Figure 2–1 The absorption, distribution, biotransformation (metabolism), and excretion of a typical drug after its oral administration.
DRUG ABSORPTION
Drug absorption refers to the passage of drug molecules from the site of administration into the circulation. The process of drug absorption applies to all routes of administration, except for the topical route, where drugs are applied directly on the target tissue, and intravenous administration, where the drug is already in the circulation. Drug absorption requires that drugs cross one or more layers of cells and cell membranes. Drugs injected into the subcutaneous tissue and muscle bypass the epithelial barrier and are more easily absorbed through spaces between capillary endothelial cells. In the gut, lungs, and skin, drugs must first be absorbed through a layer of epithelial cells that have tight junctions. For this reason, drugs face a greater barrier to absorption after oral administration than after parenteral administration.
Processes of Absorption
Most drugs are absorbed by passive diffusion across a biologic barrier and into the circulation. The rate of absorption is proportional to the drug concentration gradient across the barrier and the surface area available for absorption at that site, known as Fick’s Law. Drugs can be absorbed passively through cells either by lipid diffusion or by aqueous diffusion. Lipid diffusion is a process in which the drug dissolves in the lipid components of the cell membranes. This process is facilitated by a high degree of lipid solubility of the drug. Aqueous diffusion occurs by passage through aqueous pores in cell membranes. Because aqueous diffusion is restricted to drugs with low molecular weights, many drugs are too large to be absorbed by this process.
A few drugs are absorbed by active transport or by facilitated diffusion. Active transport requires a carrier molecule and a form of energy, provided by hydrolysis of the terminal high-energy phosphate bond of ATP. Active transport can transfer drugs against a concentration gradient. For example, the antineoplastic drug, 5-fluorouracil, undergoes active transport. Facilitated diffusion also requires a carrier molecule, but no energy is needed. Thus drugs or substances cannot be transferred against a concentration gradient but diffuse faster than without a carrier molecule present. Some cephalosporin antibiotics, such as cephalexin, undergo facilitated diffusion by an oligopeptide transporter protein located in intestinal epithelial cells.
Effect of pH on Absorption of Weak Acids and Bases
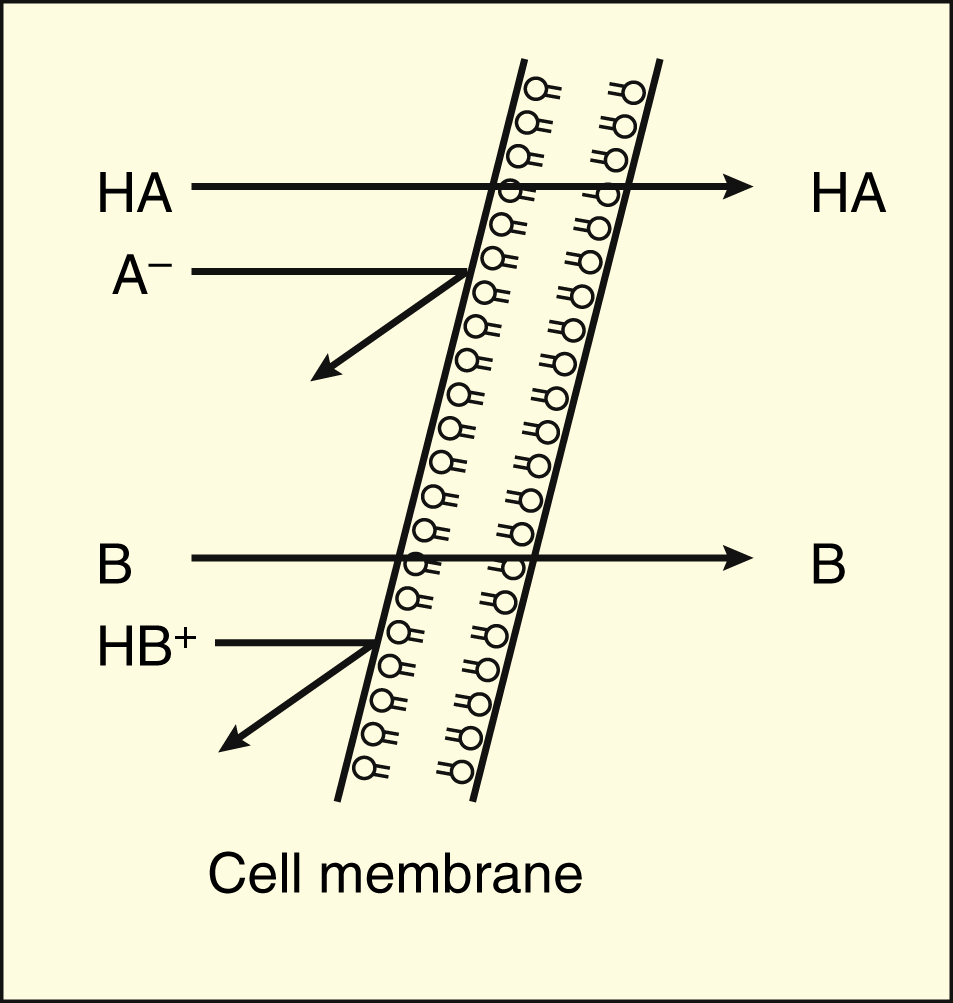
Many drugs are weak acids or bases that exist in both ionized and non-ionized forms in the body. Only the non-ionized form of these drugs is sufficiently soluble in membrane lipids to cross cell membranes (Box 2–1). The ratio of the two forms at a particular site influences the rate of absorption and is also a factor in distribution and elimination.
BOX 2–1 EFFECT OF pH ON THE ABSORPTION OF A WEAK ACID AND A WEAK BASE
Weak acids (HA) donate a proton (H+) to form anions (A−), whereas weak bases (B) accept a proton to form cations (HB+).

Only the non-ionized form of a drug can readily penetrate cell membranes.
The pKa of a weak acid or weak base is the pH at which there are equal amounts of the protonated form and the nonprotonated form. The Henderson-Hasselbalch equation can be used to determine the ratio of the two forms:
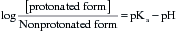
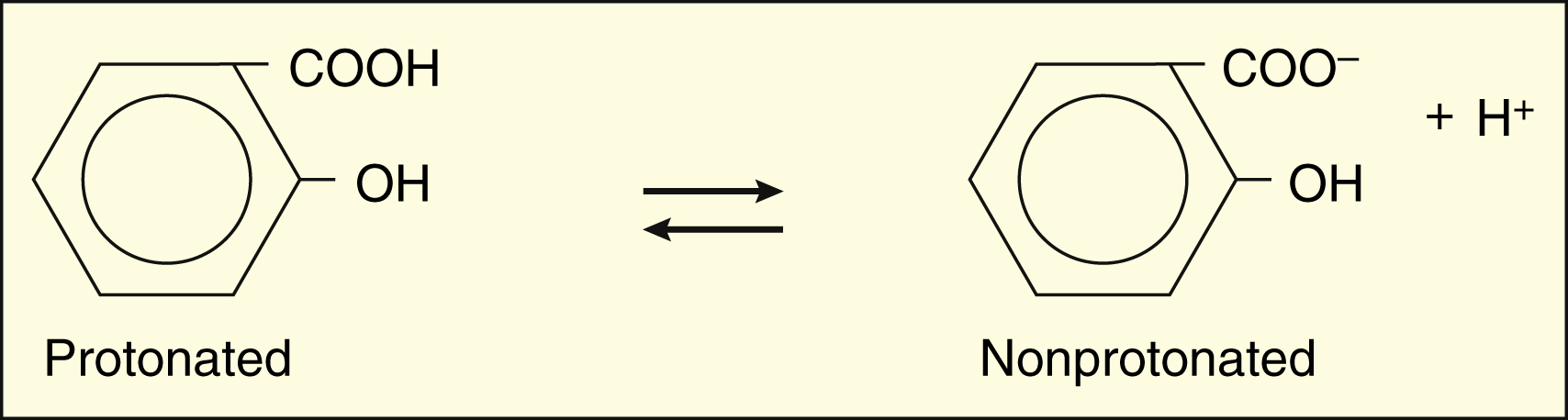
For salicylic acid, which is a weak acid with a pKa of 3, log [HA]/[A−] is 3 minus the pH. At a pH of 2, then, log [HA]/[A−] = 3 − 2 = 1. Therefore, [HA]/[A−] = 10/1.
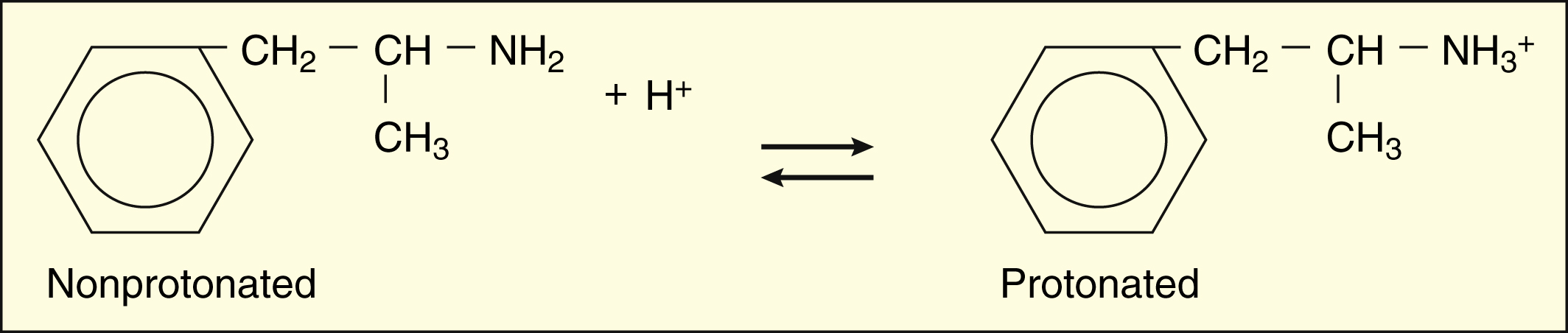
For amphetamine, which is a weak base with a pKa of 10, log [HB+]/[B] is 10 minus the pH. At a pH of 8, then, log [HB+]/[B] = 10 − 8 = 2. Therefore, [HB+]/[B] = 100/1.
The following are the ratios of the protonated form to the nonprotonated form at different pH levels:
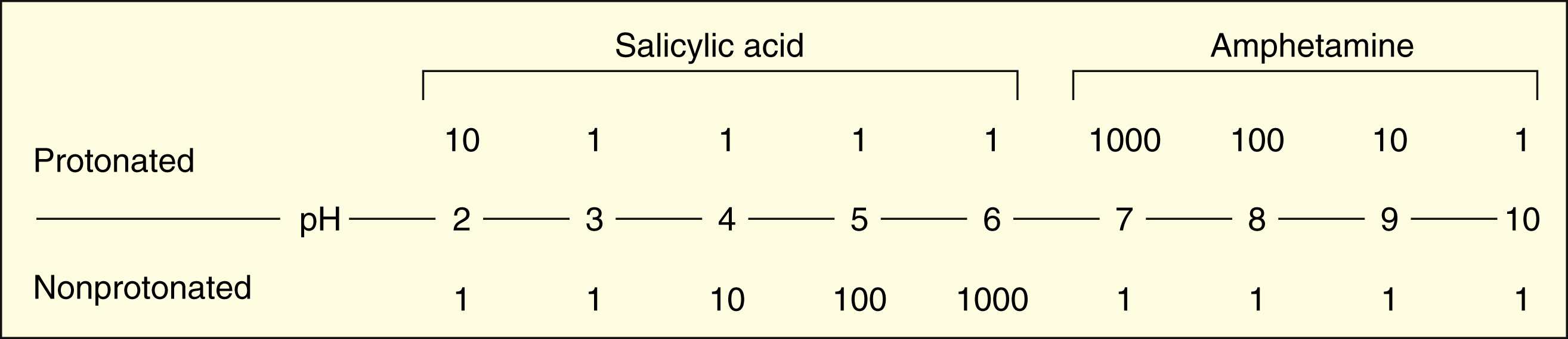
The protonated form of a weak acid is non-ionized, whereas the protonated form of a weak base is ionized. The ratio of the protonated form to the nonprotonated form of these drugs can be calculated using the Henderson-Hasselbalch equation (see Box 2–1). The pKa is the negative log of the ionization constant, particular for each acidic or basic drug. At a pH equal to the pKa, equal amounts of the protonated and nonprotonated forms are present. If the pH is less than the pKa, the protonated form predominates. If the pH is greater than the pKa, the nonprotonated form predominates.
In the stomach, with a pH of 1, weak acids and bases are highly protonated. At this site, the non-ionized form of weak acids (pKa = 3–5) and the ionized form of weak bases (pKa = 8–10) will predominate. Hence, weak acids are more readily absorbed from the stomach than are weak bases. In the intestines, with a pH of 7, weak bases are also mostly ionized, but much less so than in the stomach, and weak bases are absorbed more readily from the intestines than from the stomach.
However, weak acids can also be absorbed more readily from the intestines than from the stomach, despite their greater ionization in the intestines, because the intestines have a greater surface area than the stomach for absorption of the non-ionized form of a drug, and this outweighs the influence of greater ionization in the intestines.
DRUG DISTRIBUTION
Drugs are distributed to organs and tissues via the circulation, diffusing into interstitial fluid and cells from the circulation. Most drugs are not uniformly distributed throughout total body water, and some drugs are restricted to the extracellular fluid or plasma compartment. Drugs with sufficient lipid solubility can simply diffuse through membranes into cells. Other drugs are concentrated in cells by the phenomenon of ion trapping, which is described further below. Drugs can also be actively transported into cells. For example, some drugs are actively transported into hepatic cells, where they may undergo enzymatic biotransformation. In the intestines, drug transport by P-glycoprotein (Pgp) in the blood-to-lumen direction leads to a secretion of various drugs into the intestinal tract, thereby serving as a detoxifying mechanism. The Pgp proteins also remove many drugs from tissues throughout the body, including anticancer agents. Inhibition of Pgp by amiodarone, erythromycin, propranolol, and other agents can increase tissue levels of these drugs and augment their pharmacologic effects.
Factors Affecting Distribution
Organ Blood Flow
The rate at which a drug is distributed to various organs after a drug dose is administered depends largely on the proportion of cardiac output received by the organs. Drugs are rapidly distributed to highly perfused tissues, namely the brain, heart, liver, and kidney, and this enables a rapid onset of action of drugs affecting these tissues. Drugs are distributed more slowly to less perfused tissues such as skeletal muscle and even more slowly to those with the lowest blood flow, such as skin, bone, and adipose tissue.
Plasma Protein Binding
Almost all drugs are reversibly bound to plasma proteins, primarily albumin, but also lipoproteins, glycoproteins, and β globulins. The extent of binding depends on the affinity of a particular drug for protein-binding sites and ranges from less than 10% to as high as 99% of the plasma concentration. As the free (unbound) drug diffuses into interstitial fluid and cells, drug molecules dissociate from plasma proteins to maintain the equilibrium between free drug and bound drug. In general, acid drugs bind to albumin and basic drugs to glycoproteins and β globulins.
Plasma protein binding is saturable, and a drug can be displaced from binding sites by other drugs that have a high affinity for such sites. However, most drugs are not used at high enough plasma concentrations to occupy the vast number of plasma protein binding sites. There are a few agents that may cause drug interactions by competing for plasma protein binding sites, as highlighted in Chapter 4.
Molecular Size
Molecular size is a factor affecting the distribution of extremely large molecules, such as those of the anticoagulant heparin. Heparin is largely confined to the plasma compartment, although it does undergo some biotransformation in the liver.
Lipid Solubility
Lipid solubility is a major factor affecting the extent of drug distribution, particularly to the brain, where the blood-brain barrier restricts the penetration of polar and ionized molecules. The barrier is formed by tight junctions between the capillary endothelial cells and also by the glial cells that surround the capillaries, which inhibit the penetration of polar molecules into brain neurons.
DRUG BIOTRANSFORMATION
Drug biotransformation and excretion are the two processes responsible for the decline of the plasma drug concentration over time. Both of these processes contribute to the elimination of active drug from the body, and as discussed later in the chapter, clearance is a measure of the rate of elimination. Biotransformation, or drug metabolism, is the enzyme-catalyzed conversion of drugs to their metabolites. Most drug biotransformation takes place in the liver, but drug-metabolizing enzymes are found in many other tissues, including the gut, kidneys, brain, lungs, and skin.
Role of Drug Biotransformation
The fundamental role of drug-metabolizing enzymes is to inactivate and detoxify drugs and other foreign compounds (xenobiotics) that can harm the body. Drug metabolites are usually more water soluble than is the parent molecule and, therefore, they are more readily excreted by the kidneys. No particular relationship exists between biotransformation and pharmacologic activity. Some drug metabolites are active, whereas others are inactive. Many drug molecules undergo attachment of polar groups, a process called conjugation, for more rapid excretion. As a general rule, most conjugated drug metabolites are inactive, but a few exceptions exist.
Formation of Active Metabolites
Many pharmacologically active drugs, such as the sedative-hypnotic agent diazepam (Valium), are biotransformed to active metabolites. Some agents, known as prodrugs, are administered as inactive compounds and then biotransformed to active metabolites. This type of agent is usually developed because the prodrug is better absorbed than its active metabolite. For example, the antiglaucoma agent dipivefrin (Propine) is a prodrug that is converted to its active metabolite, epinephrine, by corneal enzymes after topical ocular administration. Orally administered prodrugs, such as the antihypertensive agent enalapril (Vasotec), are converted to their active metabolite by hepatic enzymes during their first pass through the liver.
First-Pass Biotransformation
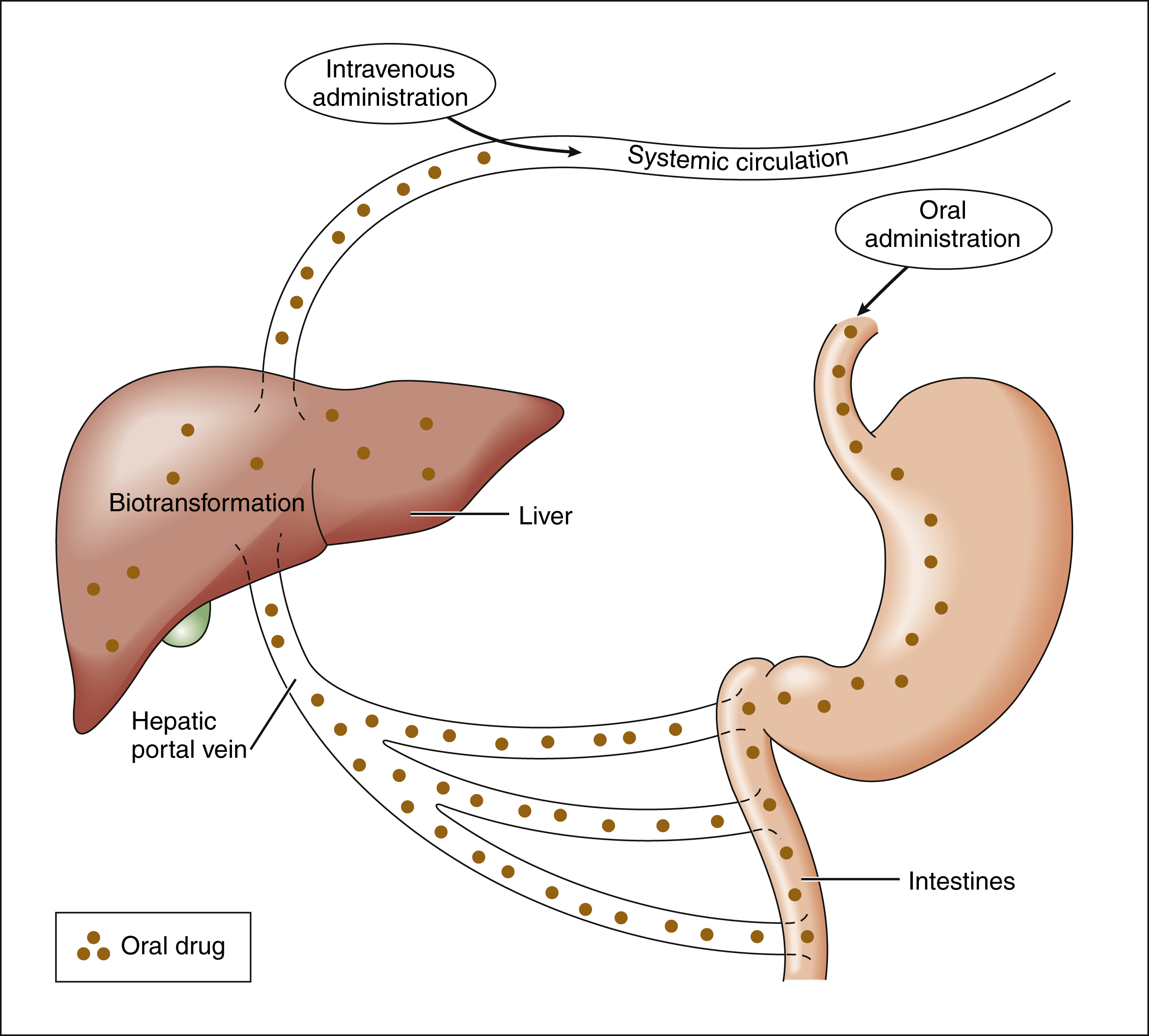
Drugs that are absorbed from the gut reach the liver via the hepatic portal vein before entering the systemic circulation (Fig. 2–2). Many drugs, such as the antihypertensive agent felodipine (Plendil), are extensively converted to inactive metabolites during their first pass through the gut wall and liver, and have low bioavailability (see below) after oral administration. This phenomenon is called the first-pass effect. Drugs administered by the sublingual or rectal route undergo less first-pass metabolism and have a higher degree of bioavailability than do drugs administered by the oral route.
Phases of Drug Biotransformation
Drug biotransformation can be divided into two phases, each carried out by unique sets of metabolic enzymes. In many cases, phase I enzymatic reactions create or unmask a chemical group required for a phase II reaction. In some cases, however, drugs bypass phase I biotransformation and go directly to phase II. Although some phase I drug metabolites are pharmacologically active, most phase II drug metabolites are inactive.
Phase I Biotransformation
Phase I biotransformation includes oxidative, hydrolytic, and reductive reactions (Fig. 2–3).
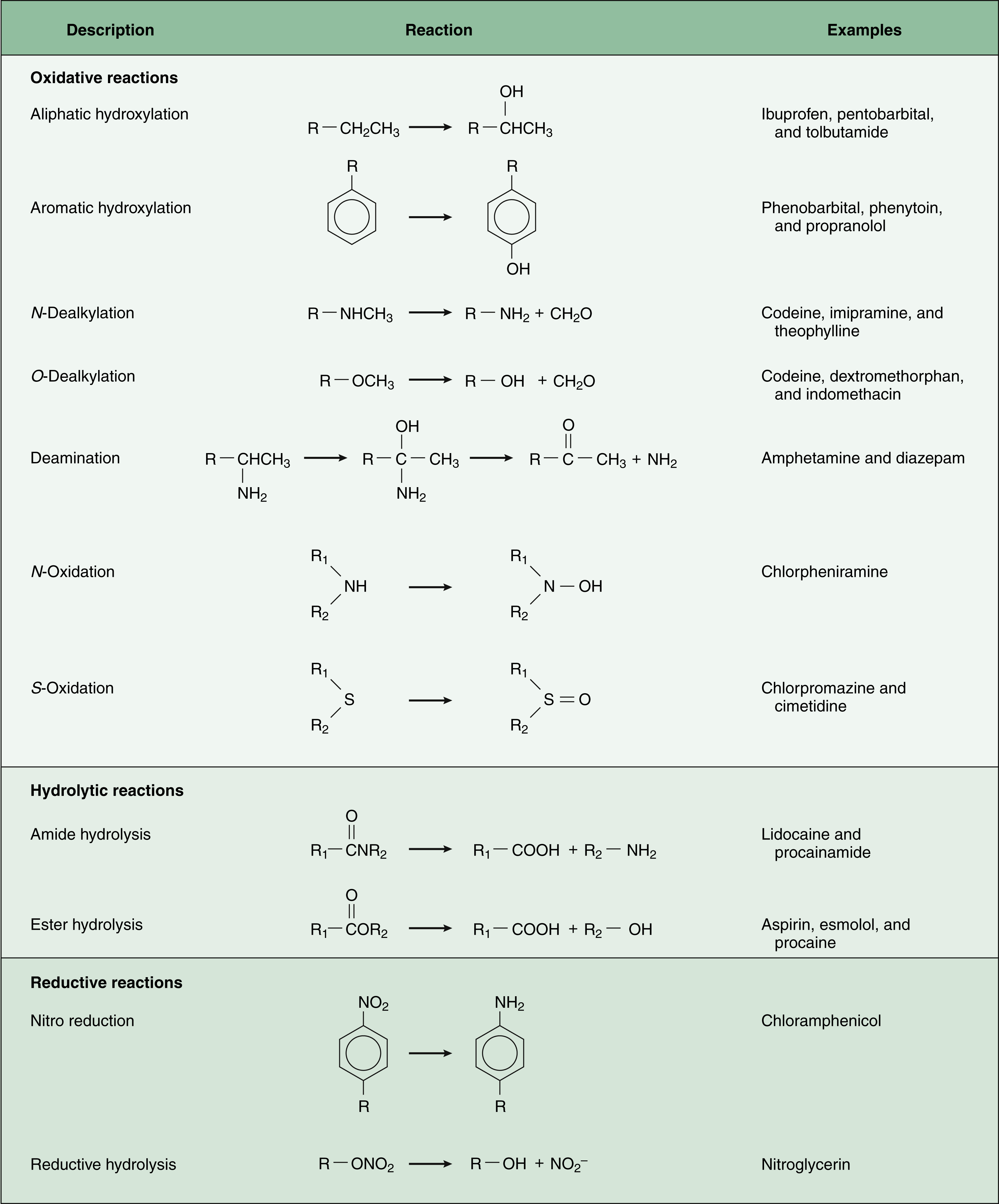
Figure 2–3 Phase I drug biotransformation. Many drugs are biotransformed by oxidative, hydrolytic, or reductive reactions and then undergo conjugation with endogenous substances. A few drugs bypass phase I reactions and directly enter phase II biotransformation.
OXIDATIVE REACTIONS
Oxidative reactions are the most common type of phase I biotransformation. They are catalyzed by enzymes isolated in the microsomal fraction of liver homogenates (the fraction derived from the endoplasmic reticulum) and by cytoplasmic enzymes.
The microsomal cytochrome P450 (CYP) monooxygenase system is a family of enzymes that catalyzes the biotransformation of drugs with a wide range of chemical structures. The microsomal monooxygenase reaction requires the following: CYP (a hemoprotein); a flavoprotein that is reduced by nicotinamide adenine dinucleotide phosphate (NADPH), called NADPH CYP reductase; and membrane lipids in which the system is embedded. In the drug-oxidizing reaction, one atom of oxygen is used to form a hydroxylated metabolite of a drug, as shown in Figure 2–4, whereas the other atom of oxygen forms water when combined with electrons contributed by NADPH. The hydroxylated metabolite may be the end product of the reaction or serve as an intermediate that leads to the formation of another metabolite.
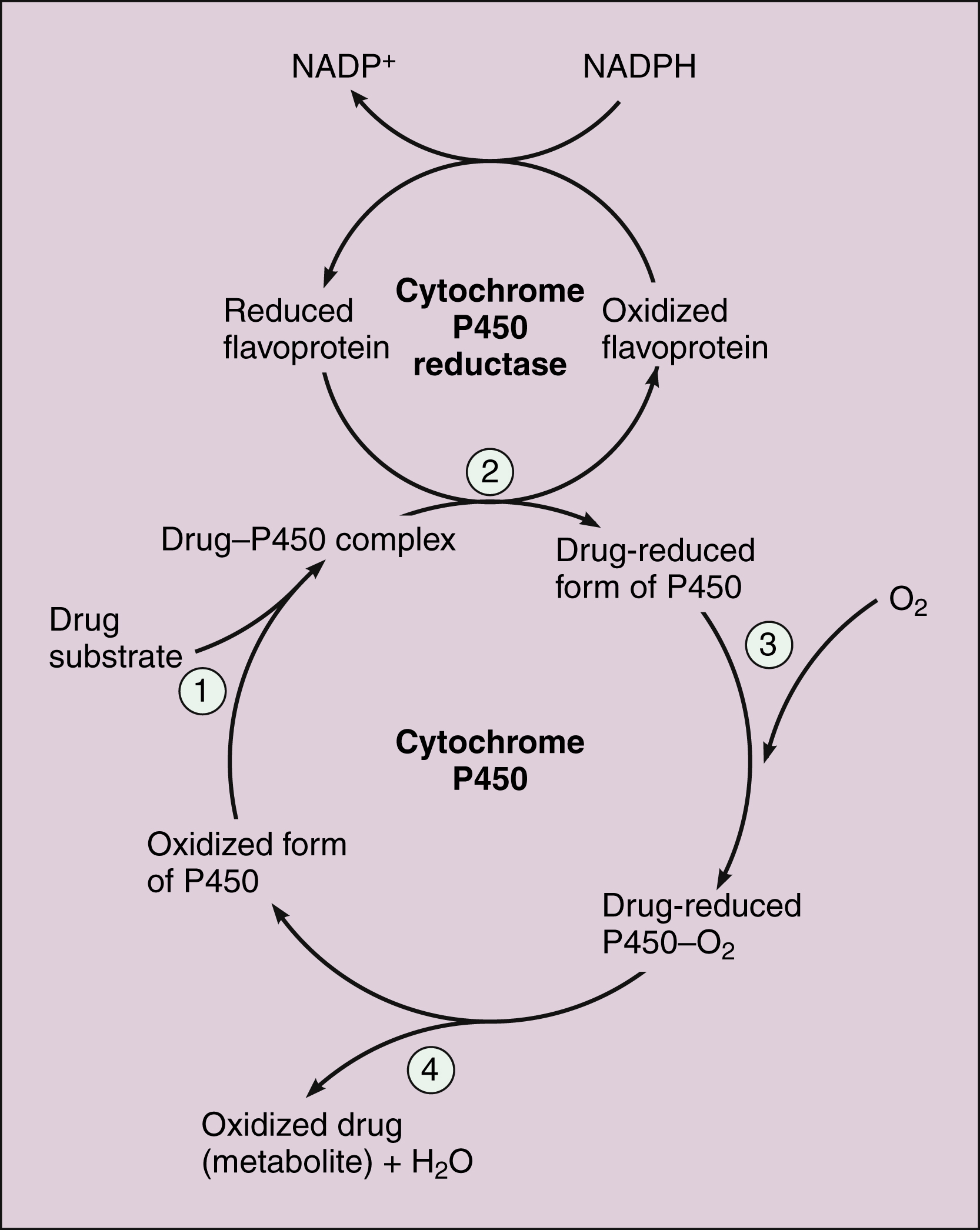
Figure 2–4 The CYP reductase mechanism for drug oxidation. Four steps are involved in the CYP reaction: First, the drug substrate binds to the oxidized form of P450 (i.e., Fe3+). Second, the drug P450 complex is reduced by CYP reductase, using electrons donated by the reduced form of NADPH. Third, the drug-reduced form of P450 (i.e., Fe2+) interacts with oxygen. Fourth, the oxidized drug (metabolite) and water are produced.
The most common chemical reactions catalyzed by CYP enzymes are aliphatic hydroxylation, aromatic hydroxylation, N-dealkylation, and O-dealkylation.
Many CYP isozymes have been identified and cloned, and their role in metabolizing specific drugs elucidated. Each isozyme catalyzes a different but overlapping spectrum of oxidative reactions. Most drug biotransformation is catalyzed by three CYP families named CYP1, CYP2, and CYP3. The different CYP families are likely related by gene duplication and each family is divided into subfamilies, also clearly related by homologous protein sequences. The CYP3A subfamily catalyzes more than half of all microsomal drug oxidations.
Many drugs alter drug metabolism by inhibiting or inducing CYP enzymes, and drug interactions can occur when these drugs are administered concurrently with other drugs that are metabolized by CYP (see Chapter 4). Two examples of inducers of CYP are the barbiturate, phenobarbital, and the antitubercular drug, rifampin. The inducers stimulate the transcription of genes encoding CYP enzymes, resulting in increased messenger RNA (mRNA) and protein synthesis. Drugs that induce CYP enzymes activate the binding of nuclear receptors to enhancer domains of CYP genes, increasing the rate of gene transcription.
A few drugs are oxidized by cytoplasmic enzymes. For example, ethanol is oxidized to aldehyde by alcohol dehydrogenase, and caffeine and the bronchodilator theophylline are metabolized by xanthine oxidase. Other cytoplasmic oxidases include monoamine oxidase, a site of action for some psychotropic medications.
HYDROLYTIC REACTIONS
Esters and amides are hydrolyzed by a variety of enzymes. These include cholinesterase and other plasma esterases that inactivate choline esters, local anesthetics, and drugs such as esmolol (Brevibloc), an agent for the treatment of tachycardia that blocks cardiac β1-adrenoceptors. There are few CYP enzymes that carry out hydrolytic reactions.
REDUCTIVE REACTIONS
Reductive reactions are less common than are oxidative and hydrolytic reactions. Chloramphenicol, an antimicrobial agent, and a few other drugs are partly metabolized by a hepatic nitro reductase, and this process involves CYP enzymes. Nitroglycerin, a vasodilator, undergoes reductive hydrolysis catalyzed by glutathione-organic nitrate reductase.
Phase II Biotransformation
In phase II biotransformation, drug molecules undergo conjugation reactions with an endogenous substance such as acetate, glucuronate, sulfate, or glycine (Fig. 2–5). Conjugation enzymes, which are present in the liver and other tissues, join various drug molecules with one of these endogenous substances to form water-soluble metabolites that are more easily excreted. Except for microsomal glucuronosyltransferases, these enzymes are located in the cytoplasm. Most conjugated drug metabolites are pharmacologically inactive.
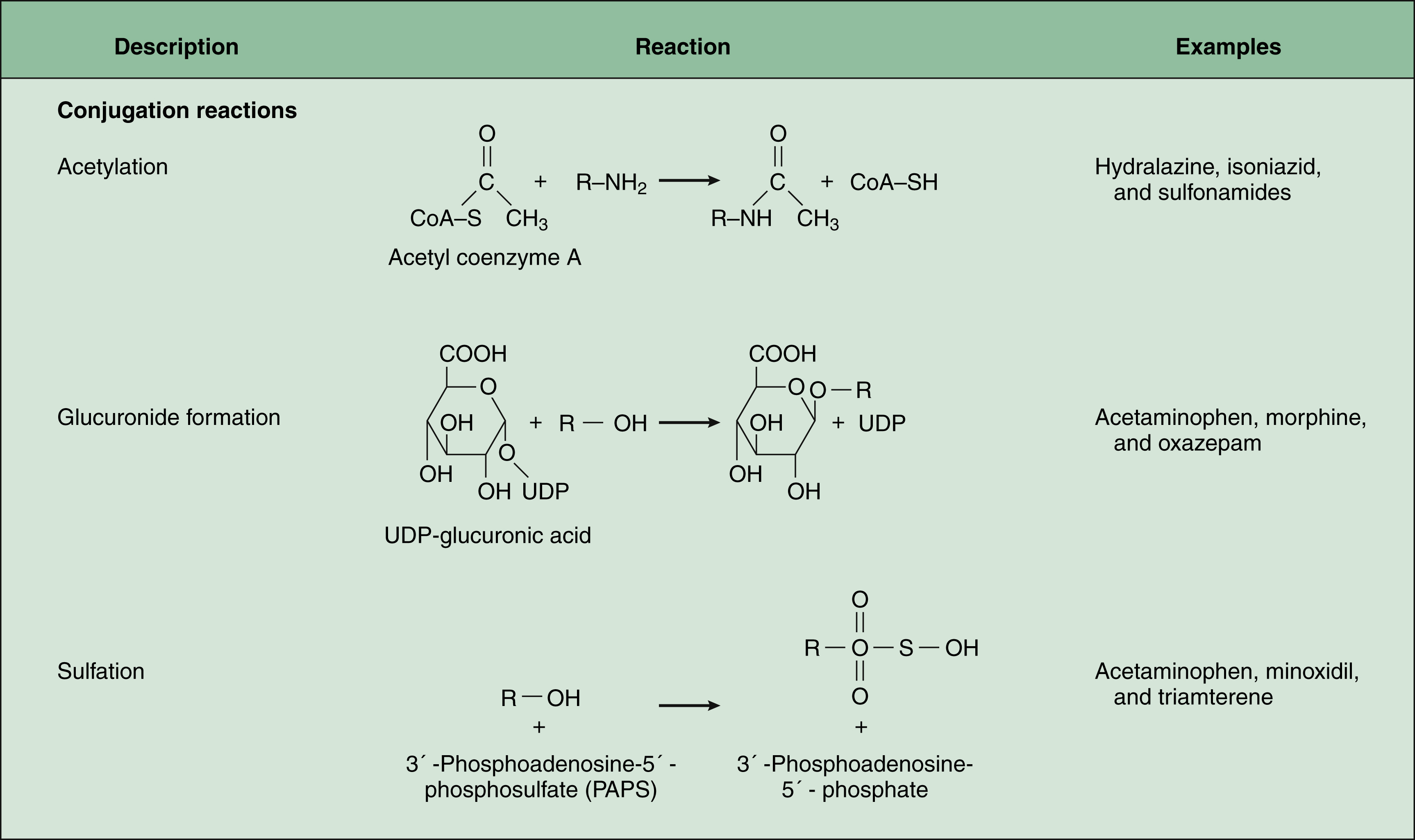
GLUCURONIDE FORMATION
Glucuronide formation, the most common conjugation reaction, utilizes glucuronosyltransferases to conjugate a glucuronate molecule with the parent drug molecule.
ACETYLATION
Acetylation is accomplished by N-acetyltransferase enzymes that utilize acetyl coenzyme A (acetyl CoA) as a source of the acetate group.
Pharmacogenomics
Since the completion of the human genome, it is now fully realized that there is a great degree of individual variation, called polymorphism, in the genes coding for drug-metabolizing enzymes. Modern genetic studies were triggered by rare fatalities in children being treating for leukemia using the thiopurine agent, 6-mercaptopurine (6-MP). It was discovered that the children died as a result of drug toxicity because they expressed a faulty variant of thiopurine methyltransferase, the enzyme that metabolizes 6-MP.
Variations in Acetyltransferase Activity
Individuals exhibit slow or fast acetylation of some drugs because of genetically determined differences in N-acetyltransferase. Slow acetylators (SAs) were first identified by neuropathic effects of isoniazid, a drug to treat tuberculosis. These patients had higher plasma levels of isoniazid compared to other patients classified as rapid acetylators (RAs). The SA phenotype is autosomal recessive, although there are more than 20 allelic variants of the gene for N-acetyltransferase identified. In individuals with one wild-type enzyme and one faulty variant, an intermediate phenotype is observed. The distribution of these phenotypes varies from population to population. About 15% of Asians, 50% of Caucasians and Africans, and more than 80% of Mideast populations have the SA phenotype. Other drugs that may cause toxicity in the SA patient are sulfonamide antibiotics, the antiarrhythmic agent procainamide, and the antihypertensive agent hydralazine.
Variations in CYP2D6 and CYP2C19 Activity
Variations in oxidation of some drugs have been attributed to genetic differences in certain CYP enzymes. Genetic polymorphisms of CYP2D6 and CYP2C19 enzymes are well characterized, and human populations of “extensive metabolizers” and “poor metabolizers” have been identified. These differences are caused by more than 70 identified variants in the CYP2D6 gene and more than 25 variants of the CYP2C19 genes, resulting from point mutations, deletions, or additions; gene rearrangements, or deletion or duplication of the entire gene. This gives rise to an increase, reduction, or complete loss of enzyme activity and to different levels of enzyme expression that result in altered rates of enzymatic reactions.
Most individuals are extensive metabolizers of CYP2D6 substrates, but 10% of Caucasians and a smaller fraction of Asians and Africans are poor metabolizers of substrates for CYP2D6. Psychiatric patients who are poor metabolizers of CYP2D6 drugs have been found to have a higher rate of adverse drug reactions than do those who are extensive metabolizers because of higher psychotropic drug plasma levels. In addition, poor metabolizers of CYP2D6 drugs have a reduced ability to metabolize codeine to morphine sufficiently to obtain adequate pain relief when codeine is administered for analgesia.
Poor metabolizers of CYP2C19 substrates have higher plasma levels of proton pump inhibitors, such as omeprazole (Prilosec), whereas some extensive metabolizers of CYP2C19 drugs require larger doses of omeprazole to treat peptic ulcer.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


