1.5
Prostatic carcinoma therapy
Somatostatin
3.1
Growth inhibition
Calcitonin
3.4
Ca2+ regulation
Glucagon
3.5
Diabetes therapy
Parathyroid hormone (1–34)
4.3
Ca2+ regulation
Insulin
6
Diabetes therapy
Parathyroid hormone (1–84)
9.4
Ca2+ regulation
Interferon-gamma
16 (dimer)
Antiviral agent
Tumour necrosis factor-α
17.5 (trimer)
Antitumour agent
Interferon-α2
19
Leukaemia, hepatitis therapy
Interferon-β1
20
Lung cancer therapy
Growth hormone
22
Growth acceleration
DNase
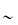 32
32
Cystic fibrosis therapy
α1-Antitrypsin
45
Cystic fibrosis therapy
Albumin
68
Plasma volume expander
Bovine IgG
150
Immunisation
Catalase
230
Treatment of wounds and ulcers
Cationic ferritin
400+
Anaemias
Reproduced with permission from Niven RW. Delivery of biotherapeutics by inhalation aerosols. Pharm Technol 1993;July: 72.
13.1 Structure and solution properties of peptides and proteins
13.1.1 Structure of peptides and proteins
The primary structure of a protein is the order in which the individual amino acids are sequenced. This tells us little about the shape that the protein will assume in solution, although the primary structure determines the secondary, tertiary and even quaternary forms. These amino acid building blocks (Table 13.2) give the key to structure and behaviour. The standard three-letter (Glu, Arg, Trp, etc.) and one-letter (G, A, W, etc.) abbreviations for amino acids are also listed: their use makes structural descriptions more accessible.
Table 13.2 Nomenclature and structure of the principal amino acids. The common name, the three-letter code and the single-letter code are given
|
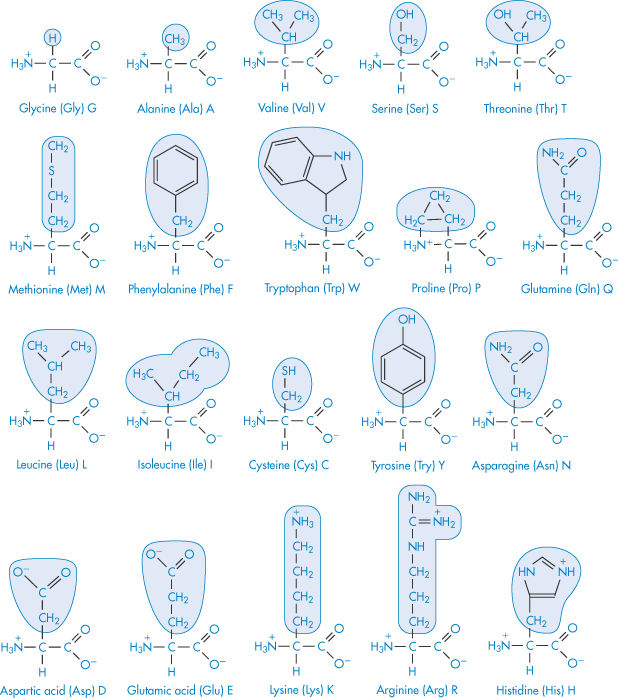
Secondary structures include the coiled α-helix, and pleated sheets, discussed below and shown in Figs 13.1 and 13.2. Chain folding, arising from crosslinking through hydrogen bonding, the hydrophobic effect and salt bridges, leads to a definition of tertiary structure, and association of these structural units leads to quaternary forms.
Figure 13.1 (a) The α-helix forms, because ‒NH and ‒C=O groups interact through hydrogen bonding, pulling the backbone into a spiral, as shown in this diagram in a 9-amino peptide: in (A) here only the central carbon atoms are shown for clarity; in (B) the nitrogen and carbons of the backbone are shown, and in (C) the oxygen and hydrogen atoms have been added. All the ‒NH groups point in the same direction ‘up’ and the ‒C=O groups point ‘down’ to allow the formation of ‒C=O … H‒N‒ bonds. In (D) the side-chains are added and point outwards from the α-helix. Hydrophobic interactions between some helix-promoting side-chains (the more hydrophobic chains: Table 13.3) help to stabilise the helix. (b) An idealised α-helix, drawn as a ribbon, showing typical stabilising intrahelical interactions. Specific hydrogen-bonded interactions are said to ‘cap’ the ends of the chain, known specifically as the N-terminal and C-terminal ends of the helix. Free energies (ΔG) of the interactions, compared with the free energy of hydrophobic bonding involving an isobutyl side-chain (−4.18 kJ mol−1), are:
N-cap, 4.2–8.4 kJ mol−1
C-cap,  2 kJ mol−1
2 kJ mol−1
Side-chain–side-chain electrostatic interactions, 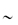 2 kJ mol−1
2 kJ mol−1
Reproduced with permission from Bryson JW et al. Protein design: a hierarchic approach. Science 1995;270:935.
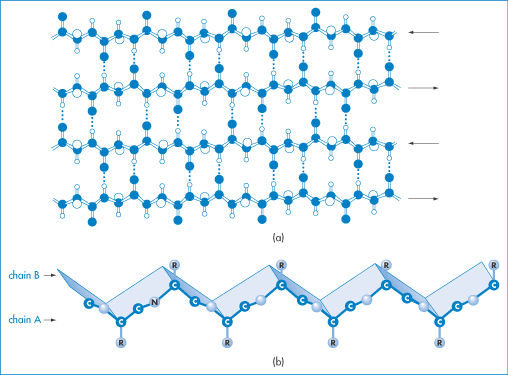
Figure 13.2 (a) A representation of β-sheet formation by four antiparallel polypeptide chains (to be visualised as being in and out of the plane on the paper). The interactions that determine β-sheet stability are more variable than those determining α-helix stability. Different amino acids have different propensities for forming β-sheets. (b) A representation of the concertina shape of a β-sheet showing two antiparallel polypeptide chains, A and B, associated by hydrogen bonding as in (a). Exposed amides at the edge of a β-sheet can hydrogen-bond to other sheets, leading to the formation of insoluble aggregates.
Representations in a stylised diagrammatic form of three proteins (interleukin-1β, zinc insulin dimers and the Fc fragment of immunoglobulin) are shown in Fig. 13.3. The nature of the three-dimensional structure shows how difficult it is to define proteins in conventional ways, and how they must be considered in a new light as pharmaceutical entities. They are in many ways fragile molecules. Loss of the unique tertiary or quaternary structure, through denaturation, can occur from a variety of insults that would not affect smaller molecules.
Figure 13.3 Ribbon diagrams of (a) interleukin-1β, (b) zinc–insulin dimer and (c) the Fc fragment of immunoglobulin.
Reproduced from Lesk AM. Protein Architecture. Oxford: IRL Press; 1991. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Complex structures are formed in solution because of interactions between the structural amino acids. Unfortunately, knowing the amino acid sequence of a protein is not enough to enable us to predict its physical behaviour. Function is determined by the way the linear chains of amino acids fold in solution to give specific three-dimensional structures comprising the coils, sheets and folds (Figs 13.1 and 13.2). Strides are being made in understanding the ‘grammar’ of protein folding, through the synthesis of synthetic peptides and sequences and observation of their structures. Some of the primary structures are so complex that it is not possible to predict their physicochemical properties, although modern molecular modelling techniques have made great inroads into understanding tertiary structure and behaviour.
13.1.2 Hydrophobicity of peptides and proteins
Amino acids have a range of physical properties, each having a greater or lesser degree of hydrophilic or hydrophobic nature. Naturally, if amino acids are spatially arranged in a molecule so that distinct hydrophobic and hydrophilic regions appear, then the polypeptide or protein will have an amphiphilic nature. Table 13.3 lists the relative hydrophobic character of a range of amino acids, where Gly is considered to have a value of zero. The amino acids range from very hydrophobic to very hydrophilic. The side-chains of selected amino acids are shown in the table where their relative hydrophobic properties are shown. Some values of log P (octanol/water) are also given, demonstrating the trend in hydrophobic parameters.
Table 13.3 Relative hydrophobic character of amino acid side-chains (Gly = 0)
Side-chain | Amino acid | Hydrophobic charactera | Log Pb |
(Most hydrophobic) | |||
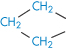
CH3CH2CH(CH3)‒ | Isoleucine | 1.83 | –1.72 |
CH3CH(CH3)CH2‒ | Leucine | 1.80 | –1.61 |
C6H5CH2‒ | Phenylalanine | 1.69 |
|
| Tryptophan | 1.35 |
|
CH3CH(CH3)‒ | Valine | 1.32 | –2.08 |
CH3‒S‒CH2CH2‒ | Methionine | 1.10 |
|
| Proline | 0.84 |
|
HSCH2‒ | Cysteine | 0.76 |
|
HOC6H5CH2‒ | Tyrosine | 0.39 |
|
CH3‒ | Alanine | 0.35 | –2.89 |
(Standard) | Glycine | 0 |
|
CH3CH(OH)‒ | Threonine | –0.27 |
|
HOCH2‒ | Serine | –0.63 |
|
| Histidine | –0.65 |
|
H2NCOCH2CH2‒ | Glutamine | –0.93 |
|
H2NCOCH2‒ | Asparagine | –0.99 |
|
H2NCH2CH2CH2‒ | Ornithine | –1.50 |
|
H3N+‒CH2CH2CH2CH2‒ | Lysine | –1.54 | –3.31 |
HOOCCH2‒ | Aspartic acid | –2.15 | –3.38 |
(Least hydrophobic) | |||
a High positive values indicate very hydrophobic amino acids, and negative values indicate amino acids with hydrophilic character.
b From Pliska V et al. J Chromatogr 1981;216:79.
Reproduced with permission from Dressler D, Potter H. Discovering Enzymes. New York: Scientific American; 1991.
An idea of the overall hydrophobicity of a peptide or protein may be gained from the use of indices of the hydrophobicity of the individual amino acids. Secondary and tertiary structures are important in determining the actual hydrophobic nature of the polypeptide, however, and this complicates the prediction of their physicochemical properties such as solubility and adsorption.
If alternating hydrophilic and hydrophobic amino acid sequences in synthetic peptides are at the right distances in space, the molecule coils with the hydrophobic amino acids on the inside of each coil and the hydrophilic ones to the outside. There are still, however, many structural mysteries: the interior of many protein structures with myriad side-chains, and the way in which metal ions can stabilise three-dimensional structures, have been likened to a terra incognita.
13.1.3 Solubility of peptides and proteins
As most proteins are delivered parenterally in solution it is important to understand their properties. In physiological conditions the aqueous solubilities of proteins vary enormously from the very soluble to the virtually insoluble. In section 4.2.4 we discussed the solubility profiles of zwitterions, including tryptophan, which all have biphasic solubility–pH profiles. One would expect proteins with terminal ‒NH2 and ‒COOH groups to behave similarly, although the effect will be complicated by the behaviour of the multitude of the intermediate amino acids, as can be seen from the pH–solubility profile of insulin in Chapter 9, Fig. 9.5. Figure 13.4 shows the general solubility behaviour of a protein ( -lactoglobin) as a function of pH at two ionic strengths.
-lactoglobin) as a function of pH at two ionic strengths.
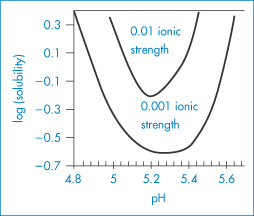
Figure 13.4 A plot of the logarithm of the aqueous stability of β-lactoglobulin versus pH at two different ionic strengths, 0.001 and 0.01 mol kg−1. When the protein is charged at pH values below and above pH 5.3, solubility increases from its lowest point at the isoelectric point. Increased ionic strength shifts the isoelectric point to lower pH values, around 5.2. See also Fig. 13.6.
The solubility of globular proteins increases as the pH of the solution moves away from the isoelectric point, which is the pH at which the molecule has a net zero charge and does not migrate in an electric field. Some examples of the isoelectric points of amino acids are shown in Table 13.4. At its isoelectric point a protein has no net charge and has a greater tendency to self-associate. As the net charge increases, the affinity of the protein for the aqueous environment increases and the protein molecules also exert a greater electrostatic repulsion. However, extremes of pH can cause protein unfolding, which not infrequently exposes further non-polar groups.
Table 13.4 Values of pK and isoelectric point (IP) of common L-amino acids
Amino acid | pK1 (COOH) | pK2 (NH3+) | pK3 | IP |
Alanine | 2.34 | 9.69 | – | 6.00 |
Asparagine | 2.02 | 8.80 | – | 5.41 |
Aspartic acid | 1.88 | 3.65 (COOH) | 9.60 (NH3+) | 2.77 |
Cysteine | 1.96 | 8.18 | 10.28 (SH) | 5.07 |
Glutamic acid | 2.19 | 4.25 (COOH) | 9.67 (NH3+) | 3.22 |
Glutamine | 2.17 | 9.13 | – | 5.65 |
Glycine | 2.34 | 9.60 | – | 5.97 |
Histidine | 1.82 | 6.00(imidazole) | 9.17 (NH3+) | 7.59 |
Isoleucine | 2.36 | 9.68 | – | 6.02 |
Leucine | 2.36 | 9.60 | – | 5.98 |
Lysine | 2.18 | 8.95(α) | 10.53 | 9.74 |
Methionine | 2.28 | 9.21 | – | 5.74 |
Phenylalanine | 1.83 | 9.13 | – | 5.48 |
Proline | 1.99 | 10.96 | – | 6.30 |
Serine | 2.21 | 9.15 | – | 5.68 |
Threonine | 2.71 | 9.62 | – | 6.16 |
Tryptophan | 2.38 | 9.39 | – | 5.89 |
Tyrosine | 2.20 | 9.11 | 10.07 (OH) | 5.66 |
Valine | 2.32 | 9.62 | – | 5.96 |
The relative hydrophilicities of the side-chains of the amino acids correlate well with the hydration of the side-chain; proteins are surrounded by a hydration layer, equivalent to about 0.3 g H2O per gram of protein, which represents about two water molecules per amino acid residue. Disturbing this layer has an impact on the behaviour of the protein in solution.
The phase behaviour of protein solutions is affected by pH, ionic strength and temperature. Protein–water solutions sometimes exhibit critical solution temperatures (see Chapter 4 for a discussion of phase separation). Phase transitions are important not only in manufacture and formulation but also because they have some pathophysiological implications. Phase separation of gamma-crystallins causes opacification of the lens of the eye in certain cataracts; Fig. 13.5 shows the phase diagram obtained.
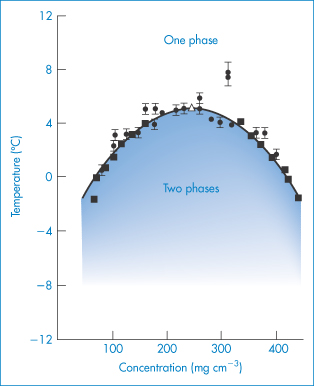
Figure 13.5 The temperature–concentration phase diagram for aqueous gamma-crystallin (mol wt 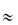 20 000 Da) systems (pH = 7, I = 0.24 mol kg−1). ●, cloud point measurements; ◼, concentration measurements of separated phases;
20 000 Da) systems (pH = 7, I = 0.24 mol kg−1). ●, cloud point measurements; ◼, concentration measurements of separated phases; 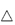 , critical point.
, critical point.
Reproduced with permission from Thompson JA et al. Binary liquid phase separation and critical phenomena in a protein/water solution. Proc Natl Acad Sci USA 1987;84:7079.
In section 4.2.3 we discussed the effect of salts on the solubility of organic electrolytes. The parabolic effects of salts on protein solubility (Fig. 13.6) might, at first sight, seem unexpected. Data on haemoglobin solubility, produced many decades ago (Fig. 13.6b), show a general increase in solubility with increasing ionic strength of salts such as NaCl, KCl and (NH4)2SO4. However, decrease in solubility occurs at higher ionic strengths – a salting-out effect. Several effects are responsible: (1) the preferential interaction of salts with bulk water; and (2) the effect of the salts on the surface tension of water, which is related to the energy of cavity formation. In fact, the degree to which a salt increases the surface tension of water is proportional to its tendency to salt out proteins.
Figure 13.6 (a) A generic plot of log(solubility) as a function of square root of ionic strength (I1/2) for proteins. (b) The plot for haemoglobin at 25°C, indicating the influence of the nature of the salt. Solubility is expressed as grams per 1000 grams H2O.
Reproduced from Green AA. J Biol Chem 1932;95:47–66.
The presence of other polymers in the solution will also tend to reduce protein solubility because of the volume exclusion effect. The addition of a water-soluble polymer (such as polyoxyethylene glycol (PEG)) can lead to the formation of two distinct liquid phases, owing to changes in the energy required to accommodate the protein molecules and to the unfavourable interaction of PEG with charges on the surface of the protein.
PEG molecules grafted to proteins, however, favourably change the properties of therapeutic proteins, as we will discuss later.
|
primary structure – the order in which the individual amino acids are arranged secondary structures – including coiled α-helix and pleated sheets tertiary structure – the three-dimensional arrangement of helices and coils, and sometimes quaternary forms, which are the association of ternary forms (e.g. the hexameric form of insulin).
|

13.2 The stability of proteins and peptides
In Chapter 3 we examined how the stability of drug formulations may be improved from a knowledge of the principal routes of degradation and the kinetics of breakdown. It has been implicitly assumed that the drugs under discussion were typical low-molecular-weight compounds and it is clear that for this type of drug there is a large body of knowledge relating to all aspects of their stability. The formulation of this important class of peptides and proteins presents more of a challenge.
Protein pharmaceuticals can suffer both physical and chemical instability: the major pathways are summarised in Fig. 13.7. Physical instability refers to changes in the higher-order structure (secondary and above), whereas chemical instability can be thought of as any kind of modification of the protein via bond formation or cleavage, yielding a new chemical entity. For each degradation route we will look at the effect of formulation parameters such as pH and ionic strength on stability and also discuss the application of accelerated stability-testing procedures. We will concentrate on the physical aspects of the problem.
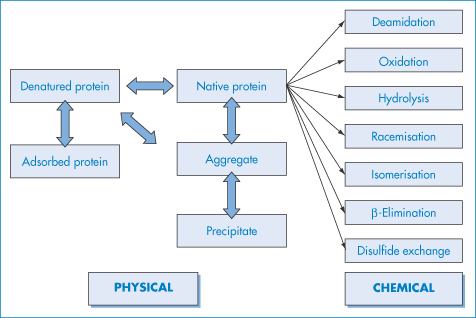
Figure 13.7 Schematic of the major pathways of protein degradation, which may be physical or chemical in origin.
13.2.1 Physical instability
Physical instability is a phenomenon that is rarely encountered with small organic molecules but that arises in peptides and proteins because of the many conditions under which proteins can lose their native three-dimensional structure. Unfolding of stable forms can lead to adsorption, aggregation or further chemical reactivity. Aggregation can lead to precipitation. Loss of the unique biologically active three-dimensional structure (denaturation) can be caused by heating and, conversely, by cooling or freezing, extremes of pH and contact with organic chemicals and denaturants. Most of these act through their influence on solubility or conformation, hence the importance of understanding protein–solvent interactions. It is generally thought that denaturation must first occur before the other processes such as aggregation, adsorption and precipitation can proceed.
Denaturation: reversible and irreversible
Denaturation refers to a disruption of the tertiary and secondary structure of the protein molecule. The denaturation can be reversible or irreversible. When caused by an increase of temperature, the process is said to be reversible if the native structure is regained on decreasing the temperature. Irreversible denaturation implies that the unfolding process is such that the native structure cannot be regained simply by lowering the temperature. In many cases the protein may become more compact, exposing hydrophobic residues that are normally buried within its core. It is the interaction between these hydrophobic residues in the protein interior that determines the physical stability of the proteins. When they become exposed to the solvent, these residues can interact with other hydrophobic surfaces such as the container walls or the exposed residues of other denatured proteins, leading to the localised accumulation of denatured protein molecules that results in the formation of aggregates. Increase of temperature results in greater flexibility of the proteins and an increased tendency for collision, leading to aggregate formation. An increase of the ionic strength may lead to neutralisation of the surface charge on the protein molecules that is responsible for their mutual repulsion, again resulting in aggregation. Charge neutralisation and subsequent aggregation can also occur when the pH of the solution approaches the isoelectric point of the protein. In both cases, molecules in the aggregated state can undergo denaturation with time as a result of subtle changes in their conformation and the aggregation process becomes irreversible.
Two basic pathways have been observed for proteins during denaturation and folding. The simplest is a two-state model. If we refer to the native state as N and the unfolded, denatured state as D, we can write

Once critical hydrophobic residues are exposed to solvents they can interact with other surfaces (containers or the air–water interface). The process may involve the formation of intermediate conformations (I) which may be stable and may self-associate during folding. The second general pathway for denaturation includes these stable intermediate species and may be written
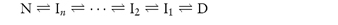
The equation shows the unfolding of a native protein to form an intermediate, In, which then unfolds to form other intermediates. If the two-state model is assumed, the concentration of denaturant required to achieve equal concentrations of N and D will give some indication of the process. If the fraction of denatured protein, fd, is calculated under given conditions (temperature, concentration of additive, etc.) we can write
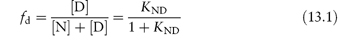
where KND is the equilibrium constant for denaturation (KND = [D]/[N]), related to the free energy of denaturation 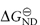 by
by
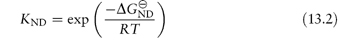
Hence,
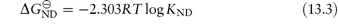
When 50% of the molecules are unfolded [D] = [N] and KND = 1, therefore 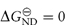 . The temperature at which this occurs is referred to as the melting temperature, Tm. An increase of Tm is indicative of an increase of stability; for example, T4 lysozyme (lysozyme from bacteriophage T4) is more stable at pH 6.5 where Tm is 65°C than at pH 2 where Tm is 42°C.
. The temperature at which this occurs is referred to as the melting temperature, Tm. An increase of Tm is indicative of an increase of stability; for example, T4 lysozyme (lysozyme from bacteriophage T4) is more stable at pH 6.5 where Tm is 65°C than at pH 2 where Tm is 42°C.
Aggregation/association
Some proteins self-associate in aqueous solution to form oligomers. Insulin, for example, exists in several associated states; the zinc hexamer of insulin is a complex of insulin and zinc that dissolves slowly into dimers and eventually monomers following its subcutaneous administration, so giving it long-acting properties. In most cases, however, it is desirable to prevent association such that only monomeric or dimeric forms are present in the formulations and a more rapid absorption is achieved. Recent studies have been directed towards engineering insulin molecules which are not prone to association,1,2 or the prevention of association through the addition of surfactants.3 Protein self-association is a reversible process, i.e. alteration of the solvent properties can lead to the re-formation of the monomeric native protein. There is an important distinction between this association process and the aggregation of proteins, which relates to the usually non-reversible interaction of protein molecules in their denatured state. Aggregation therefore implies that the proteins have undergone some form of denaturation prior to their interaction.
If an intermediate forms that has a solubility less than that of N or D, this can lead to aggregation and eventually to precipitation. For example, the addition of moderate amounts of denaturant to bovine growth hormone can generate a partially unfolded intermediate of low solubility that aggregates. Similarly, γ-interferon is inactivated by acid treatment or by the addition of salt because the dimeric native state is converted into monomers, which are partially denatured. For both proteins the formation of intermediates leads to inactivation.
Surface adsorption and precipitation
The adsorption of proteins such as insulin on surfaces such as glass or plastic in giving sets is important as it can reduce the amount of agent reaching the patient. It can also lead to further denaturation, which can then cause precipitation and the physical blocking of delivery ports in insulin pumps, for example. Insulin adsorption on the surface of the containers and the subsequent ‘frosting’ effect, due to the presence of a finely divided precipitate on the walls (Fig. 13.8), is accelerated by the presence of a large headspace allowing a greater interaction of the insulin with the air–water interface, which facilitates denaturation.

Figure 13.8 A vial of insulin showing the frosted appearance of flocculated and adsorbed protein.
Reproduced from diabetesindogs.com
13.2.2 Formulation and protein stabilisation
There are several possible ways in which the physical stability of the protein can be improved through formulation. We will examine methods for minimising this and chemical degradation in the following sections.
Prevention of adsorption
Some measures can be taken to eliminate, or at least minimise, protein denaturation resulting from surface adsorption. The surface of glass is conducive to adsorption and it is preferable in principle to use more hydrophilic surfaces, although this may not be feasible in practice. Alternatively, when the use of glass cannot be avoided, components may be added to the protein solution to prevent adsorption to the glass surface. These additives can act by coating the surface of glass or by binding to the proteins. For example, serum albumin can be included in the formulation since this will compete with the therapeutic protein for the binding sites on the glass surface and so reduce its adsorption. A similar effect can be achieved by the addition of surfactants such as poloxamers and polysorbates to the protein solution. Consideration must be given, however, to the effects of the surfactants on the pharmacology of the protein and to the toxicological effects of the surfactant itself.
Minimisation of exposure to air
Significant denaturation of proteins can occur when the protein solutions are exposed at the air–solution interface. In this respect the air interface is behaving as a hydrophobic surface and the extent of denaturation is found to be dependent on the time of exposure of the protein at the interface. Agitation of protein solutions in the presence of air or application of other shear forces, such as those that occur when the solutions are filtered or pumped, may also cause denaturation. Again, the inclusion of surfactants can reduce denaturation arising from these processes. Stability testing of protein-containing formulations often involves subjecting the solutions to shaking for several hours and the subsequent assessment of the protein configuration. If the protein has retained its native state and has not aggregated, the formulation is considered to be stable against surface- or shear-induced denaturation.
Addition of cosolvents
Some excipients and buffer components added to the protein solution are able to minimise denaturation through their effects on solvation. These compounds, including polyethylene glycols and glycerol, are referred to as cosolvents. They may act either by causing the preferential hydration of the protein or alternatively by binding to the protein surface. Preferential hydration results from an exclusion of the cosolvent from the protein surface due to steric effects (as in the case of polyethylene glycols); surface tension effects (as with sugars, salts and amino acids) or some form of chemical incompatibility such as charge effects. As a result, more water molecules pack around the protein in order to exclude the additive and the protein becomes fully hydrated and stabilised in a compact form (Fig. 13.9). Alternatively, the cosolvent may stabilise the protein molecule either by binding to it non-specifically or by binding to specific sites on its surface.
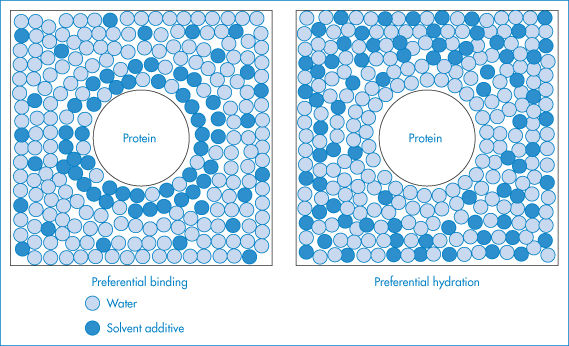
Figure 13.9 Schematic illustration of preferential binding and preferential hydration by solvent additives. In preferential binding the additive occurs in the solvation shell of the protein at a greater local concentration than in the bulk solvent, while preferential hydration results from the exclusion of the additive from the surface of the protein.
Reproduced with permission from Timasheff SN, Arakawa T. In: Creighton TE (ed.) Protein Structure: A Practical Approach. Oxford: IRL Press; 1989: 331–345.
Optimisation of pH
In order to avoid stability problems arising from charge neutralisation and to ensure adequate solubility, a pH must be selected that is at least 0.5 pH units above or below the isoelectric point. This is often difficult to achieve, however, since a pH range of 5–7 is usually required to minimise chemical breakdown and this frequently coincides with the isoelectric point.
Characterisation of degradation
If the formulation does not prevent denaturation and aggregation of the protein, then the pharmacology, immunogenicity and toxicology of the denatured or aggregated protein must be studied to determine its safety and efficacy. Several studies must be performed to determine the extent of degradation that is acceptable for administration. If the aggregates are soluble there may be a significant effect on the pharmacokinetics and immunogenicity of the protein. On the other hand, insoluble aggregates are generally unacceptable and instructions are usually given not to administer protein solutions containing precipitates.
|
avoiding contact with glass (if possible) or adding compounds (e.g. serum albumin or surfactants) that coat the glass surface or bind to the proteins, so minimising surface adsorption avoiding exposure at the air–solution interface, agitation of protein solutions in the presence of air or application of other shear forces; inclusion of surfactants can reduce denaturation arising from these processes addition of cosolvents such as polyethylene glycols and glycerol minimises denaturation by causing preferential hydration of the protein or by binding to the protein surface buffering at a pH at least 0.5 pH units above or below the isoelectric point to avoid stability problems arising from charge neutralisation and to ensure adequate solubility. |

13.2.3 Chemical instability
Chemical instability can involve one or more of a variety of chemical reactions, including:
- deamidation, in which the side-chain linkage in a glutamine (Gln) or asparagine (Asn) residue is hydrolysed to form a carboxylic acid
- oxidation: the side-chains of histidine (His), methionine (Met), cysteine (Cys), tryptophan (Trp) and tyrosine (Tyr) residues in proteins are potential oxidation sites
- racemisation: all amino acid residues except glycine (Gly) are chiral at the carbon atom bearing the side-chain and are subject to base-catalysed racemisation.
- proteolysis, involving the cleavage of peptide (CO-NH) bonds
- beta elimination: high-temperature treatment of proteins leads to destruction of disulfide bonds as a result of beta elimination from the cystine residue
- disulfide formation: the interchange of disulfide bonds can result in an altered three-dimensional structure.
Table 13.5 lists the amino acids or sequences that are subject to chemical degradation and the formulation approaches used to overcome the problems.
Table 13.5 Amino acids or sequences susceptible to chemical degradation, together with formulation strategies to reduce degradation
Amino acid or sequence | Mechanism of degradation | Formulation strategy |
Cysteine–cysteine | Aggregation | Addition of surfactants, polyalcohols |
Glutamine, asparagine | Deamidation | pH 3–5 |
Tryptophan, methionine, cysteine, tyrosine, histidine | Oxidation | pH < 7 |
Methionine | Oxidation | Protect from oxygen |
Tryptophan | Photodecomposition | Protect from |
Lysine-threonine | Copper-induced cleavage | Chelating |
Asparagine-proline, asparagine-tyrosine | Hydrolysis | pH > 7 |
Reproduced with permission from Parkins DA, Lashmar UT. The formulation of biopharmaceutical products. Pharm Sci Technol Today 2000;3:129–137.
13.3 Protein formulation and delivery
It has been said that ‘drug delivery represents the potential Achilles’ heel of biotechnology’s peptide drug industry’. The reasons for this include the range of instabilities discussed above, the inherent low membrane transport by diffusion because of molecular size and hydrophilicity, and often the need for temporal and site control of delivery.
13.3.1 Protein and peptide transport
For a series of 11 model peptides in an in vitro intestinal cell monolayer system, a good correlation was found between the permeability coefficient, P, and the log of the partition coefficient of the peptides between heptane and ethylene glycol (rather than octanol and water) (Fig. 13.10), results that also suggest that the principal deterrent to peptide transport is the breaking of hydrogen bonds. Molecular volume (or size) will increasingly be a factor as the molecular weight of the peptide increases.
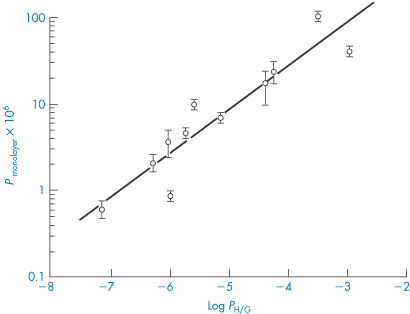
Figure 13.10 A plot of the permeability coefficient P across Caco-2 cell monolayers versus the log P (heptane/glycol) for a series of synthetic peptides to illustrate relationships between lipophilicity and transport.
Reproduced with permission from Paterson DA et al. Quant Struct–Action Relationships 1994;13:4–10.
The diffusion of proteins and peptides in solution is dictated by the same considerations as those discussed in Chapter 2, section 2.6. The rate of translational movement depends on the size of the molecule, its shape and interactions with solvent molecules. The rate of translational movement is often expressed by a frictional coefficient, f, defined in relation to the diffusion coefficient D, by equation (13.4):

where kB is the Boltzmann constant and T is the temperature in kelvins. Many proteins are nearly spherical in solution, but if their shape deviates from sphericity this is reflected in a frictional ratio, f/f0, above unity, where f0 is the rate of diffusion of a molecule of the same size but of spherical shape. The frictional ratio of lysozyme is 1.24 and that of trypsin is 1.187. Globular proteins have values of f/f0 in the range 1.05–1.38.
The diffusion coefficients and translational movements of proteins are important in considering the release of proteins from hydrogel matrix devices and other delivery vehicles, and in membrane transport, as far as this can be considered to be a passive diffusion process. Changes in shape during membrane transport in a lipid environment may also have to be considered. Table 13.6 gives some values of the diffusion coefficients of a number of therapeutic peptides and proteins.
Table 13.6 Diffusion coefficients and molecular weights of peptides and proteins
Compound | Molecular weight (Da) | 106 D (cm2 s–1) |
Oxytocin | 1007 | 4.30 |
Lys-vasopressin | 1056 | 4.18 |
Arg-vasopressin | 1084 | 4.27 |
Somatostatin | 1638 | 3.74 |
Calcitonin | 3418 | 2.76 |
Insulin | 5807 | 1.14 |
Calculated (insulin) | ||
Monomer | 5807 | 2.03 |
Hexamer | 34 845 | 0.93 |
Reproduced from Hosoya O et al. J Pharm Pharmacol 2004;56:1501–1507. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
13.3.2 Lyophilised proteins
Because of their potential instability in solution, therapeutic proteins are often formulated as lyophilised powders. Even in this state several suffer from moisture-induced aggregation. Proteins such as insulin, tetanus toxoid, somatotropin and human albumin aggregate in the presence of moisture, which can lead to reduced activity, stability and diffusion.
In the presence of water vapour at 37°C, lyophilised recombinant human albumin experiences intermolecular thiol–disulfide interchange and forms high-molecular-weight, water-insoluble aggregates.4 The use of excipients (e.g. low- and high-molecular-weight sugars, organic acids) to prevent such effects leads to the conclusion that the stabilising effect is correlated with the ability of the additives preferentially to absorb water. Figure 13.11 illustrates the effects on recombinant human albumin solubility and the influence of sorbitol on the changes in solubility.
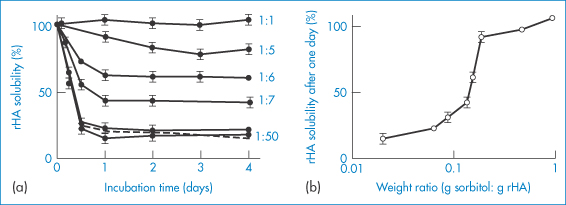
Figure 13.11 Stabilisation of recombinant human albumin (rHA) against aggregation afforded by co-lyophilised sorbitol. (a) The time-dependent change of solubility of rHA co-lyophilised with sorbitol at various sorbitol-to-rHA weight ratios, as indicated (the dashed line depicts the time course of rHA aggregation in the absence of sorbitol). (b) rHA solubility after a 1-day incubation as a function of sorbitol-to-rHA weight ratio, taken from the data in plot (a).
Reproduced with permission from Costantino JR et al. Aggregation of a lyophilized pharmaceutical protein, recombinant human albumin: effect of moisture and stabilization by excipients. Biotechnology 1995;13:493–496.
13.3.3 Water adsorption
Because of the sensitivity of proteins to moisture, the ability to measure sorption of water is vital in preformulation. Water sorption isotherms from recombinant bovine somatotropin (rbST), a protein with 191 amino acids, molecular weight 22 000 Da, show water content versus the relative vapour pressure (p/p0) for the sodium salt and the ‘internal’ salt of rbST (Fig. 13.12). These were fitted to modified BET isotherms which have an additional state of interacting water intermediate to the free water and the strongly bound water states.5,6 Calculations reveal that a monolayer of water is formed from 88 moles of H2O per mole of protein for the lyophilised sodium salt of rbST, which is equivalent to about 7.3 g per g of protein. The much higher apparent surface areas obtained using water (as opposed to nitrogen) adsorption isotherms (typically 264 m2 g−1, compared with 1.3 m2 g−1 with nitrogen) suggest that water penetrates the powder and that the isotherm represents adsorption and absorption. The practical implications of moisture load are seen in Fig. 13.13, which shows the rate of decomposition of rbST (albeit at 47°C) following incubation in sealed vials.
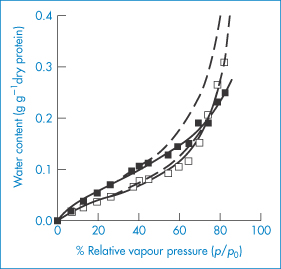
Figure 13.12 Comparison of fits obtained by the Brunauer–Emmett–Teller (BET: dashed lines) and Guggenheim–Anderson–de Boer (GAB: solid lines) models for the sorption isotherms of the sodium (◼) and internal (◻) salts of bovine somatotropin.
Reproduced with permission from Hageman MJ et al. Prediction and characterization of the water sorption isotherm for bovine somatotropin. J Agric Food Chem 1992;40:342–347. Copyright 1992 American Chemical Society.
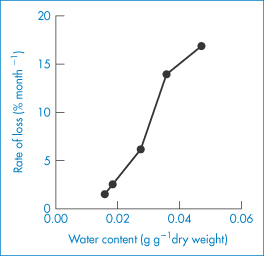
Figure 13.13 The increasing rate of decomposition of a lyophilised recombinant bovine somatotropin formulation with increasing water content following incubation in sealed vials at 47°C.
Reproduced from reference 5.
Both solubility and stability have to be addressed, as well as the mode of delivery. The wide range of biodegradable polymers used for the controlled delivery of proteins and peptides includes natural substances, starch, alginates, collagen and a variety of proteins such as crosslinked albumin, a range of synthetic hydrogels, polyanhydrides, polyesters or orthoesters, poly(amino acids) and poly(caprolactones). Poly(lactide-glycolide) (PLGA) is one of the commonest polymers used in microsphere form to deliver, inter alia, growth hormone-releasing factor, a somatostatin analogue, ciclosporin and luteinising hormone-releasing hormone antagonists.7,8
13.3.4 PEGylation of peptides and proteins
PEG is a versatile molecule. It is used pharmaceutically in molecular weight ranges from 200 to 10 000 Da. The polyoxyethylene chain forms an integral part of many non-ionic surfactants (polysorbate 80, Triton X-100). PEG is used as a vehicle for drug delivery: higher-molecular-weight fractions are used in suppository bases and lower-molecular-weight species (200–300 Da) that are liquid at room temperature are useful drug solvents. The molecule can be coupled in a range of molecular weights and hence lengths to peptides and proteins, enhancing their native properties by creating a hydrophilic ‘envelope’ around proteins and antibodies (Fig. 13.14). A variety of techniques is also used to modify the properties of liposomes and nanoparticles to reduce their uptake by the reticuloendoethelial system. The PEG chains act to prevent opsonisation and hence reticuloendoethelial system scavenging, thus increasing half-life in vivo.
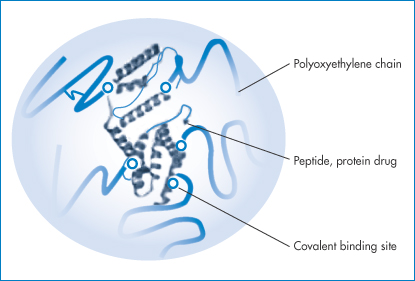
Figure 13.14 A schematic representation of a PEGylated protein, showing the protein surrounded by a polyoxyethylene glycol (PEG) protective sheath.
Pegylation is the term used to describe this modification of proteins, peptides or, indeed, non-peptide molecules by linking one or more PEG chains to the molecule.9 PEG is non-toxic, non-immunogenic, non-antigenic and highly soluble in water. The PEG–drug conjugates also have a prolonged circulation time after intravenous administration; they are less susceptible to degradation by metabolic enzymes; and their immunogenicity is reduced. The hydrophilic halo is thus important, sometimes increasing the water solubility of the molecule and potentially its biological activity through modification of interactions with biological target sites. The section on colloid stability should be consulted (Chapter 6, section 6.2) to see how PEG molecules affect the physical stability of emulsions and particles.
Naturally the physical properties of the PEGylated molecules are altered by their acquired hydrophilic chains. Clearly their molecular radius increases: haemoglobin has a molecular weight of 64 kDa and a radius of 3.1 nm. As the molecular weight of the attached PEG chains increases, so too does the radius, more than doubling to 7 nm with large PEG chains of 450 (‒OCH2CH2‒) units. The viscosity of solutions increased by nearly fivefold over native haemoglobin solutions. Because of the variety of possible linkages between the PEG molecules and the protein concerned, and because of the choice of PEG molecular size and indeed number of attached molecules, it is vital that the exact structural properties of these modified products are known, especially when generic versions of therapeutic proteins are available. The structure and properties of proteins and MAbs prepared by different methods, and processed in different ways with non-identical excipients, are rarely identical. Thus generic macromolecular products are termed ‘biosimilars’ to make this point. Many of the issues surrounding biosimilars are dealt with in Chapter 16.
Pegylated therapeutic proteins
As discussed above proteins have been modified by the covalent addition of PEG chains to their structures. While the intrinsic activity of the protein is generally unaffected, other parameters change so that a pegylated protein must be considered to be a new chemical entity. Pegylation of therapeutic proteins changes their pharmacokinetics and dynamics. It improves the performance of therapeutic proteins, providing them with a longer circulation time in vivo, and a reduction in immune responses. There is often also enhanced solubility of the peptide or protein (as might be expected from the addition of these very hydrophilic chains) or improved stability. Increased circulation time decreases the dose of protein necessary for biological action. Reduced antigenicity, immunogenicity and proteolysis are some of the benefits claimed for pegylated over non-pegylated forms of proteins and peptides.
Why is this? The barrier produced by the PEG chains is both a physical and a thermodynamic one. The polyoxyethylene molecules shield the protein, reducing uptake and loss by the reticuloendothelial system. The resulting larger size of the molecule may, however, confer slower diffusional characteristics on the molecules. The addition of these water-soluble entities not only increases solubility but can also decrease the pH dependency of protein solubility. Above all, the PEG layer shields the protein from opsonisation in vivo and thus the molecules are not scavenged by the reticuloendothelial system and hence circulation times are enhanced. As might be anticipated, the length of the PEG chain is important: the longer the chain, the longer the circulation time. Figure 13.15 shows the kinetics of free (unattached) PEG molecules as a function of molecular size.10
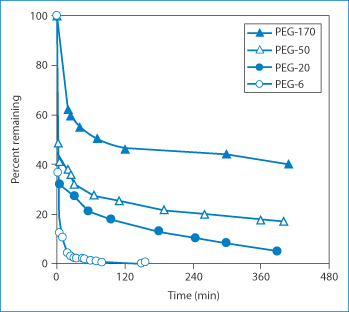
Figure 13.15 The percentage of polyethylene glycols (PEG) remaining in the circulation as a function of their size (molecular weight: 6 = 6 kDa, 20 = 20 kDa etc.).
Reproduced from Mehrar R. Modulation of the pharmacokinetics and pharmacodynamics of proteins by polyoxyethylene glycol conjugation. J Pharm Pharm Sci 2000;3:125–136.
Pegfilgastim is a pegylated recombinant methionyl human granulocyte-stimulating factor indicated in the reduction of the duration of neutropenia and incidence of febrile neutropenia during cancer chemotherapy. It is administered by intramuscular injection, and has a half-life in human subjects of 15–80 hours compared to the 3–4 hours of the parent compound.
PEG-Intron (peginterferon alfa) (Fig. 13.16) is a covalent conjugate of recombinant interferon alfa-2b with monomethoxy polyethylene glycol. Compared to standard interferon alfa, this modified molecule has a longer half-life after injection, allowing once-weekly injections and superior antiviral efficacy in the treatment of hepatitis C when used in combination with ribavirin. A case of a local blistering reaction developing in a patient receiving pegylated interferon alfa-2b has been reported.11 Dalmau et al.12 state that, although injection site pain is infrequent (2–3%), site inflammation and skin reactions (e.g. bruises, itchiness, irritation) occur at approximately twice the incidence with pegylated interferon alfa-2b treatment (in up to 75% of patients) compared with ordinary recombinant interferon alfa-2b. Dalmau and co-workers suggest that ‘because pegylated interferon alfa-2b has almost completely replaced interferon for its most frequent indications, increased awareness of the possibility of cutaneous necrosis is necessary for early diagnosis to prevent continuation of pegylated interferon alfa-2b injections at the involved area’. There are hints that any toxicity at the site of injection might be due to its higher molecular weight and slower diffusion from the site.
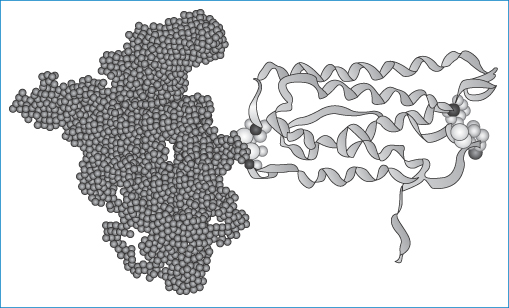
Figure 13.16 Pegylated interferon (Mire Zloh, The School of Pharmacy, University of London). The right-hand portion of the molecule represents the interferon while the left-hand part represents the polyoxyethylene chains.
Methoxy polyethylene glycol-epoetin beta (pegzerepoetin alfa; Mircera) is given by subcutaneous injection or intravenous infusion. Its half-life is extended over other forms of epoetin alfa or epoetin beta, as shown in Fig. 13.17.
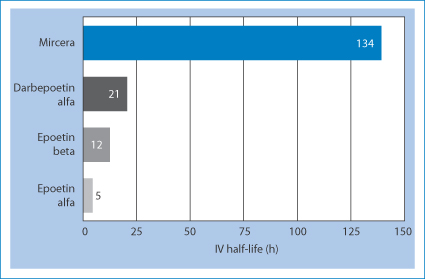
Figure 13.17 The half-life of pegzerepoetin alfa (Mircera) compared with darbepoetin alfa, epoetin alfa and epoetin beta.
Pegvisomat (Somavert), a pegylated analogue of human growth hormone structurally altered to act as a growth hormone receptor antagonist, comprises 191 amino acids with an average of 4–6 PEG molecules covalently bound to lysine residues and one bound to the terminal phenylalanine. It acts by binding to growth hormone receptors on cell surfaces. The molecular weight of the protein is 21 998 Da and the molecular weight of each PEG chain is 5 000 Da; hence there are three predominant molecular sizes in the product, with molecular weights of 42 000, 47 000 and 52 000 Da. Thus it is not a homogeneous product, but it should be a consistent product in terms of the ratios of the predominant species. Clearly, when generic versions become available, information on the molecular weight of species will be an important parameter. Peak concentrations after subcutaneous administration are achieved 33–77 hours after administration, a result likely to be due to its high molecular weight.
13.3.5 Peptide–albumin complexes
Other strategems to increase the circulation time of peptides include the formation of adducts with serum albumin, an example of which is shown in Fig. 13.18. This is of albuferon, an albumin–interferon-alpha fusion product. While on a molar basis it is less active than the native interferon-alpha, its overall activity is higher, and its half-life is prolonged, which reduces the burden of frequent dosing.
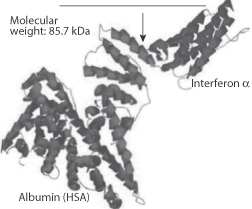
Figure 13.18 An interferon-human serum albumin (HSA) conjugate molecule (Albuferon) combining the activity of interferon-alpha with the longevity in the circulation of albumin.
Reproduced with permission from www.natap.org (2005).
Albumin–drug complexes
To alter the in vivo behaviour (such as half-life and stability) of some drugs their conjugates with albumin have been prepared. Hence the albumin with its own circulation and half-life characteristics acts as a carrier for the drug substance. A methotrexate–albumin complex has been studied, as has a doxorubicin conjugate. Another strategy is to create prodrugs with exposed hydrophobic groups which cause the drug to bind to circulating albumin. One such case, the myristic acid derivative of insulin, Levemir, is discussed below in section 13.4.1.
13.3.6 Routes of peptide and protein delivery
Table 13.7 lists the invasive and non-invasive routes of delivery for peptides and proteins, involving direct injection of solutions, depot systems and a variety of oral, nasal, topical and other formulations. Figure 13.19 summarises some of the issues involved in delivery of peptides and proteins by different routes. Cyclic peptides such as ciclosporin (I) can be absorbed orally; the bioavailability of ciclosporin is of the order of 15%. Such cyclic peptides, although molecularly large, bear a closer resemblance to conventional drugs in many ways, but absorption is quite compound-specific.
Table 13.7 Routes of delivery for proteins and peptides
Delivery routes | Formulation and device requirements | Commercial productsa |
Invasive | ||
Direct injection: intravenous, subcutaneous, intramuscular, intracerebral vein | Liquid or reconstituted solid, (syringe) | Activase (alteplase) |
Depot system (subcutaneous or intramuscular) | Biodegradable polymers, liposomes, permeable polymers (not degradable), microspheres, implants | Lupron Depot (leuprolide) |
Non-invasive | ||
Pulmonary | Liquid or powder formulations, nebulisers, metered-dose inhalers, dry-powder inhalers | Pulmozyme (dornase alfa) |
Oral | Solids, emulsions, microparticulates, with or without absorption enhancers |
|
Nasal | Liquid, usually requires permeation enhancers | Synarel (nafarelin) |
Topical | Emulsions, creams or pastes (liposomes) |
|
Transdermal | Electrophoretic (iontophoresis), electroporation, chemical permeation enhancers, prodrugs, ultrasonics |
|
Buccal, rectal, vaginal | Gels, suppositories, bioadhesives, particles |
|
Modified with permission from Cleland JL, Langer R. Formulation and delivery of proteins and peptides: design and development strategies. In: Cleland JL, Langer R (eds) Formulation and Delivery of Proteins and Peptides. Washington, DC: American Chemical Society; 1994: 1–19. Copyright 1994 American Chemical Society.
aActivase (recombinant human tissue plasminogen activator), Nutropin (recombinant human growth hormone) and Pulmozyme (recombinant human deoxyribonuclease I) are all products of Genentech; RecombiVax (recombinant hepatitis B surface antigen) is produced by Merck; Lupron Depot (leuprolide acetate – poly(lactide-glycolide) (PLGA)) is a product of Takeda Pharmaceuticals; Zoladex (goserelin acetate – PLGA) is produced by AstraZeneca; Decapeptyl is produced by Debiopharm; Synarel (nafarelin acetate) is produced by Roche.
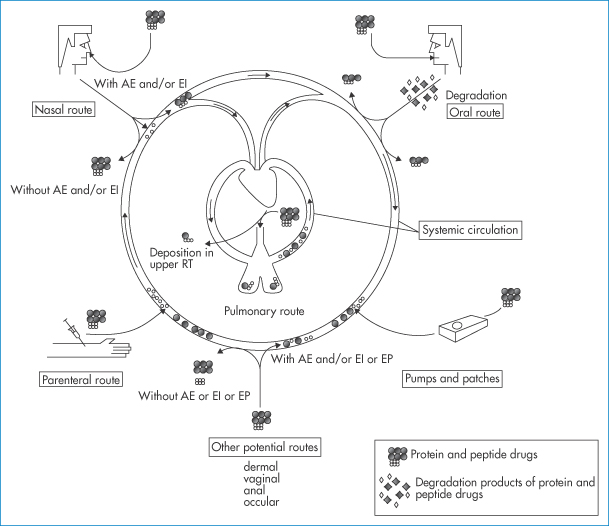
Figure 13.19 A schematic diagram referring to the different routes of administration of protein and peptide drugs, namely the parenteral, nasal, oral and pulmonary routes and by pumps and patches. Of all these, the oral route is the most problematic. Other potential routes, dermal, vaginal, anal and ocular are listed. AE = absorption enhancers; EI = enzyme inhibitors; EP = electroporation or iontophoresis; RT = respiratory tract.
Reproduced with permission from Agu RU et al. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res 2001;2:198–202.
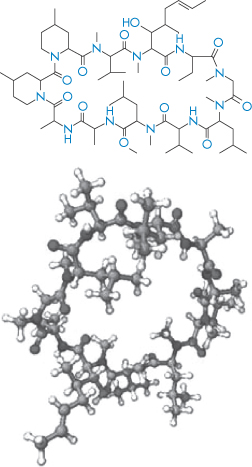
Structure I Ciclosporin in two different modes of representation
Other cyclic peptides include the antibiotics Tyrocidin A and gramicidin S, which are both cyclic decapeptides.
13.4 A therapeutic protein and a peptide
13.4.1 Insulin
It is appropriate that the protein therapeutic substance with the longest pedigree is discussed here. There are three main types of insulin preparations:
- those with a short duration of action that have a relatively rapid onset (soluble insulin, insulin lispro, insulin aspart, insulin glulisine)
- those with an intermediate duration of action (isophane insulin and insulin zinc suspension)
- those with a slow action, slower in onset and lasting for long periods (crystalline insulin zinc suspension; insulin glargine, insulin detemir, insulin degludec).
Some aspects of insulin were dealt with in section 9.2.5. Tables 13.8a and b list some of the insulin formulations designed to produce different durations of onset and action. Insulin is generally self-administered subcutaneously with injection pens, needle-free devices or pumps.
Table 13.8a Effect of insulin formulation on its pharmacokinetics after subcutaneous injection
Producta | Formulation | Pharmacokineticsb |
Humulin R | Zinc-insulin crystalline suspension | Rapid onset, short duration |
Humulin N | Isophane suspension protamine, zinc crystalline insulin (buffer water for injection) | Intermediate-acting, slower onset, longer duration than regular insulin |
|
| Start, 1.5 h; peak, 4–12 h; end, up to 24 h |
Humulin 70/30c | 70% isophane suspension | Intermediate-acting, faster onset |
|
| Start, 0.5 h; peak, 2–12 h; end, up to 24 h |
Humulin U | Extended zinc-insulin suspension – all crystalline | Slow-acting, slow onset, longer, less-intense duration than R or N forms |
Humulin L | 70% zinc-insulin crystalline suspension | Intermediate-acting, slower onset, longer duration |
Humulin BR | Zinc crystalline insulin dissolved in sodium diphosphate buffer | Rapid onset, short duration; use in pumps only |
a Humulin products (Eli Lilly & Company) contain recombinant human insulin derived from Escherichia coli; Novolin products (Novo Nordisk) are recombinant human insulin derived from Saccharomyces cerevisiae; both companies also sell other forms of recombinant human insulin and may have additional forms (formulations or new drugs) in clinical trials.
b The pharmacokinetics of each formulation may vary greatly among individuals; the onset of therapeutic levels of insulin is referred to as the start of the effect, the maximum serum levels is the peak, and the time when the levels of insulin are below therapeutic levels is listed as the end of the therapeutic time course.
c Solution consists of 70% N form and 30% R form for both products.
Table 13.8b Comparison of the onset peak and duration of action of insulin products
Examples | Products
| Onset of action (min) | Peak action | Duration of action (h) |
Insulin aspart | Novorapid | 10–20 | 30–180 min | 2–5 |
Human sequence soluble insulin | Actrapid, Humulin S | 30–90 | 2–4 h | 6–8 |
Isophane insulin | Insulatard | 30–90 | 6–8 h | 11–24 |
Insulin glargine | Lantus | 60–120 | No peak | 20–26 |
Reproduced with permission from Jacques N, Hackett E. Clin Pharmacist 2013;5:75.
There is discussion of some of the characteristics of these products below. The pharmacokinetics of each formulation may vary greatly among different individuals; in these tables the onset of therapeutic levels of insulin is referred to as the start of the effect, the maximum serum level of insulin is denoted the peak, and the time at which the insulin levels are below therapeutic levels is listed as the end of the therapeutic time course.
Precipitation of insulin and other proteins
Precipitation of insulin in pumps due to the formation of amorphous particles, crystals or fibrils of insulin can lead to changes in release pattern or to blockage that prevents insulin release. ‘Amorphous’ or ‘crystalline’ precipitates can be caused by the leaching of divalent metal contaminants or lowering of pH (due to CO2 diffusion or leaching of acidic substances), but can be prevented. More difficult to solve is the tendency of insulin to form fibrils, as illustrated in Fig. 13.20.
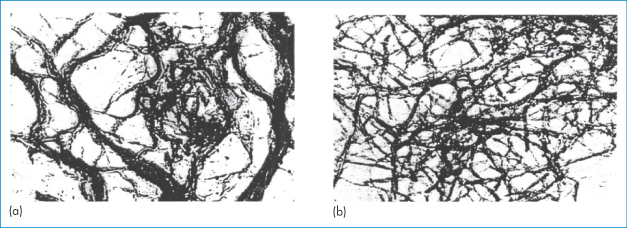
Figure 13.20 Transmission electron micrographs of insulin fibrils formed (a) at 37°C and (b) at 80°C (×100 000).
Reproduced from Brange J. Galenics of Insulin. Berlin: Springer; 1987, with kind permission from Springer Science and Business Media.
The interactions leading to fibril formation result from the monomeric form, and from changes in the monomer conformation and hydrophilic attraction of the parallel β-sheet forms. Fibril formation is also encouraged by contact of the insulin solution with hydrophobic surfaces.
Propylene glycol, glycerol, non-ionic and ionic surfactants and calcium ions have been used in formulations to achieve greater stability, reducing fibril formation, but the most successful strategy is the addition of calcium ions or zinc, which appear to protect the hexameric form of the insulin (see section 9.2.5).
Recombinant human insulin: insulin lispro and insulin aspart
Insulin lispro and insulin aspart are mentioned in Table 13.8b. Chemical modifications to an endogenous protein, however minor, can lead to significant differences in properties and activity. Species differences can also be great. Salmon calcitonin (SCT) is 10 times more potent than human calcitonin (hCT), for example. Recombinant human protein analogues may be subtly different, as in the case of insulin lispro, in which the sequence of proline and lysine at positions 28 and 29 in the B protein chain has been reversed (Fig. 13.21a). This sole difference leads to formation of hexamers, which more rapidly dissociate to monomers on injection, giving a faster onset of action than human insulin, in which the B28 is proline and B29 is lysine (such as the recombinant product Humulin S, which is injected up to an hour before meals).
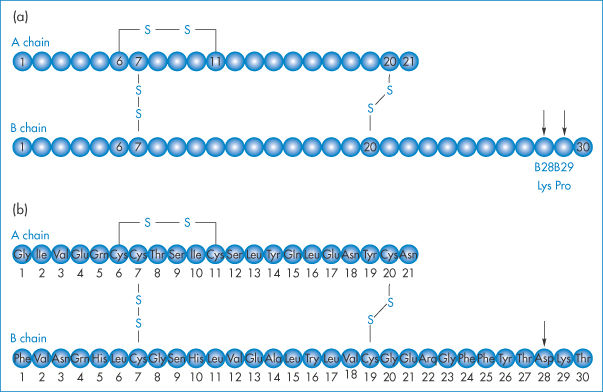
Figure 13.21 (a) Insulin lispro, a product of recombinant DNA technology, is an analogue of human insulin in which the natural sequence of proline and lysine at positions B28 and B29 (arrowed) of human insulin has been reversed. (b) Insulin aspart (Novo Nordisk) has an aspartame at B28 (arrow) replacing the proline of the natural insulin. Insulin glargine has a glycine at position A21 and two arginines at B30.
Insulin glargine (Lantus, Aventis) has two arginines added to the B30 position (Fig. 13.21b). Its isoelectric point changes from a pH of 5.4 to 6.7, making it a soluble (and thus clear) solution in the acid medium of pH 4 in which it is supplied. When it is injected subcutaneously, however, glargine precipitates at the physiological pH of 7.4, which delays its absorption and prolongs its action. Substituting glycine for asparagine at the A21 position and adding zinc stabilises the hexamer and delays absorption further.
Glargine has a similar insulin receptor affinity to that of NPH (Neutral Protamine Hagedorn, an intermediate-acting insulin that initially achieves slower action through the addition of protamine to short-acting insulin) so that, once absorbed in the circulation, glargine is equipotent to NPH. The result is a relatively constant supply of insulin with an onset of approximately 2 hours and a duration of action of approximately 24 hours.
Insulin detemir (Levemir) is a long-acting human insulin analogue for maintaining the basal level of insulin and is an example of a modified therapeutic agent which allows it to bind to plasma proteins. It is an analogue in which a fatty acid (myristic acid) is bound to the lysine amino acid at position B29. It is quickly absorbed, after which it binds to albumin in the blood through its fatty acid at position B29. It then slowly dissociates from this complex. Figure 13.22 aids the visualisation of the chemistry of this and human and porcine insulin.
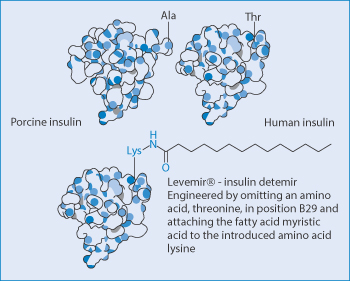
Figure 13.22 The chemical modification by the addition of the hydrophobic myristic acid group to insulin to encourage its binding in vivo to albumin and the change in it pharmacokinetics.
Reproduced from Elsadek B, Kratz F. J Control Release 2012;157:4–28. Copyright Elsevier 2012.
13.4.2 Calcitonin
Calcitonin, a peptide hormone of 32 amino acids having a regulatory function in calcium and phosphorus metabolism, is used in various bone disorders such as osteoporosis. Salmon, human, pig and eel calcitonin are used therapeutically. hCT has a tendency to associate rapidly in solution and, like insulin, form fibrils, resulting in a viscous solution.13 The fibrils are 8 nm in diameter and often associate with one another. Heating fibrillated hCT solutions in 50% acetic acid–water converts the system back to soluble monomers. hCT has a pK of 8.7, while sCT has a pK of 10.4. This accounts for the high stability of sCT at pH 7.4, as electrostatic interactions between calcitonin monomers play a role. At pH 7.4, sCT monomers will be charged and will repel each other.
13.5 DNA and oligonucleotides
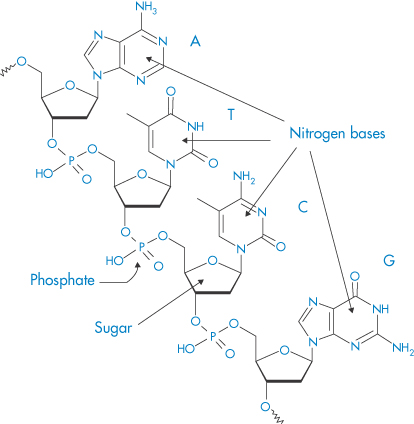
13.5.1 DNA
DNA of varying molecular weights is used in gene therapy. Figure 13.23 shows the well-known primary structure of DNA and the charge-determining phosphate groups connected to the deoxyribose rings. As DNA is a large hydrophilic, polyanionic and sensitive macromolecule, its successful delivery to target cells and the nucleus within these cells is an issue. Shearing of high-molecular-weight DNA while stirring in solution can lead to breakage of the molecule. One approach to delivery is to complex the DNA with polymers or particles of opposite charge to produce, by condensation, more compact species. Many studies have been conducted to condense the DNA with cationic polymers (e.g. polylysine and chitosan), cationic liposomes and dendrimers, to produce nano-sized complexes that retain an overall positive charge and are able to transfect cells more readily than native or naked DNA. The interaction of DNA with polycationics like chitosan is akin to a coacervation process14 (Fig. 13.24).
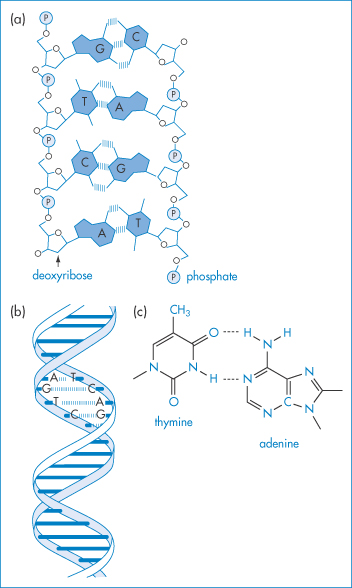
Figure 13.23 (a) Diagrammatic representation of the interactions between two oligonucleotide strands. ////// represents hydrogen-bonding interactions between the base pairs G–C, T–A, C–G, A–T. P represents the phosphate groups, which confer the negative charge on the molecule. (b) Representation of the double helix, again illustrating the hydrogen-bonding interaction that is key to the helical conformation. (c) The detailed primary structure showing the phosphate–deoxyribose–base point sequence for T–A.
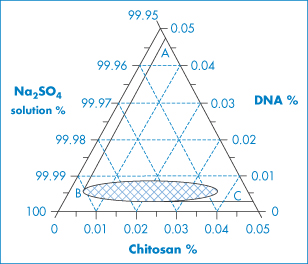
Figure 13.24 Ternary-phase diagram of complex coacervation between a plasmid DNA and chitosan at 55°C in 50 mmol dm−3 Na2SO4. Sodium sulfate solution was regarded as one component, since the concentration change in the experiment range was minimal. The region to the right of the line ABC depicts the conditions under which phase separation occurs. The concentration ranges in the small grid area yield distinct particles.
Reproduced from Mao H-Q et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterisation and transfection efficiency. J Control Release 2001;70:399–421. Copyright Elsevier 2001.
Through the formation of particulates (Fig. 13.25) cell-uptake characteristics and stability are improved, although transfection inside the cell depends ultimately on the successful release of DNA from the complex and its uptake into the nucleus.
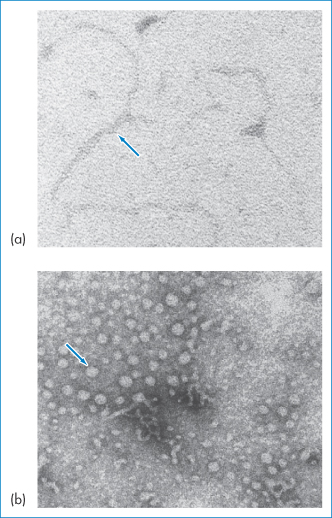
Figure 13.25 (a) Electron micrograph of naked plasmid DNA; (b) the same sample after condensation with a cationic partial dendrimer. The size, shape, charge and stability of such complexes depend on the ionic strength of the medium, lipid composition, lipid/DNA ratio and concentration, and even on mixing procedures. Manufacturing issues include those of reproducibility, avoiding aggregation of the resulting colloidal suspension and lyophilisation, as discussed by RI Mahato et al. (Pharm Res 1997;14:853–859).
Reproduced from Ramaswamy C, Florence AT. Self-assembly of some amphipathic dendrons. J Drug Del Sci Tech 2005;15:307–311. Copyright Elsevier 2005.
13.5.2 Oligonucleotides
Antisense oligonucleotides (II) (used for the sequence-specific inhibition of gene expression) are polyanionic single-strand molecules comprising 10–25 nucleotides. They resemble single-stranded DNA or RNA. They have molecular weights ranging from 3000 to 8000 Da and are hydrophilic, having a log P of approximately −3.5. Like DNA, they clearly do not have the appropriate properties for transfer across biological membranes. They are also sensitive to nucleases and non-specific adsorption to biological surfaces, and hence require to be formulated to achieve delivery and nuclear uptake. Chemical modification of oligonucleotides, which is outside of the scope of this book, can increase the stability and activity of the molecules.
Structure II An oligonucleotide
13.6 Therapeutic monoclonal antibodies
There are growing numbers of MAbs (antibodies produced from a single clone of cells) available for use in the clinic, including MAb–drug conjugates. Typical MAbs include anti-HER2 MAb, trastuzumab (Herceptin, Roche) for breast cancer and rituximab for malignant lymphoma. Although MAbs were first produced in the mid-1970s, there have been several problems with their clinical use, for, in spite of their target specificity, their poor distribution and tissue penetration result in the need for high doses. The latter leads to various pharmaceutical issues common to the group. The restricted penetration results from the fact that MAbs are large molecules with molecular weights of the order of 150 kDa, so that they diffuse slowly in tissues, especially in tumours or inflamed sites. Antibodies with a molecular size greater than 150 kDa are prevented from passage through the blood–brain barrier, for example, hence the cerebrospinal fluid level of one MAb, rituximab, is known to be only 0.1% of its serum levels. While there has been evidence suggesting that antibodies against beta-amyloid plaques can reverse cognitive deficits in early Alzheimer disease, promising constructs do not cross the blood–brain barrier. Their physical chemistry can explain in part the properties of MAbs, such as blocking the action of target antigens, their binding to antigens and their diffusion and translocation in tissues.15
Many MAbs have doses in the range of mg kg–1 when administered by intravenous or subcutaneous routes. Used subcutaneously there is the need to minimise the volume administered, hence the use of concentrated solutions. Such solutions often have an elevated viscosity and the concentrated antibodies tend to be more prone to self-association and aggregation.16 These large protein molecules have complex structures and complex solution behaviour, as might be expected, often sensitive to impurities or additives: even small amounts of silicone oil used as lubricants for syringes can lead to antibody aggregation,17 as do some preservatives also. Aggregation generally implies partial loss of activity.
Figure 13.26 shows the simplified structure of a typical MAb with its light and heavy chains. The variable and constant regions of the model structure are also shown. An MAb is represented interacting with a target, and a simplified representation is given of a MAb–drug conjugate (ADC) and a drug conjugate showing the labile disulfide linkage, one of several that can be employed, to control the rate of release of the drug from the conjugate, a key element in its biological activity. See Box 13.1.
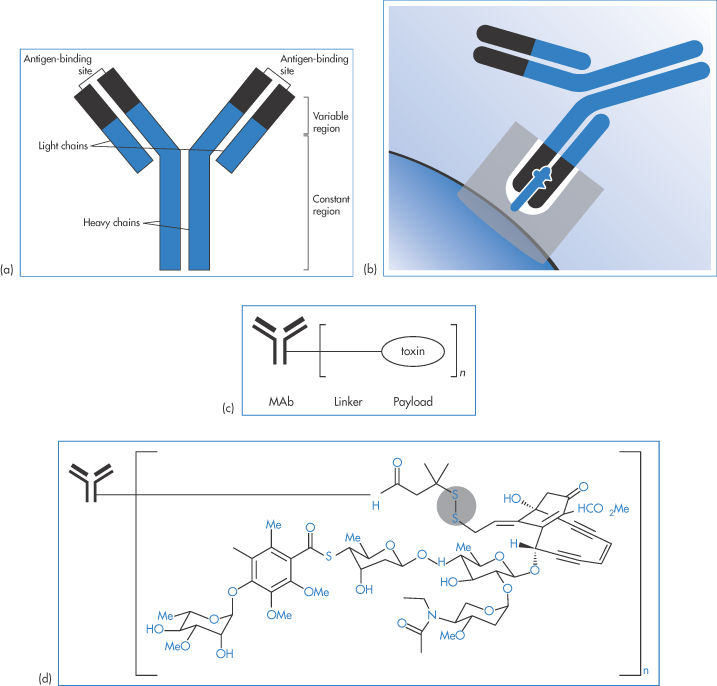
Figure 13.26 (a) Representation of a typical monoclonal antibody (MAb) showing the light and heavy chains. The variable and constant regions of the model structure are also shown. (b) An MAb interacting with a target. (c) A much simplified representation of an MAb–drug conjugate, the form that many therapeutic approaches now take. (d) (not to scale) A conjugate showing the labile S–S– linkage, one of several that can be employed. The rate of release of the drug from the conjugate can be a key element in its biological activity.
Box 13.1 Nomenclature of MAbs |
The International Non-proprietary Names of all drugs should convey meaning about the class to which each drug belongs. Trade names do not serve this purpose. All MAbs thus have the -mab suffix in their name. Other elements of the name provide further information. A source suffix (before -mab) reveals the cell origin : -u- (human), -o- (mouse), -xi- chimeric, -zu- (humanised), -xi-zu- (hybrid of chimeric and humanised) are examples, as shown in Fig. 13.27. Thus, trastuzumab is a humanised MAb. Disease targets can also be included in the name, e.g. -lim- (immune), -les- (infectious diseases), -vir- (viral), -mel- (melanoma), -col- (colon). |
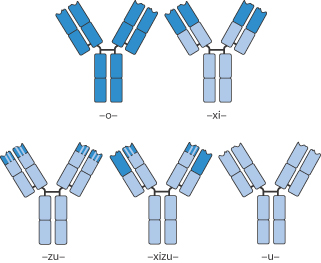
Figure 13.27 Illustrations of mouse (-o-) to human (-u-) monoclonal antibodies with intermediate forms such as chimeric (-xi-), humanised (-zu-) and (-xizu-), a hybrid form derived from chimeric and humanised sources.
13.6.1 Antibody–drug conjugates
Figure 13.28 illustrates many of the vital features of drug conjugates: the antibody itself, the active drug such as a cytotoxic agent attached to the antibody and the linker between antibody and drug. The MAb provides the potential for specific targeting; in other words, it is the vector. The link between drug and MAb can be designed to control payload release. Two examples of ADCs are trastuzumab emtansine (MAb linked to the cytotoxic mertansine) and brentuximab vedotin (MAb linked to monomethyl auristatin E), whose payloads are inactive until binding to their target receptors, the conjugate internalised in lysosomes, linkers cleaved and drug released within the target cells.* This is the theory, but there are constraints.
A key point is the ratio of drug to antibody. It is tempting to think that increasing the payload enhances activity, but the attached molecules can begin to interfere sterically with the behaviour of the antibody: increased numbers of drug molecules change the physical properties of the construct by changing the surface properties, hydrophobicity, charge, shape, and so on. The position of the linker and thus the drug is also important to activity, as are the degradation pathways of the complex. Premature breakdown of the linker will clearly lead to release of drug before the optimal target is reached – a problem discussed in Chapter 14 in relation to many drug carriers such as liposomes and nanoparticles.
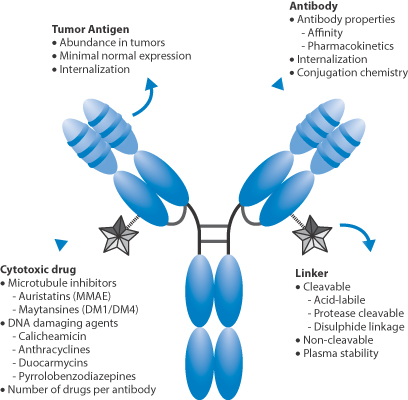
Figure 13.28 Key features of antibody–drug conjugates: the cytotoxic drug, the nature of the link between drug and antibody, the affinity of the antibody to sites of action such as tumour antigen, pharmacokinetics and internalisation.
Reproduced with permission from Panowski S et al. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014;6:34–45.
13.6.2 Aggregation and other forms of instability: the pharmaceutical issues
The antibody constructs can associate in solution, forming dimers, tetramers or larger aggregates. Such changes in the form of the construct can lead to decreased therapeutic activity and enhanced immunogenicity and will affect the transport properties of the MAbs or ADCs, as size matters! Figure 13.29 schematically illustrates the potential for change in solution behaviour:
- unfolding of chains
- clustering of unfolded antibodies
- aggregation into soluble clusters
- formation of insoluble particles.
The several processes that contribute to the instability of antibodies are outlined in Figure 13.30, showing chemical instability through oxidation, deamidation, hydrolysis and proteolysis to the physical manifestations of instability, conformational change and aggregation, already discussed.
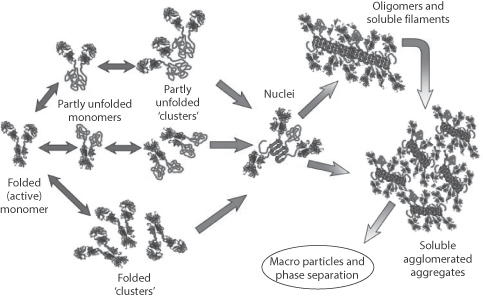
Figure 13.29 A comprehensive survey of the possibilities of the association and aggregation of monoclonal antibodies (MAbs) in solution, starting from the folded active MAb monomers, through partly unfolded, to oligomers and particles. Increasing the net charge on proteins minimises these effects, as does decreasing ionic strength.
Reproduced from Roberts CJ. Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol 2014;32:372–380. Copyright Elsevier 2014.
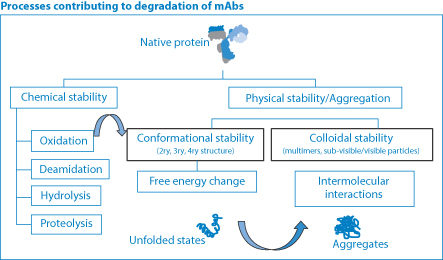
Figure 13.30 The many processes contributing to degradation of monoclonal antibodies (MAbs). Chemical degradation is applied to most actives, coupled with the conformational stability of proteins and macromolecules and physical instability, leading to aggregate formation.
Courtesy of M Connolly, University of Bath.
13.6.3 Rheological changes
Higher concentrations of MAbs increase the viscosity of their solutions, as one would expect from earlier discussions of rheology of colloids (see Chapter 6). But beyond the normal enhanced viscosity due to increased volume fraction, the aggregation of the antibodies has a marked effect on viscosity, as shown in Fig. 13.31, where there is a linear increase in viscosity with the average number of antibodies in each cluster, rising from around 10 cP to more than 400 cP with aggregates containing nine antibodies. This can result in difficulties not only in manufacture and processing but also in the filling of syringes, and their subsequent dispersal from intramuscular and subcutaneous injection sites.
13.6.4 Formulations
As examples we can cite palivizumab (Synagis) or efalizumab (Raptiva) for intramuscular injection, both with a concentration of 100 mg mL–1, while omalizumab (Xolair) for subcutaneous injection exceeds this at 125 mg mL–1. Antibody formulations often contain surfactants such as polysorbate 20 and polysorbate 80 to aid stabilisation of the protein: altering the nature of the solution can influence viscosity also.
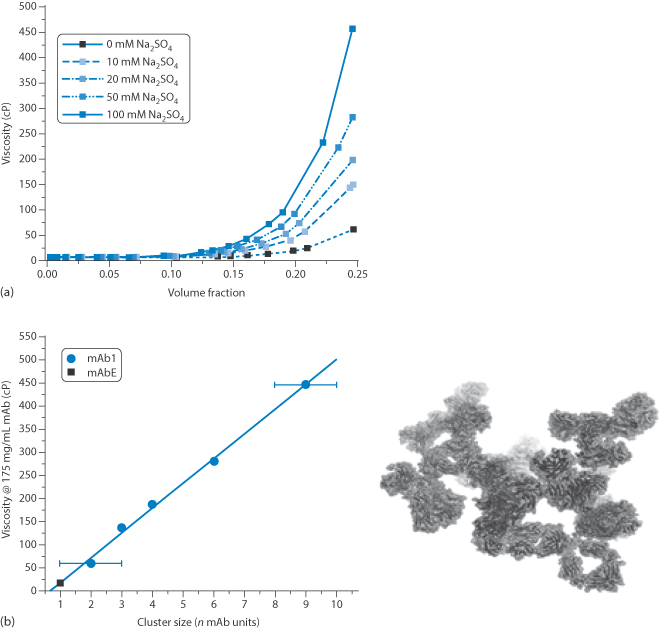
Figure 13.31 (a) The solution viscosity of an Ig1 monoclonal antibody solution as function of concentration and electrolyte (Na2SO4) concentration, showing the sensitivity to both parameters above certain concentrations. (b) The viscosity of 175 mg mL–1 solutions as a function of the cluster size determined for each system with a computer model of cluster formation.
Reproduced with permission from Lilyestrom WG et al. Monoclonal antibody self-association, cluster formation and rheology at high concentrations. J Phys Chem B 2013;117:6373–6384. Copyright 2013 American Chemical Society.
13.6.5 Diffusion coefficients and transport
Aggregation may be reversible, as shown in the examples above, by the addition of electrolyte, or surfactant or other formulation excipients. One consequence of: (1) the size of the MAb and (2) its aggregation is the effect these parameters have on the diffusion coefficient which, as the Stokes–Einstein equation dictates, is a function of the radius of spherical particles, and determines the ability of the molecules or their aggregates to negotiate the body as has been discussed in Chapters 2 and 14.
The aqueous diffusion coefficient of glucose (molecular weight 192 Da) is 6.6 × 10–6 cm2 s–1; that of insulin (5734 Da) is 2.10 × 10–6 cm2 s–1, serum albumin (66,500 Da) is 0.6 × 10–6cm2 s–1 (its radius is 6 nm) while human IgM (970 000 Da), with a radius of 15 nm, has a diffusion coefficient of 0.32 × 10–6 cm2 s–1. We know that:

where t is the diffusion time (the elapsed time after diffusion has begun), D is the diffusion coefficient, x is the mean distance travelled in one axis after time t. Hence the time taken for glucose, insulin and IgM to travel 1 cm is 21 hours, 66 hours and 434 hours respectively. Diffusion in more complex media, e.g. in tumours, mucus and tissues, is in general affected also by (1) binding of the diffusant to biological molecules and tissues and (2) the obstruction effect, where size and surface properties of the molecule and possible flexibility come into play as the molecules move through a complex fluid milieu with impenetrable obstacles like fibres and cellular components.
Botulinum toxin diffusion
Botulinum toxins, which can decrease the release of acetylcholine, and thus elicit a neuromuscular blocking effect, are used in many cases to relieve unwanted muscular contractions as occur in strabismus, blepharospasm, hemifacial spasm and spasticity. There are a number of products, processed and purified in different ways, providing a variety of clinical products with different protein content, excipients and so on. As botulinum toxin is obviously a toxic product (in inappropriate concentrations), one characteristic of interest is the diffusion of the toxin molecules,18 not least when the products are used in cosmetic treatments. Naturally the diffusion, spread and migration of the botulinum toxin A are vital and it is the limited diffusion of the molecule that has allowed this potent molecule to be used safely!
Three of the main products are:
1. Botox/Vistabel (Allergan, NPN onabotulinumtoxinA)
2. Dysport/Azzalure (Ipsen, NPN abobotulinumtoxinA)
3. Xeomin/Bocouture (Merz, NPN incobotulinumtoxinA).
These have markedly different specific activities and cannot be considered to be bio-equivalent. For further discussion of so-called biosimilars, see Chapter 16. A detailed review of the literature is available.19
13.7 Vaccine formulation
The field of immunology is complex. Vaccines stimulate production of antibodies and other components of the immune system and comprise four types, which, in the words of the British National Formulary, are:
1. live attenuated forms of a virus (e.g. measles, mumps and rubella vaccine) or bacteria (e.g. Bacillus Calmette-Guérin (BCG) vaccine)
2. inactivated preparations of a virus (e.g. influenza vaccine) or bacteria
3. detoxified exotoxins produced by a microorganism (e.g. tetanus vaccine)
4. extracts of a microorganism, which may be derived from the organism (e.g. pneumococcal vaccine) or produced by recombinant DNA technology (e.g. hepatitis B vaccine).
|
Live attenuated vaccines usually produce a durable immunity, but not always as long-lasting as that resulting from natural infection. Inactivated vaccines may require a primary series of injections of vaccine to produce an adequate antibody response, and in most cases booster (reinforcing) injections are required; the duration of immunity varies from months to many years. Some inactivated vaccines are adsorbed onto an adjuvant (such as aluminium hydroxide) to enhance the antibody response. Source: BNF, through Medicines Complete December 2014. |

This short account will discuss some of the issues related to the pharmaceutics of vaccines. There are components and excipients in some vaccines which can cause problems.
|
Vaccines are contraindicated in those who have a confirmed anaphylactic reaction to a preceding dose of a vaccine containing the same antigens or vaccine component (such as antibacterials in viral vaccines). The presence of the following excipients should be noted in some vaccines. GelatinPenicillins GentamicinPolymyxin B KanamycinStreptomycin NeomycinThiomersal Source: BNF, through Medicines Complete, December 2014. |

The two main areas to be dealt with here are adjuvants,† used to boost immune response and delivery options, some of which have already been dealt with earlier in this book. The roles adjuvants play are fascinating, but still offer us some mysteries.
13.7.1 Routes of administration
Most vaccines are given by the intramuscular route, while some are given by the intradermal, deep subcutaneous or oral routes, for example:
- intradermal route – BCG
- deep subcutaneous route – varicella
- oral route – cholera, poliomyelitis, rotavirus and live typhoid vaccines (where the gut-associated lymphoid tissue plays its part in achieving activity).
The UK Department of Health has advised against the use of jet guns for vaccination owing to the risk of transmitting blood-borne infections, such as human immunodeficiency virus (HIV), because of the potential for production of sprays through impact on the skin.
13.7.2 Vehicles
Some of the materials used or under study today are represented in Fig. 13.32. These include a number of systems discussed elsewhere in the book: neutral and cationic liposomes, microspheres and nanoparticles (such as PLGA), salts such as alum (aluminium hydroxide), immunostimulating complexes (ISCOMs), water-in-oil and oil-in-water emulsions and saponins.
Alum
Alum (aluminium hydroxide) was first introduced as a vaccine adjuvant in the 1920s and its mode of action in enhancing the action of antigens is still being debated – one of the ‘slowest processes in the history of medicine,** according to De Gregorio et al.20 One hypothesis is that alum, by combining with a soluble antigen, forms a precipitate and that slow release of the antigen from the precipitate on injection encourages a prolonged, strong antibody response. However, the desired effects of aluminum-containing adjuvants have also been concluded not to derive from direct antigen adsorption and retention, but rather from other contributions to immunopotentiation.
Since then, a number of adjuvant materials and formulations have been described with different modes of action, but alum is still the only adjuvant licensed in the USA, while the European authorities have approved two oil-in-water emulsions, MF59 and AS03.
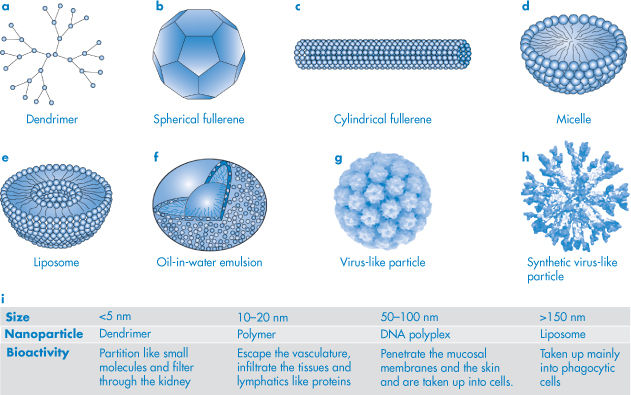
Figure 13.32 Representation of types of carriers for vaccines acting (left to right) mainly as depots, or in dual roles as immunostimulants and carriers or as immunomodulators. Immunostimulating complexes (ISCOMs) are relatively stable but non-covalently-bound complexes of the saponin adjuvant Quil-A from Quillaia saponaria Mollina, cholesterol and amphipathic Ag in a molar ratio of approximately 1:1:1. ISCOMs have been shown to induce cytotoxic T-lymphocytes.
Reproduced from Smith DM, Simon JK, Baker JR Jr. Nat Rev Immunol 2013;13:592–605.
ISCOMs
ISCOMs are relatively stable but non-covalently bound complexes of the saponin adjuvant Quil-A from Quillaia saponaria Mollina, cholesterol and amphipathic Ag in a molar ratio of approximately 1:1:1. The adjuvanticity of Quil-A saponin (III) depends on its structure; it is composed of hydrophilic sugar side-chains and a hydrophobic aglycone backbone.
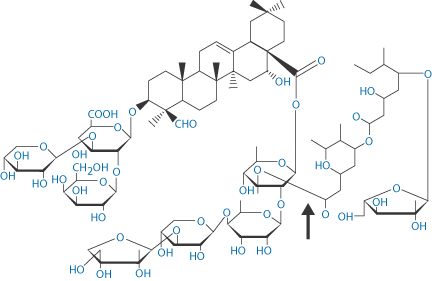
Structure III Quil-A saponin
Saponins
Saponins derive from many plant sources. Like many adjuvant components they have an amphipathic structure. It is claimed that the overall hydrophile–lipophile balance of the saponins is more important for its adjuvanticity than the structures involved. Being surface active, micelles can be formed by the saponins and these must play a part in the protection and release properties of the adjuvant.
Emulsions
Freund’s incomplete adjuvant (Statens Seruminstitut, Copenhagen, Denmark) is a water-in-oil emulsion containing antigen in the aqueous phase, light paraffin oil and an emulsifying agent, mannide monooleate. On injection, this mixture (Freund’s incomplete adjuvant) induces strong persistent antibody formation. The addition of killed, dried mycobacteria, e.g., Mycobacterium butyricum, to the oil phase (Freund’s complete adjuvant) elicits cell-mediated immunity (delayed hypersensitivity), as well as humoral antibody formation.
MF59 (Novartis) is a squalene (IV)-in-water emulsion with a mean globule size of 160 nm, stabilised by sorbitan trioleate and polysorbate. It is used in influenza vaccines. A related product is AS03 or ‘Adjuvant System 03’, a squalene-based adjuvant used in various vaccine products by GlaxoSmithKline. A dose of AS03 adjuvant contains 10.69 mg squalene, 11.86 mg DL-α-tocopherol and 4.86 mg polysorbate 80.
Structure IV Squalene
Adjuvant 65 is a marketed water-in-oil emulsion containing antigen in peanut oil with Arlacel A (mannide monooleate) and aluminum monostearate as the emulsifying agents.
Summary
This chapter has not attempted to provide an overview of proteins and peptides and other complex therapeutics such as DNA, oligonucleotides or MAbs. It has concentrated on giving a flavour of some of the unique physical properties that the substances discussed here have. It has tried to give some feeling for the properties of peptides and proteins and how their physical properties are dictated not only by the properties of their individual amino acids but also by the spatial arrangement of the amino acids in their polypeptide chains, and of course their interaction with water and other ingredients that might be in formulations. Hydrophobic amino acids alternating with hydrophilic, or charged amino acids can produce tertiary and quaternary structures that are quite different from those induced by the spatial separation of hydrophobic and hydrophilic amino acids. The stability of proteins and peptides is of prime pharmaceutical significance. Maintenance of chemical and physical stability is a prerequisite for clinical use. The macromolecular and generally hydrophilic character of all but the smallest peptides makes this class of compound one that is challenging in terms of delivery for therapeutic ends. Some of the issues involved in the formulation of proteins and vaccines, such as botulinum toxin A vaccine, are also discussed.
References
1. Brange J et al. Monomeric insulins and their experimental and clinical implications. Diabetes Care 1990;13:923–954.
2. Brems DN et al. Altering the self-association and stability of insulin by amino acid replacement. In: Cleland J (ed.) Protein Folding: In Vivo and In Vitro. ACS Symposium Series 526. Washington, DC: American Chemical Society; 1993: 254–269.
3. Shao Z et al. Differential effects of anionic, cationic, nonionic, and physiologic surfactants on the dissociation, alpha-chymotryptic degradation, and enteral absorption of insulin hexamers. Pharm Res 1993;102:43–51.
4. Costantino JR et al. Aggregation of a lyophilized pharmaceutical protein, recombinant human albumin: effect of moisture and stabilization by excipients. Biotechnology 1995;13:493–496.
5. Hageman MJ et al. Prediction and characterization of the water sorption isotherm for bovine somatotropin. J Agric Food Chem 1992;40:342–347.
6. Zografi G. States of water associated with solids. Drug Dev Ind Pharm 1988;14:1905–1906.
7. Cleland JL, Langer R. Formulation and delivery of proteins and peptides: design and development strategies. In: Cleland JL, Langer R (eds) Formulation and Delivery of Proteins and Peptides. Washington, DC: American Chemical Society; 1994: 1–19.
8. Ye M et al. Issues in long term protein delivery using biodegradable microparticles. J Control Rel 2010;146:241–260.
9. Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Deliv Today 2005;10:1451.
10. Mehrar R. Modulation of the pharmacokinetics and pharmacodynamics of proteins by polyoxyethylene glycol conjugation. J Pharm Pharm Sci 2000;3:125–136.
11. Gallina K et al. Local blistering reaction complicating subcutaneous injection of pegylated interferon in a patient with hepatitis C. J Drugs Dermatol 2003;263–67.
12. Dalmau J et al. Cutaneous necrosis after injection of polyethylene glycol-modified interferon alfa. J Am Acad Dermatol 2005;53:62–66.
13. Arvinte T et al. The structure and mechanism of formation of human calcitonin fibrils. J Biol Chem 1993;268:6415–6422.
14. Mao HQ et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterisation and transfection efficiency. J Control Release 2001;70:399–421.
15. Breedveld FC. Therapeutic monoclonal antibodies. Lancet 2000;355:735–740.
16. Yadav S et al. Factors affecting the viscosity of high concentration solutions of different monoclonal antibodies. J Pharm Sci 2010;99:4812–4829.
17. Thirumangalathu R et al. Silicone oil- and agitation-induced aggregation of a monoclonal antibody in aqueous solution. J Pharm Sci 2009;98:3167–3181.
18. Brodksy MA et al. Diffusion of botulinium toxins. Tremor and other hyperkinetic movements, 2012. Available online at: http://www.tremorjournal.org.
19. Brin MF et al. (2014) Botulinum toxin type A products are not interchangeable: a review of the evidence Biologics: Targets Ther 2014;8:227–241.
20. De Gregorio E et al. Alum adjuvanticity: Unraveling a century old mystery. Eur J Immunol 2008;38:2068–2071.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


