Dasatinibb | Maalox 30 mL 2 h ptda | NSE |
|
| Maalox 30 mL coadministered |
|
|
Dasatinibc | Omeprazole 40 mg daily for 5 days and on day 5 with dose |
|
|


a ptd = prior to dose.
b Eley T et al. J Clin Pharmacol 2009;49:700–709.
c Clinical study report 2009.
d NSE = no significant effect.
Reproduced from Budha NR et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther 2012;92:203. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
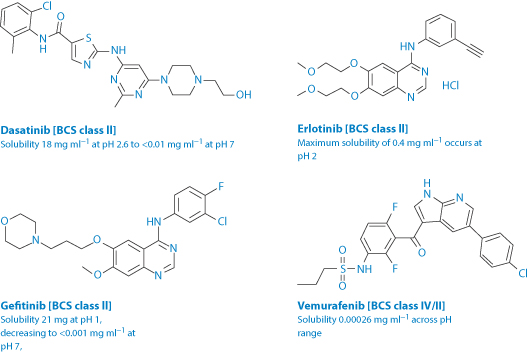
Figure 11.25 The structures, solubilities and Biopharmaceutics Classification System (BCS) class of four anticancer agents of related activity (protein kinase inhibitors). The pKa values of these drugs are: dasatinib: 3.1, 6.8 and 10.8; erlotinib: 5.4; gefitinib: 5.4 and 7.2; vemurafenib: 7.9.
Omeprazole reduces the AUC of erlotinib by 46% and the Cmax by 61%. These are significant effects.
The effects on absorption of acid-reducing agents on drugs in the four different Biopharmaceutics Classification System and Biopharmaceutical Drug Disposition Classification System classes are summarised in Table 11.12.
Table 11.12 The impact of acid-reducing agents on the absorption of 15 molecular targeted anticancer drugs
Class | BCS description | BDDCS description | Impact on absorption with acid-reducing therapy |
I | High solubility | High solubility | Minimal to none |
II | Low solubility | Low solubility | Moderate to high |
III | High solubility | High solubility | Minimal to none |
IV | Low solubility | Low solubility | Moderate to high |
Reproduced from Budha NR et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther 2012; 92:203. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
BCS = Biopharmaceutics Classification System; BDDCS = Biopharmaceutical Drug Disposition Classification System.
In a comprehensive examination of approved kinase inhibitors,24 it is emphasised that ‘Oral exposure involves interplay of multiple factors, including solubility, dissolution rate, permeability, disposition properties and food effects. Therefore, decent clearance, moderate/high permeability and appropriate formulation vehicles may overcome the effect of low intrinsic solubility, and result in an adequate oral exposure to achieve efficacy.’
Vemurafenib
In examining the data on these kinase inhibitors and the effect of acid-reducing agents, other issues around bioavailability also appear. This demonstrates the pharmaceutical difficulties encountered in drug development. Vemurafenib is now available as Zelboraf (Roche), formulated as a coprecipitate with hypromellose acetate succinate. The early formulations of a crystalline form demonstrated poor oral bioavailability dosed in capsules which were reported to be ‘challenging to manufacture and store’, because the crystal form with the highest solubility was unstable. To improve solubility and stability, it was reformulated as a coprecipitate with hypromellose acetate succinate, which resulted in a sixfold increase in bioavailability compared to the crystalline formulation.25 The amorphous nature of the coprecipitate aids dissolution (Fig. 11.26).

Figure 11.26 Photomicrograph of a coprecipitated bulk powder of a drug prepared with soluble polymers such as hypromellose acetate succinate.
Reproduced from Shah N et al. Development of novel microprecipitated bulk powder (MBP) technology for manufacturing stable amorphous formulations of poorly soluble drugs. Int J Pharm 2012;438:53–60. Copyright Elsevier 2012.
The effect of acid-reducing agents begs the question of the effect of food. A study in 201426 showed that a high-fat/high-calorie meal intake prolonged the Tmax of vemurafenib from 4 to 8 hours, but increased the AUC threefold.
The work to determine the optimum conditions in which to administer these anticancer agents continues. As the authors Ribas et al.26 state, ‘The exact mechanism of how a high-fat meal increased the single-dose exposure of vemurafenib is unknown, but it is likely the result of a combination of factors that influence the in vivo dissolution of the tablet and the absorption of vemurafenib. The predominant effect of food after a single oral dose of 960-mg vemurafenib tablets is on the absorption phase, with little or no effect on the terminal drug elimination. The variability associated with PK [pharmacokinetic] parameters seems greater in the fasted condition than in the fed condition.’
Summary
This chapter has dealt with the wide range of physicochemical factors that might cause changes in formulations or drug absorption processes. These have included effects of pH and solvent, interactions between cations and anions, salting in and salting out, ion-exchange reactions, adsorption and interactions with plastics, as well as protein binding, often mediated by hydrophobic interactions. It is, as always, difficult to anticipate the exact quantitative nature of the events that follow from these effects, especially in the body, where there are many compensating and contradictory influences. However, a knowledge of the theoretical possibilities allows us to rationalise what has been observed. By analogy we should therefore be able to predict likely effects with new drugs. One has to be cautious, and aware that what we anticipate theoretically may not take place in vivo, where a dynamic situation exists. But the physicochemical basis of interactions and incompatibilities must be appreciated if we are to rationalise interactions.
The Appendix contains tables of drug interactions that could be considered to be physicochemically driven. However, it is important that continuously updated sources are consulted, such as Stockley’s Drug Interactions (e.g. via Medicines Complete, which is updated frequently online).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


