Figure 3–1 Structure of a typical GPCR. All GPCRs consist of a long polypeptide chain of amino acids threaded through the cell membrane with seven transmembrane (TM) domains. These TM domains are arranged in α-helices composed of hydrophobic residues. The N-terminal of the receptor protein is outside the cell and the C-terminal is on the inside. Three extracellular loops (ELs) and three intracellular loops (ILs) are formed by this configuration. The protein in the cell membrane forms a circle with TM1 and TM7 in close proximity but is shown here in a two-dimensional view for clarity.
A number of ligands inhibit the function of specific enzymes by competitive or noncompetitive inhibition. A ligand that binds to the same active, catalytic site as the endogenous substrate is a competitive inhibitor. Ligands that bind at a different site on the enzyme and alter the shape of the molecule, thereby reducing its catalytic activity, are called noncompetitive inhibitors.
Drugs also target membrane transport proteins, including ligand- and voltage-gated ion channels and neurotransmitter transporters. At ligand-gated ion channels, drugs can bind at the same site as the endogenous ligand and directly compete for the receptor site. Drugs can also bind at a different site, an allosteric site, that alters the response of the endogenous ligand binding to the ligand-gated ion channel and increase or decrease the flow of ions. Some drugs directly bind and inactivate voltage-gated ion channels; these are ion channel proteins that do not have an endogenous ligand (as ligand-gated ion channels do) but open or close as a function of the membrane voltage potential. Neurotransmitter transporter proteins are large, 12-transmembrane domain proteins that transfer neurotransmitter molecules out of the synapse and back into the neuron. A large group of agents, known generally as re-uptake inhibitors, target these transport proteins.
Steroid hormone receptors are intracellular proteins that translocate to the nucleus upon ligand (steroid) binding. In the nucleus, the steroid-receptor complex alters the transcription rate of specific genes. DNA is also a receptor site for ligands that bind directly to nucleic acids, most notably the antineoplastic agents. Other macromolecules that serve as receptors include the various lipids and phospholipids that make up the membrane. Some of the effects of general anesthetics and alcohol are due to interaction with membrane lipids.
Receptor Classification
Drug receptors are classified according to drug specificity, tissue location, and, more recently, by their primary amino acid sequence. For example, adrenoceptors were initially divided into two types (α and β), based on their affinity for norepinephrine, epinephrine, and other agents in different tissues. Subsequently, the distinction between the types was confirmed by the development of selective antagonists that blocked either α-adrenoceptors or β-adrenoceptors. Later, the two types of receptors were divided into subtypes, based on more subtle differences in agonist potency, tissue distribution, and varying effects.
At present, most receptors for drug targets and endogenous ligands are cloned and their amino acid sequences determined. There are also numerous other receptor-like proteins predicted from the human genome for which an endogenous ligand is not identified, called orphan receptors. The orphan receptors are of great interest to pharmaceutical companies as they represent targets for the development of new drugs. Families of receptors types are grouped by their sequence similarity using bioinformatics and this classification supports results from earlier in vivo and in vitro functional studies. In many cases, each type of receptor corresponds to a single, unique gene with subtypes of receptors arising from different transcripts of the same gene by the process of alternative splicing.
DRUG-RECEPTOR INTERACTIONS
Receptor Binding and Affinity
To initiate a cellular response, a drug must first bind to a receptor. In most cases, drugs bind to their receptor by forming hydrogen, ionic, or hydrophobic (Van der Waals) bonds with a receptor site (Fig. 3–2). These weak bonds are reversible and enable the drug to dissociate from the receptor as the tissue concentration of the drug declines. The binding of drugs to receptors often exhibits stereospecificity, so that only one of the stereoisomers will form a three-point attachment with the receptor. In a few cases, drugs form relatively permanent covalent bonds with a specific receptor. This occurs, for example, with antineoplastic drugs that bind to DNA and with drugs that irreversibly inhibit the enzyme cholinesterase.
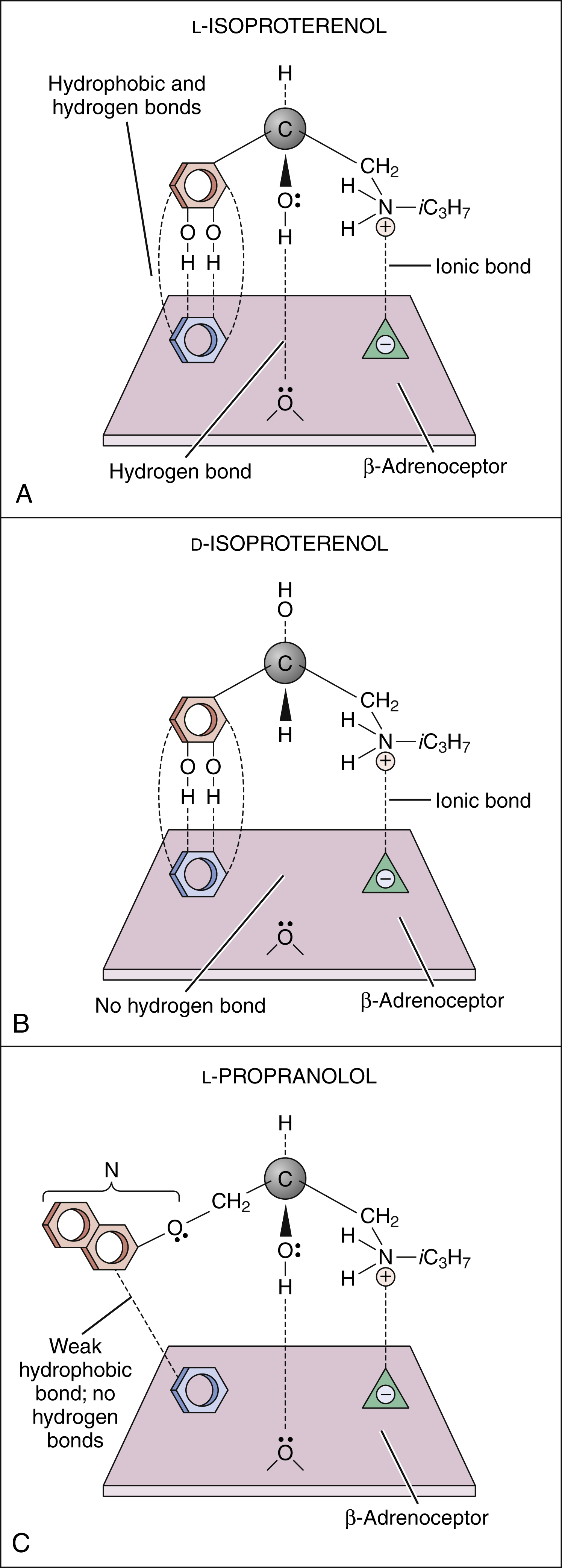
Figure 3–2 Drug binding to receptors. (A) l-isoproterenol, a β-adrenoceptor agonist, forms hydrogen, ionic, and hydrophobic (Van der Waals) bonds with three sites on the β-adrenoceptor. (B) d-isoproterenol binds to two sites on the β-adrenoceptor but is unable to bind with the third site. (C) l-Propranolol, a β-adrenoceptor antagonist, binds to two sites on the receptor in the same way that l-isoproterenol does. The naphthyloxy group (N) forms weak bonds with the third receptor site, but these are not sufficiently strong for the drug to have intrinsic (agonist) activity. iC3H7 = isopropyl.
The tendency of a drug to combine with its receptor is called affinity, which is a measure of the strength of the drug-receptor complex. According to the law of mass action, the number of receptors [R] occupied by a drug depends on the drug concentration [D] and the drug-receptor association and dissociation rate constants (k1 and k2):
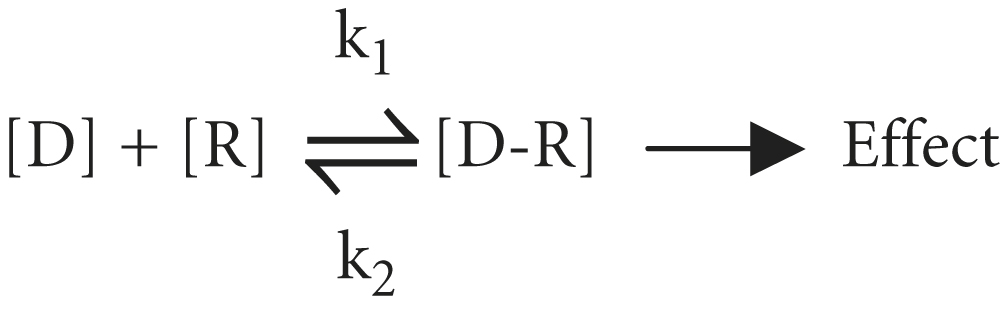
The ratio of k2 to k1 is known as the KD and represents the drug concentration required to saturate 50% of the receptors. The lower the KD is, the greater is the drug’s affinity for the receptor. Most drugs have a KD in the micromolar to nanomolar (10−6 to 10−9 M) range of drug concentrations. As discussed below, receptor affinity is the primary determinant of drug potency.
Signal Transduction
Signal transduction describes the pathway from ligand binding to conformational changes in the receptor, receptor interaction with an effector molecule (if present), and other downstream molecules called second messengers. This cascade of receptor-mediated biochemical events ultimately leads to a physiologic effect (Table 3–2).
TABLE 3–2 Examples of Receptors and Signal Transduction Pathways
| Family and Type of Receptor | Mechanism of Signal Transduction | Example of Effect in Tissue or Cell |
|---|---|---|
| G Protein–Coupled Receptors | ||
| α1-Adrenoceptor | Activation of phospholipase C | Vasoconstriction |
| α2-Adrenoceptor | Inhibition of adenylyl cyclase | Release of norepinephrine decreased |
| β-Adrenoceptor | Stimulation of adenylyl cyclase | Heart rate increased |
| Muscarinic receptor | Activation of phospholipase C | Glandular secretion increased |
| Ligand-Gated Ion Channels | ||
| GABAA receptors | Chloride ion flux | Hyperpolarization of neuron |
| Nicotinic receptors | Sodium ion flux | Skeletal muscle contraction |
| Membrane-Bound Enzymes | ||
| Atrial natriuretic factor receptors | Stimulation of guanylyl cyclase | Sodium excretion increased |
| Insulin receptors | Activation of tyrosine kinase | Glucose uptake stimulated |
| Nuclear Receptors | ||
| Steroid receptors | Activation of gene transcription | Reduced cytokine production |
| Thyroid hormone receptors | Activation of gene transcription | Oxygen consumption increased |
GABA = γ-aminobutyric acid.
G PROTEIN–COUPLED RECEPTORS (GPCRS)
The signal transduction pathway for GPCRs is well understood. These receptors constitute a superfamily of receptors for many endogenous ligands and drugs, including receptors for acetylcholine, epinephrine, histamine, opioids, and serotonin. Figure 3–3 illustrates signal transduction for a receptor that is coupled with G proteins.
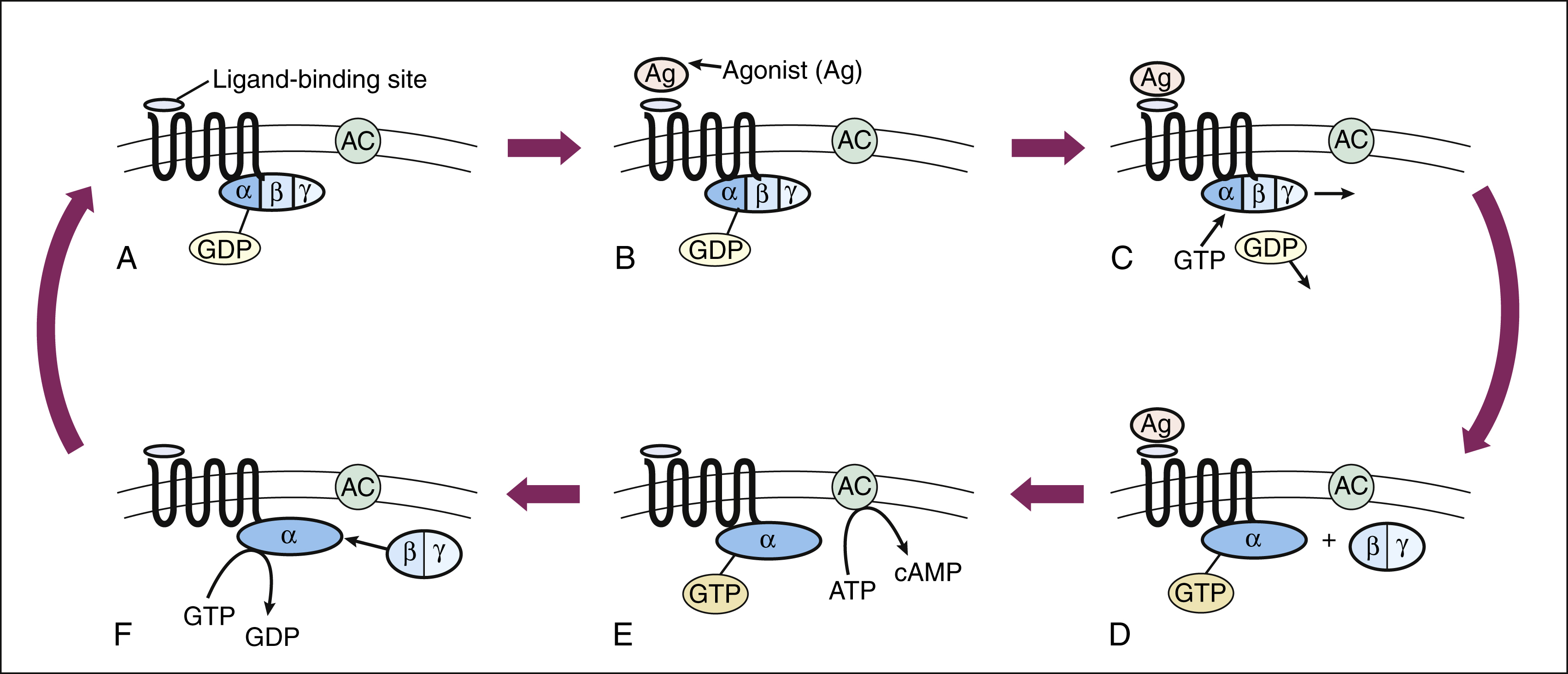
Figure 3–3 Signal transduction with a G protein–coupled receptor. (A) A typical G protein–coupled receptor contains a ligand-binding site on the external surface of the plasma membrane and a G protein–binding site on the internal surface. In the inactive state, guanosine diphosphate (GDP) is bound to the Gα subunit of the G protein. (B) and (C) When the agonist (Ag) binds to the receptor, guanosine triphosphate (GTP) binds to the G protein and causes the dissociation of GDP. (D) Activation of the Gα subunit by GTP causes the dissociation of the Gβ and Gγ subunits. (E) The Gα subunit is then able to activate adenylyl cyclase (AC) and thereby stimulate the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP). (F) GTP hydrolysis, catalyzed by Gα subunit GTPase, leads to reassociation of the Gα and the Gβ and Gγ subunits.
The heterotrimeric G proteins have three subunits, known as Gα, Gβ, and Gγ. The Gα subunit serves as the site of guanosine triphosphate (GTP) hydrolysis, a process catalyzed by innate GTPase activity, which acts to terminate the signal (see Fig. 3–3). Several types of Gα subunits exist, each of which determines a specific cellular response. For example, the Gαs (stimulating) subunit increases adenylyl cyclase activity and thereby stimulates the production of cyclic adenosine monophosphate (cyclic AMP, or cAMP). The Gαi (inhibitory) subunit decreases adenylyl cyclase activity and inhibits the production of cAMP. Another G protein (Gαq) activates phospholipase C and leads to the formation of inositol triphosphate (IP3) and diacylglycerol (DAG) from membrane phospholipids. IP3 and DAG further cause an elevation of Ca+2 ions inside the cell. Several other types of Gα subunits are also present in cells and activated by receptors. The Gβ, and Gγ subunits are so tightly bound together that they do not dissociate and are therefore written as Gβγ. The Gβγ also has signaling function when separated from Gα upon ligand-receptor activation, for example, by altering K+ or Ca+2 channel conductance.
The second messengers cAMP, IP3, DAG, and Ca+2 activate or inhibit unique cellular enzymes in each target cell. Cyclic AMP activates a number of tissue-specific cAMP-dependent protein kinases. These kinases phosphorylate other enzymes or proteins that ultimately affect intracellular processes such as ion channel activity, release of neurotransmitter, regulation of transcription, and numerous other processes. For example, one of the best studied kinases, protein kinase A, is activated by the increase of cAMP produced by epinephrine binding to β2-adrenoceptors in muscle. Protein kinase A phosphorylates the enzyme glycogen phosphorylase, which then increases the breakdown of glycogen to free glucose, providing the fuel needed by the muscles to respond to the event that initiated the release of epinephrine.
IP3 and DAG evoke the release of calcium from intracellular storage sites and thereby augment calcium-mediated processes such as muscle contraction, glandular secretion, and neurotransmitter release. The increased intracellular Ca+2 ions also activate calcium-dependent kinases and a number of other enzyme cascades.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


