Clinical point
IA injection of BCNU (carmustine; bis-chloronitrosourea) in the treatment of malignant glioma results in levels of drug that are four times greater than those following IV infusion, but arguments persist about the relative clinical risks and benefits of IA administration of this drug. The rationale in liver cancer treatment for IA administration is that liver tumours derive their blood supply from the hepatic artery, unlike normal tissue. Two commonly used drugs are fluorouracil and floxuridine.
Microspheres loaded with drugs such as doxorubicin have been used experimentally to optimise delivery by this route.
9.1.1 Implantable infusion pumps
Such devices are implanted under the skin in the lower abdomen and designed to deliver infusate containing the appropriate drug at a constant rate (usually 1 cm3 per day) into an artery or vein. The Infusaid implantable pump was originally devised for the long-term administration of heparin but has since found application for a wide variety of drugs.1 The device consists of a relatively small (9 × 2.5 cm) titanium disc that is divided into two chambers by cylindrical titanium bellows that form a flexible but impermeable barrier between the compartments (Fig. 9.1). The outer compartment contains Freon (chlorofluorocarbon propellants); the inner compartment contains the infusate and connects via a catheter to a vein or artery through a series of filters and flow-regulating resistance elements. The vapour pressure above the liquid propellant remains constant because of the relatively constant temperature of the body, and hence a constant pressure is exerted on the bellows, ensuring a constant rate of delivery of infusate into the blood stream. The propellant is replenished as required by a simple percutaneous injection through the skin.
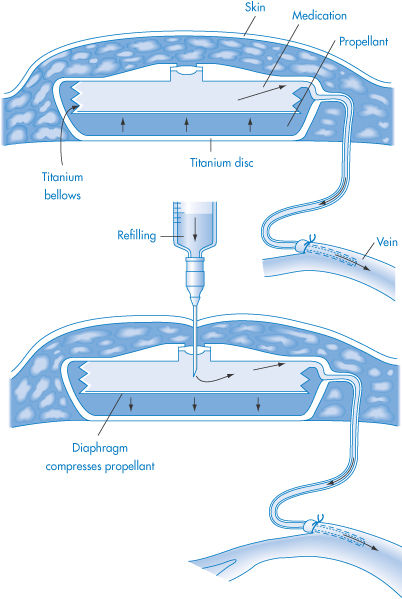
Figure 9.1 Diagrammatic representation of the Infusaid implantable infusion pump during operation (top) and during refilling (bottom).
Reproduced with permission from Blackshear PJ, Rhode TH. In: Burk SD (ed.) Controlled Drug Delivery, vol. 2, Clinical Applications. Boca Raton, FL: CRC Press; 1983: 11.
|
|

9.2 Intramuscular and subcutaneous injection
The IM and SC routes of administration have long been regarded as efficient because they bypass the problems encountered in the stomach and intestine. Early views that SC and IM administration of drugs resulted in only local action at the site were dispelled by Benjamin Bell, who wrote in 1858 in the Edinburgh Medical Journal that ‘absorption from the enfeebled stomach may not be counted on; we possess in subcutaneous injections a more direct, rapid and trustworthy mode of conveying our remedy in the desired quantity to the circulatory blood’. We now know that not all drugs are efficiently or uniformly released from IM or SC sites. Some of the drugs that are not fully absorbed from their IM sites are listed in Table 9.1.
Table 9.1 Widely used drugs that may be incompletely absorbed after intramuscular injection
Ampicillin | Digoxina |
Cefaloridine | Insulina |
Cefradine | Phenylbutazone |
Chlordiazepoxide | Phenytoina |
Diazepam | Quinidine |
Dicloxacillin |
|
a Clinically important problems have been demonstrated with these.
Where differences in bioavailability between IM, SC, oral or IV delivery occur, the clinical importance is most significant when the route of administration is changed from one to another. Such changes are most important in drugs with a low therapeutic index, such as digoxin and phenytoin.
The various regions into which injections are given are shown in Fig. 9.2. The SC region has a good supply of capillaries, although it is generally agreed that there are few, if any, lymph vessels in muscle proper. Drugs with the optimal physicochemical characteristics can diffuse through the tissue and pass across capillary walls and thus enter the circulation by way of the capillary supply.
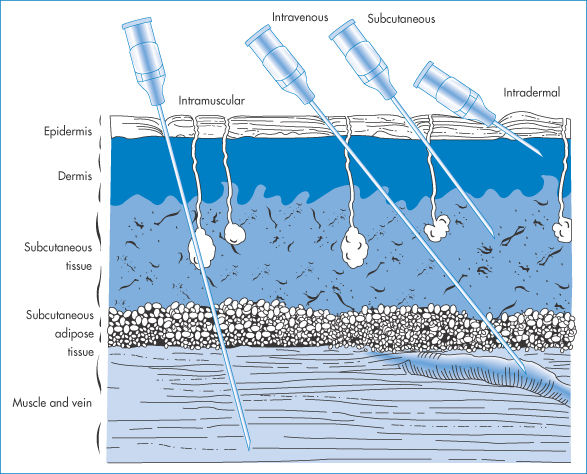
Figure 9.2 Routes of parenteral medication, showing the tissues penetrated by intramuscular, intravenous, subcutaneous and intradermal injections; the needles, with bevel up, penetrate the epidermis (cuticle), consisting of stratified epithelium with an outer horny layer, the corium (dermis or true skin) consisting of tough connective tissue, elastic fibres, lymphatic and blood vessels, and nerves, the subcutaneous tissue (tela subcutanea) consisting of loose connective tissue containing blood and lymphatic vessels, nerves and fat-forming cells, the fascia (a thin sheet of fibrous connective tissue) and the veins, arteries and muscle.
Modified with permission after Quackenbush DS. In: Martin EW (ed.) Techniques of Medication. Philadelphia: Lippincott; 1969.
If it is reasonably assumed that drug absorption proceeds by passive diffusion of the drug, it can be considered to be a first-order process. Thus the rate of absorption is proportional to the concentration, C, of drug remaining at the injection site:

where ka is the first-order rate constant. The half-life of the absorption process is

Drug absorption is 90% complete when a time equivalent to three times the half-life has elapsed.
Both dissolution and diffusion are important parameters in defining bioavailability of species by the IM or SC routes. Soluble neutral compounds disperse from IM sites according to size: Table 9.2 shows that mannitol, a small molecule, rapidly diffuses from the site of injection; inulin disperses less rapidly; and dextran (molecular weight 70 000 Da), as might be expected, disperses more slowly.
Table 9.2 Influence of molecular size on clearance from intramuscular sites
Substance | Molecular weight | Fraction cleared (5 min) |
Mannitol | 182 | 0.70 |
Inulin | 3 500 | 0.20 |
Dextran | 70 000 | 0.07 |
With smaller molecules, those with molecular weights in the range 100–1000 Da, molecular size is a factor, but a minor one, in controlling release of drugs, but it assumes greater importance with proteins and other macromolecules, as discussed in Chapter 13. To achieve slow release of low-molecular-weight drugs they may be attached to a macromolecular backbone. Hydrophilic molecules such as those listed in Table 9.2 will be transported to the blood after diffusing through muscle fibres and then through the pores in the capillary walls, being incapable of absorption through the lipid walls. The transport through the capillary wall is the rate-limiting step in most cases; the larger the molecule, the more slowly it diffuses (as seen from equation 2.64) and the greater difficulty it has in traversing the aqueous pore in the capillary walls or the cell junction. The pores account for only 1% of the available surface of the capillary wall.
Absorption of weak electrolytes across the capillary walls follows the expected patterns, uptake of more lipid-soluble agents being relatively fast. Hydrophobic drugs may bind to muscle protein, leading to a reduction in free drug and perhaps to prolongation of action. Dicloxacillin is 95% bound to protein, while ampicillin is bound to the extent of 20%; as a consequence, dicloxacillin is absorbed more slowly from muscle than is ampicillin.
The significance of the diffusion phase through muscle tissue was demonstrated by Duran Reynolds who, in 1928, observed that dye solutions injected together with hyaluronidase spread out more rapidly and over a greater distance in the tissue than in the absence of the enzyme. The enzyme achieves its effect by breaking down the hyaluronic acid in intercellular spaces, thus leading to a decrease in viscosity and so easing the passage of small molecules in the matrix.
9.2.1 The importance of site of injection
The region into which the injection is administered is complex, being composed of both aqueous and lipid components. Muscle tissue is more acidic than normal physiological fluids. Measurement of percutaneous muscle surface pH (pHm) using microelectrodes indicates a mean value of 7.38; patients with peripheral vascular disease demonstrated a mean pHm of 7.16. pHm reflects the IM ion concentrations but is stated to be higher than the actual IM pH, which may be as low as 6.4. The pH of the region will determine whether drugs will dissolve in the tissue fluids or precipitate from formulations. In some cases precipitation will occur and may be the cause of the pain experienced on injection (see Chapter 12). The rate of solution here, as in many other routes, therefore determines how quickly the drug begins to act or, in some cases, over which period of time it acts. The deliberate reduction of the solubility of a drug achieves prolonged action by both routes.
9.2.2 Vehicles
Many drugs for IM administration are formulated in water-miscible solvents such as polyethylene glycol (PEG) 300 or 400, propylene glycol or ethanol mixtures. Dilution by the tissue fluids may cause the drug to precipitate. Drugs formulated as aqueous solutions by adjustment of pH will only transiently alter the pH at the injection site. Three main types of formulation are used for IM and SC injections: aqueous solutions, aqueous suspensions and oily solutions. Rapid removal of aqueous vehicles is to be expected, the drug being left perhaps as a precipitate. If the vehicle is non-aqueous and is an oil such as sesame oil, or another vegetable or a mineral oil, the oil phase disperses as droplets in the muscle and surrounding tissue, and is eventually metabolised.
The rate-determining step in the absorption of drug esters such as fluphenazine decanoate (which has an aqueous solubility of about 1 part per million) is the hydrolysis of the drug at the surface of the oil droplet. Hydrolysis of the fluphenazine decanoate to its soluble alcohol therefore depends on the state of dispersion and surface area of the droplets.
|
Dispersing the droplets of sustained-release oil-based injections by rubbing the site of injection or by vigorous exercise can result in excessive dosage, with toxic effects. Exercise also causes increased blood flow and, as absorption is a dynamic process requiring the sweeping away of the drug from the localised absorption site, this increased flow increases the rate of drug dispersal. |

A recent addition to long-acting injections for schizophrenia, paliperidone palmitate (I) takes a different approach using nanocrystals of the insoluble drug which will be deposited in the muscle tissue and hydrolysed by esterases.
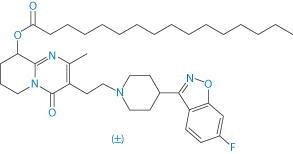
Structure I Paliperidone palmitate
9.2.3 Blood flow in muscles
Different rates of blood flow in different muscles mean that the site of IM injection can be crucial. Resting blood flow in the deltoid region is significantly greater than in the gluteus maximus muscle; flow in the vastus lateralis is intermediate. The difference between blood flow in the deltoid and gluteus is of the order of 20% and is likely to be the reason why deltoid injection of lidocaine has been found to give higher blood levels than injection into the lateral thigh. Therapeutic plasma levels for the prevention of arrhythmia with a dose of 4.5 mg kg−1 of a 10% solution of lidocaine were achieved only following deltoid injection.
|
The age of the patient should influence the behaviour of the injection, as ageing affects vascular blood flow and fatty deposits, but age has not been specifically isolated as a factor in studies to date. In some disease states it is possible to predict that the outcome of an IM injection might be different from that in other patients. For example, in patients with circulatory shock, hypotension, congestive heart failure and myxoedema, where blood flow to skeletal muscle is decreased, one might anticipate lower bioavailability. |

9.2.4 Formulation effects
Crystalline suspensions of fluspirilene, some steroids and procaine benzylpenicillin can be prepared in different size ranges to produce different pharmacokinetic profiles following IM or SC injection, as discussed above. Variability in response to a drug, or differences in response to a formulation from different manufacturers, can be the result of the nature of the formulation. Some years ago, in one trial diazepam as Valium (Roche) produced plasma levels ranging from about zero to 160 ng cm−3 90 minutes after injection into muscle (Fig. 9.3). The formulation contained diazepam dissolved in an ethyl alcohol–propylene glycol mixture. Upon injection, this solvent is absorbed and the drug precipitates (see Chapter 11). Injection of the formulation into the fatty tissue of the buttock can result in poor dispersal of drug, as the drug would have little opportunity for dissolution in that environment. Accordingly, the depth of the injection is significant. If, in addition, the blood supply to the region is limited, there will be an additional restriction to rapid removal. An alternative formulation of diazepam (Stesolid) containing a surface-active agent as a solubiliser has a greater bioavailability than a non-aqueous solution form of the drug (Diapam).
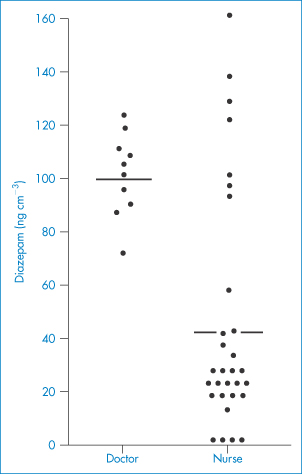
Figure 9.3 Plasma diazepam levels 90 minutes after intramuscular injection by one doctor and several nurses, showing the importance of technique and site of injection, which was variable in the latter group (individual values; horizontal lines denote average levels).
Redrawn with permission from Dundee JW et al. Plasma diazepam levels following intramuscular injection by nurses and doctors. Lancet 1974;2:1461.
|
Additives can influence dispersion, the solubiliser undoubtedly in the case of Stesolid reducing precipitation of the drug at the site of injection and thus increasing the rate of solution. Reports of marked difference in side-effects and adverse reactions to diazepam formulations are due to the additives present, either indirectly or directly. |

Controlled delivery from SC or IM sites may be achieved by dissolving a lipophilic drug in an oil phase, as discussed above, or encapsulating the drug in liposomes, niosomes or biodegradable microspheres. These techniques are discussed in Chapters 6 and 7.
9.2.5 The case of insulin
Insulin is a classic example of what can be achieved by manipulation of the properties of a drug and its formulation. Modification of the crystallinity of insulin allows control over its solubility and duration of action. An acid-soluble formulation of insulin was introduced for clinical use in 1923. It had a short duration of action and attempts were then made to prolong the action. In 1936, Hagedorn and his colleagues found that insulin complexed with zinc and protamine from the trout (Salmo irideus) formed an amorphous precipitate at neutral pH. When injected subcutaneously, the insulin was slowly released from the complex into the blood and was active for about 36 hours. The prolonged-acting insulins introduced since the advent of protamine-zinc insulin (PZI) have been designed to have intermediate durations of action (Table 9.3).
Table 9.3 Pharmaceutical injections of insulin BP
Preparation | pH | Buffer | Description | Onset (h) | Duration of effect (h) |
Insulin injection | 3.0–3.5 | – | Solution | ∼0.5–1 | 6–8 |
Neutral injection | 6.6–7.7 | Acetate | Solution | ∼0.5–1 | 6–9 |
Protamine zinc | 6.9–7.4 | Phosphate | Amorphous particles; | ∼5–7 | 36 |
Globin zinc | 3.0–3.5 | – | Solution | ∼2 | 18–24 |
Isophane | 7.1–7.4 | Phosphate | Rod-shaped crystals | ∼2 | 28 |
Zinc suspension (amorphous) ‘Semilente’ | 7.0–7.5 | Acetate | Amorphous particles | ∼1 | 12–16 |
Zinc suspension ‘Lente’ | 7.0–7.5 | Acetate | Amorphous particles (30%) Rhombohedral crystals (70%) | ∼2 | 24 |
Zinc suspension (crystalline) ‘Ultralente’ | 7.0–7.5 | Acetate | Rhombohedral crystals | ∼5–7 | 36 |
Biphasic | 6.6–7.2 | Acetate | Insulin in solution (25%) Rhombohedral crystals (75%) | ∼1 | 18–22 |
Modified from Stewart GA. Historical review of the analytical control of insulin. Analyst 1974;99:913, with permission of the Royal Society of Chemistry.
The long-acting insulins in use today are mainly protamine insulin and zinc insulins. Protamine insulins are the salt-like compounds formed between the acid polypeptide (insulin) and the polypeptide protamine, which consists primarily of arginine. They are used in the form of neutral suspensions of protamine insulin crystals (isophane insulin).
Isophane insulin is produced by titration of an acidic solution of insulin with a buffered solution of protamine at neutral pH until so-called isophane precipitation occurs; that is, no insulin or protamine is present in the supernatant. Under these conditions the precipitate consists of rod-shaped crystals. Crystalline forms of insulin free from foreign protein were developed in 1951. Under conditions of high zinc ion concentration (10 times that normally used to crystallise insulin) and in the absence of citrate and phosphate ions, rhombohedral crystals of insulin are formed in acetate buffer (IZS crystalline). By adjustment of the pH during the crystallisation stage, the insulin is produced as an amorphous precipitate (IZS amorphous) that has a duration of action of up to 16 hours. Various mixtures can be used providing modified durations of actions, as in biphasic insulin. Insulin zinc suspensions are incompatible with PZI and isophane insulin, both of which act as a buffer that destroys the zinc complex.
|
1 Variable insulin activity may result from the mixing of PZI and soluble insulin in a syringe prior to administration. It has been suggested that this variability is due to the combination of the soluble insulin with excess protamine. As a general rule, insulin formulations of different pH should not be mixed. 2 Absorption of insulin is faster from injections administered subcutaneously in the arms than into the thighs. Insulin absorption in some patients may be poor because the SC tissue can act as a mechanical barrier to diffusion and as an active site of degradation. |

Figure 9.4 is a diagrammatic representation of the events following SC administration of a soluble human insulin existing initially as a hexameric zinc–insulin complex.
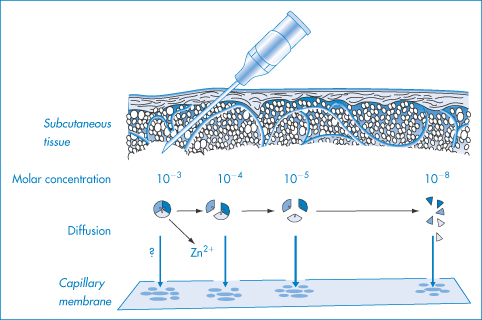
Figure 9.4 Suggested scheme of events after the subcutaneous administration of soluble human insulin: the concentration of hexameric zinc-insulin, which is the predominant form of insulin in soluble insulin (40 or 100 units – i.e. 0.6 mmol dm−3), decreases as diffusion of insulin occurs and as a result the hexamer dissociates into smaller units; to achieve monomeric insulin requires a 1000-fold dilution. The importance of the association state is that the larger species have more difficulty dispersing and passing through the capillary membrane.
Reproduced with permission from Brange J et al., Monomeric insulins and their experimental and clinical implications. Diabetes Care, 1990; 13: 923.
The use of insulins in solution obviates the potential source of error that arises when drawing a suspension into a syringe, but soluble insulins have the drawback that they must be stored at acid pH. Figure 9.5 shows the solubility–pH profiles for soluble insulin and a trilysyl derivative. Injected subcutaneously, the insulin precipitates as amorphous particles. The pH of insulin preparations influences their stability, solubility and immunogenicity.
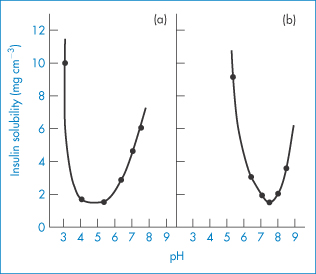
Figure 9.5 The solubility dependence of (a) insulin and (b) trilysyl insulin, a chemically modified insulin with an isoelectric point of 7.4 compared with the isoelectric point of unmodified insulin of 5.3.
Reproduced with permission from Fischel-Ghodsian F, Newton JM. Simulation and optimisation of a self-regulating insulin delivery system. J Drug Targeting 1993;1:67.
1 After IM administration, short-acting insulin is absorbed about twice as rapidly as after SC injection, and therefore the IM route is used in the management of ketoacidosis in those cases where continuous IV infusion cannot be established. After SC injection, absorption of short-acting insulin varies considerably depending in particular on the site of injection. Patient-to-patient variability is as great with these preparations as the variations in the absorption rate of intermediate-acting insulins. This can lead to difficulty in control. 2 Flocculation of Humulin N insulin and of NPH insulin leads to changes in the appearance of vials (a frosting) and signals a problem, that of loss of potency and thus control. In affected samples it was found that the insulin concentration ranged from 6 to 64 U mL–1, where in normal samples within narrow margins the concentration was 110 U mL–1. |

|
|

9.2.6 Long-acting contraceptives
There is a series of progestogen-only contraceptives which display a variety of techniques of formulation and delivery, namely:
- Depo-Provera, an aqueous medroxyprogesterone acetate suspension delivered by deep IM injection, which is repeated every 12 weeks for long-term contraception
- Noristerat, an oily injection of norethisterone acetate injection given by deep IM injection, repeated once every 8 weeks
- Sayan Press, a suspension of medroxyprogesterone acetate injected SC into the anterior thigh or abdomen, which is repeated every 13 weeks.
These show the potential for very slow release, the duration being a factor of drug, dose and formulation, which must include the control of particle size distribution for the two suspensions and drug solubility, as we have discussed elsewhere.
Nexplanon subdermal implant
To achieve even longer-term action there are implants such as Nexplanon, which allows contraceptive cover over 36 months, delivering subdermally etonogestrel from a soft, flexible sterile ethylene vinyl acetate (EVA) copolymer core, surrounded by an EVA skin. It also contains barium sulfate as a radiopaque material for location during its lifetime (Fig. 9.6).
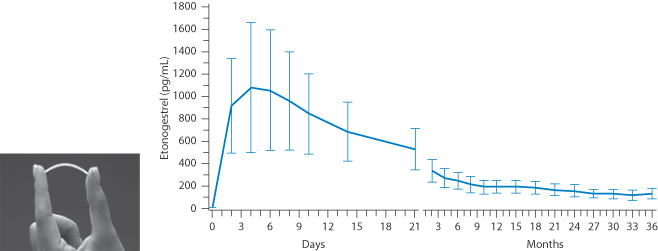
Figure 9.6 Mean (± SD) Serum concentration–time profile of etonogestrel after insertion of Nexplanon (a photograph of a subdermal implant is shown) during 36 months of use.
From data on Nexplanon, MSD.
In Chapter 8 we referred briefly to human variability in responses to drugs and medication, particualry by the oral route with the vagaries of the behaviour of the gastrointestinal tract. Apart from genetic and racial factors, and factors related to age and concomitant pathologies, one additional factor that is evident with the lipophilic steroids used in contraception is illustrated clearly in Fig. 9.7 – the effect of obesity. Plasma levels of etonogesterel from implants were much reduced in obese women.
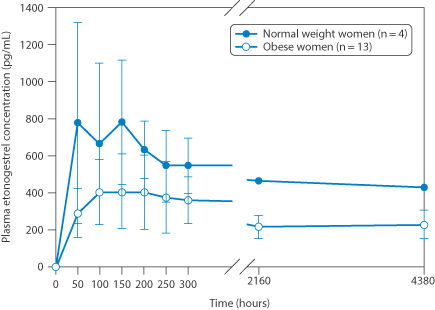
Figure 9.7 Human variability: the pharmacokinetics of etonogestrel in women of normal weight versus women categorised as obese.
Reproduced from S. Mornar et al. Am. J Obstet Gynecol 2012;207:110e1–110e6.
9.3 Transdermal delivery
A comprehensive account of the pharmaceutics and biopharmaceutics of topical preparations is not possible here. This section is restricted in scope to the physicochemical principles involved both in the process of treating the skin and in systemic medication by the transdermal or percutaneous route.
Formulation of topical vehicles for the potent drugs applied to the skin is now a more exact art. It is readily demonstrated that the vehicle in which the drug is applied influences the rate and extent of absorption. However, topical formulations can change rather rapidly once they have been spread on the skin, and absorption of some excipients and evaporative loss of water cause the breakdown of original structures.
Before any drug applied topically can act either locally or systemically it must penetrate the barrier layer of the skin, the stratum corneum (Fig. 9.8). This behaves like a passive diffusion barrier with no evidence of metabolic transport processes; drugs are absorbed by transcellular or intercellular pathways.
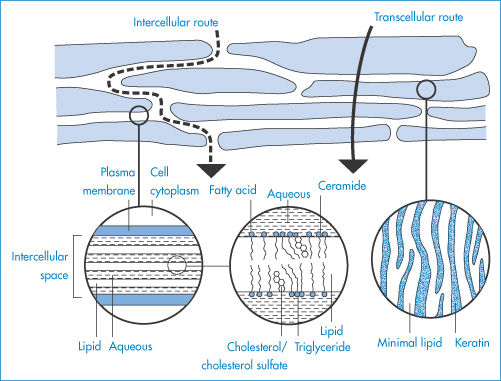
Figure 9.8 ‘Bricks and mortar’ model of the stratum corneum, illustrating possible pathways of drug permeation through intact stratum corneum (transcellular and tortuous intercellular pathways) and the lamellar structure of intercellular lipids.
Reproduced from Moghimi HR et al. A lamellar matrix model for stratum corneum intercellular lipids II. Effect of geometry of the stratum corneum on permeation of model drugs 5-fluorouracil and oestradiol. Int J Pharm 1996;131:117. Copyright Elsevier 1996.
Penetration of water and low-molecular-weight non-electrolyte solutes through the epidermis is proportional to their concentration, and to the partition coefficient of the solute between tissue and vehicle. A form of Fick’s law describes steady-state transport through the skin:

where J is the solute flux, D is the solute diffusion coefficient in the stratum corneum (values range from 1 × 10−12 to 1 × 10−17 m2 s−1 for human stratum corneum), P is the solute partition coefficient between skin and vehicle and δ is the thickness of the stratum corneum. ΔCv is the difference in solute concentration between vehicle and tissue? This relation is obtained as shown in Derivation Box 9A.
Before steady-state penetration is achieved, the rate builds up over a period of time and a lag phase will be apparent (Fig. 9.9). The lag time, τ, does not indicate the point at which steady state is achieved, which, as shown in Fig. 9.9, is obtained by extrapolation. The linear portion of the graph can be described by an equation relating the total amount absorbed at time t, Qt, to ΔC, P, δ, t and D. From equation (9.3), as Qt = Jt,

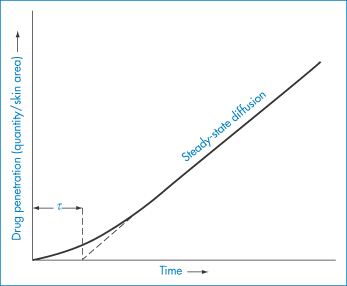
Figure 9.9 Drug penetration–time profile for an idealised drug diffusing through human skin; once steady-state diffusion occurs Qt can be obtained using equation (9.4), where t = (time elapsed − τ).
But from Fig. 9.9 it is obvious that the time to be substituted is the time over which steady-state flux has been maintained; namely, t − τ. Thus we write

As the lag time, τ, has been shown to be equal to δ2/6D, the drug’s diffusion coefficient D is readily obtained from data such as those shown in Fig. 9.7. Values of τ range from a few minutes to several days, so lag times are of obvious clinical relevance.
9.3.1 Routes of skin penetration
Drug molecules may penetrate the skin not only through the stratum corneum but also by way of the hair follicles or through the sweat ducts, but these offer only a comparatively minor route because they represent such a small fraction of the surface area. Only in the case of molecules that move very slowly through the stratum corneum may absorption by these other routes predominate. Passage through damaged skin is increased over normal skin. Skin with a disrupted epidermal layer will allow up to 80% of hydrocortisone to pass into the dermis, whereas only 1% is absorbed through intact skin.
The physicochemical factors that control drug penetration include the hydration of the stratum corneum, temperature, pH, drug concentration and the molecular characteristics of the penetrant and the vehicle. The stratum corneum is a heterogeneous structure containing about 40% protein (keratin, a disulfide-crosslinked linear polypeptide), about 40% water and 18–20% lipids (principally triglycerides and free fatty acids, cholesterol and phospholipids). Hydration of the stratum corneum is one determinant of the extent of absorption: increased hydration decreases the resistance of the layer, presumably by causing a swelling of the compact structures in the horny layer.
|
Occlusive dressings increase the hydration of the stratum corneum by preventing water loss by perspiration; certain ointment bases are designed to be self-occluding. The use of occlusive films can increase the penetration of corticosteroids by a factor of 100 and of lower-molecular-weight drugs up to fivefold. |

In the idealised model of the stratum corneum shown in Fig. 9.10, L represents the lipid-rich interstitial phase and P the proteinaceous phase. If ρ = PL/PP (the ratio of the partition coefficients of the drug between vehicle and the L and P phases) and DL and DP are the diffusion coefficients of the drug in these phases, the flux through stratum corneum of average thickness (40 μm) is found to reduce to
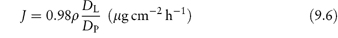
when ρ(DL/DP) is very small. When ρ(DL/DP) is very large, the flux becomes
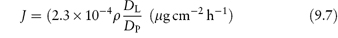
which emphasises the importance of both the partition coefficient and diffusion coefficient of the drug in the absorption process.
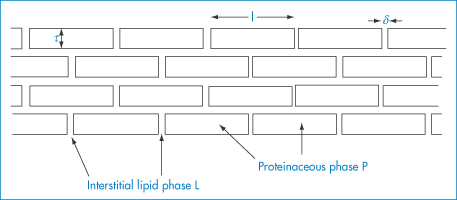
Figure 9.10 Idealised model of the stratum corneum. Lipid (L) and proteinaceous (P) parts of the stratum corneum are represented; this model is used to derive model equations for drug transport across this layer (see text).
Reproduced from Michaels AS et al. Drug permeation through human skin, theory and in vitro experimental measurement. A I Chem J 1975;21:985. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
9.3.2 Influence of the drug
The diffusion coefficient of the drug in the skin will be determined by factors such as molecular size, shape and charge; the partition coefficient will be determined not only by the properties of the drug but also by the vehicle as this represents the donor phase, the skin being the receptor phase. The quantity ρ can be approximated by experimentally determined oil/water partition coefficients. Substances that have a very low oil solubility will have lower rates of penetration.
The major pathway of transport for water-soluble molecules is transcellular, involving passage through cells and cell walls. The pathway for lipid-soluble molecules is presumably the endogenous lipid within the stratum corneum, the bulk of this being intercellular. Increase in the polar character of the molecule decreases permeability, as seen from the data on steroids in Table 9.4, in which progesterone and hydroxyprogesterone and deoxycortone and cortexolone should be compared. Each pair differs only by a hydroxyl group. The lipid/water partition coefficients of the drugs in Table 9.4 decrease as the number of hydroxyl groups increases, but a simple lipid/water partition coefficient is not an ideal guide, as the stratum corneum is a complex system, as described above. If a substance is more soluble in the stratum corneum than in the vehicle in which it is presented, however, the concentration in the first layer of the skin may be higher than that in the vehicle. If depletion of the contact layers of the vehicle occurs, then the nature of the formulation will dictate how readily these are replenished by diffusion and, therefore, will dictate the rate of absorption.
Table 9.4 Permeability constants of steroids
Steroid | Structure | Permeability constant, κP (10−6 cm−2 h−1) |
Progesterone |
| 1500 |
Hydroxyprogesterone |
| 600 |
Deoxycortone |
| 450 |
Cortexolone |
| 75 |
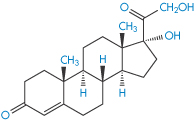
Reproduced by permission from Macmillan Publishers Ltd from Scheuplein R. Permeability of the skin: a review of major concepts and some new developments. J Invest Dermatol 1976;67:672.
The penetration rates of four steroids through intact abdominal autopsy skin were, in the order of their physiological activity, betamethasone 17-valerate (II) > desonide > triamcinolone acetonide (III) > hydrocortisone (IV).
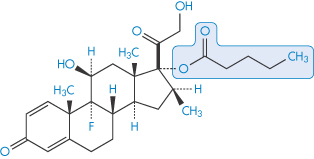
Structure II Betamethasone 17-valerate
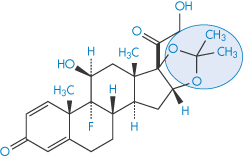
Structure III Triamcinolone acetonide
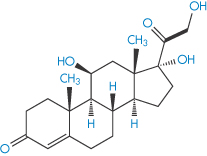
Structure IV Hydrocortisone
|
Note that the order of systemic and transdermal biological activities can be quite different. And of course the esters of certain steroids can result in startling differences in activity. 1 Triamcinolone is five times more active systemically than hydrocortisone, but has only one-tenth of its topical activity. 2 Betamethasone is 30 times as active as hydrocortisone systemically but has only four times the topical potency. 3 Triamcinolone acetonide has a topical activity 1000 times that of the parent steroid because of its favourable lipid solubility. |

Of 23 esters of betamethasone (V), the 17-valerate ester possesses the highest topical activity. The vasoconstrictor potency of betamethasone 17-valerate is 360 (fluocinolone acetonide = 100), that of betamethasone 0.8, of its 17-acetate 114, of the 17-propionate 190, and of the 17-octanoate 10. The peak activity coincides with an optimal partition coefficient, one that favours neither lipid nor aqueous phase.
Structure V Betamethasone
9.3.3 Influence of the vehicle
Consideration of equation (9.3) shows immediately that the vehicle has an influence on the absorption of the drug: if the vehicle is changed so that the drug becomes less soluble in it, P increases and so permeability increases. The vehicle is more dominant in topical therapy than in most routes of administration because the vehicle remains at the site, although not always in an unchanged form. Evaporation of water from a water-in-oil base would leave drug molecules immersed in the oily phase. Oil-in-water emulsion systems may invert to water-in-oil systems, in such a way that the drug would have to diffuse through an oily layer to reach the skin. Volatile components are driven off, probably altering the state of saturation of the drug and hence its activity. Drug may precipitate owing to lack of remaining solvent. This all means that theoretical approaches very much represent the simplest or, in thermodynamic terms, ‘ideal’ cases.
Formulations
Many modern dermatological formulations are washable oil-in-water systems. Simple aqueous lotions are also used as they have a cooling effect on the skin. Ointments are used for the application of insoluble or oil-soluble medicaments and leave a greasy film on the skin, inhibiting loss of moisture and encouraging the hydration of the keratin layer. Aqueous creams combine the characteristics of the lotions and ointments. A classification of semisolid bases is given in Fig. 9.11.
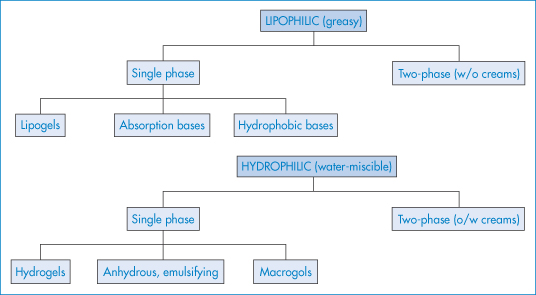
Figure 9.11 Classification of topical semisolid bases into lipophilic and hydrophilic systems in either single-phase or two-phase forms.
The descriptions ‘ointment’ and ‘cream’ have no universally accepted meaning and generally refer to the completed formulation. Ointments are generally composed of single-phase hydrophobic bases, such as pharmaceutical grades of soft paraffin or microcrystalline paraffin wax.
‘Absorption’ bases have an alleged capacity to facilitate absorption by the skin, but the term also alludes to their ability to take up considerable amounts of water to form water-in-oil emulsions. Lipogels are gels prepared by dispersion of long-chain fatty-acid soaps such as aluminium monostearate in a hydrocarbon base. Hydrogels prepared from Carbopols or cellulose derivatives are discussed in Chapter 7.
Thermodynamic activity in topical products
The complexity of many of the topical bases means that a simple physicochemical explanation of their influence on the release of medicament is not always possible. It is not difficult, however, to ascertain the effect of the base and to highlight the problem. The interactions between the physical properties of the base and the drug are exemplified by the effect of propylene glycol in simple bases, shown in Fig. 9.12, where the percentage of the glycol acting as a solvent for the drug significantly affects permeation.
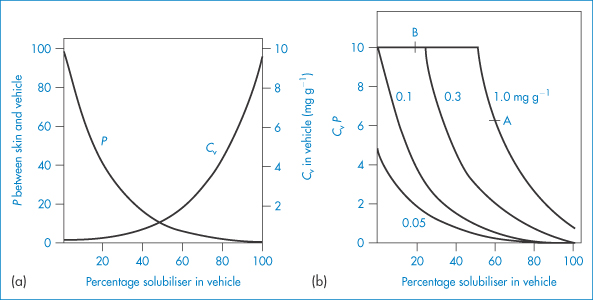
Figure 9.12 (a) Drug partition coefficient and solubility dependence on vehicle composition for the ‘ideal’ case. (b) Predicted relative steady-state penetration (CvP) as a function of vehicle composition for the ‘ideal’ case. Point A represents a formulation containing 1.0 mg g−1 and 60% solubiliser. On threefold dilution with a base containing no solubiliser, point B is arrived at, where CvP is higher.
Reproduced with permission from Poulsen BJ. In: Ariens EJ (ed.) Drug Design, vol. 4. New York: Academic Press; 1973.
The thermodynamic activity of the drug is the ultimate determinant of its biological activity. If the solubility of the drug in the base is increased by addition of propylene glycol, then its partition coefficient, P, towards the skin is reduced. On the other hand, increasing the amount that can be incorporated in the base is an advantage and the concentration (Cv) gradient is increased. It is apparent that there will be an optimum amount of solubiliser. The optimum occurs at the level of additive that just solubilises the medicament. Addition of excess results in desaturation of the system, and a decrease in thermodynamic activity. As the total amount of drug in the vehicle is increased from 0.5 to 1.0 mg g−1 (Fig. 9.12b), more propylene glycol has to be added to cause a decrease in CvP. From Fig. 9.12b one could postulate that threefold dilution of a formulation containing 1.0 mg g−1 and 60% solubiliser with a base with no solubiliser would cause the thermodynamic activity to increase. The vehicle affects penetration only when the release of drug is rate-limiting.
In situations where all of the activity gradient is in the applied phase, skin properties play no part. In these cases drug concentration in the vehicle, the diffusion coefficient of the drug in the system and the solubility of the drug are the significant factors. Where these factors are not important, the only significant factor involving the vehicle is the thermodynamic activity of the drug contained in it. Using the simplest model, for a solution of concentration C applied to an area A of the skin, the steady-state rate of penetration, dQ/dt, is given by

δ being the thickness of the barrier phase. An equivalent form of this equation expresses release in terms of the thermodynamic activity, a, of the agent in the vehicle:

where γ is the activity coefficient of the agent in the skin barrier phase. Ointments containing finely ground particles of drug where the thermodynamic activity is equal to that of solid drug will have the same rate of penetration, provided that the passage of the drug in the barrier phase is rate-limiting.
Drug release from complex vehicles
In more complex vehicles the activity a is impossible to determine and other approaches must be adopted. For example, in emulsions the relative affinity of drug for the external and internal phases of the emulsion is an important factor. A drug dissolved in an internal aqueous phase of a water-in-oil emulsion must be able to diffuse through the oily layer to reach the skin. Three cases can be considered: solution, suspension and emulsion systems.
1. Release from solutions is most readily understood and quantified by equations of the form

and

where C0 is the initial medicament concentration in solution, D is the diffusion coefficient and t is the time after application of the vehicle. As D is inversely proportional to the viscosity of the vehicle, one would expect that drug release would be slower from a viscous vehicle.
2. If a drug in suspension is to have any action it must have a degree of solubility in the base used. An aged suspension of a drug will therefore have a saturated solution of the drug present in the continuous phase. Release of medicament in these conditions is given by
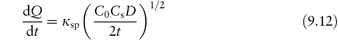
where Cs is the total solubility of the drug in the vehicle and C0 is the concentration in the vehicle. This equation applies only when Cs 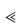 C0. Only material in solution penetrates the stratum corneum and the depleted layer in the vehicle is replenished only by solution of particles and diffusion of the drug molecules to the depleted layer.
C0. Only material in solution penetrates the stratum corneum and the depleted layer in the vehicle is replenished only by solution of particles and diffusion of the drug molecules to the depleted layer.
3. The equations for emulsion-type vehicles are discussed in Chapter 6.
9.3.4 Dilution of topical steroid preparations
|
Inappropriate dilution of carefully formulated creams and ointments may result in changes in stability and effectiveness. An example would be clobetasol propionate formulated in a base containing the optimum amount of propylene glycol. The solubilities of the steroid as a function of propylene glycol concentration are shown in Fig. 9.13. A 1 in 2 dilution of a 0.05% cream, with a vehicle containing water and no propylene glycol, will precipitate a large proportion of the steroid. The same principles apply to steroids presented in a fatty-acid propylene glycol base. |

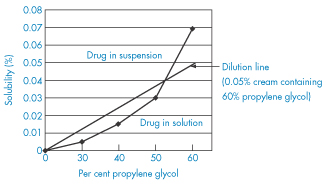
Figure 9.13 A diagram showing the solubility of clobetasol propionate as a function of propylene glycol concentration. The dilution line shows that, if a 0.05% cream containing 60% propylene glycol is diluted with a base not containing the glycol, the drug will be precipitated.
Data from Busse M. Pharm J 1978;220:25.
Enhancing skin penetration
The many techniques for enhancing the penetration of drugs through the skin are summarised in Fig. 9.14 and can be used as an aide mémoire for the whole topic of transdermal drug delivery.
Figure 9.14 Physical, passive and other means of improving the absorption of drugs or their penetration, e.g. via microneedles, iontophoresis, electroporation or by penetration enhancers or formulation techniques.
Reproduced from Hadgraft J, Lane M. Skin: the ultimate interface. Phys Chem Chem Phys 2011;13:5215–5222.
9.3.5 Transdermal medication: patches and devices
The ease with which some drugs can pass through the skin barrier into the circulating blood means that the transdermal route of medication is a possible alternative to the oral route. Theoretically there are several advantages:
- For drugs that are normally taken orally, administration through the skin can eliminate the vagaries that influence gastrointestinal absorption, such as pH changes and variations in food intake and intestinal transit time.
- A drug may be introduced into the systemic circulation without initially entering the portal circulation and passing through the liver.
- Constant and continuous administration of drugs may be achieved by a simple application to the skin surface.
- Continuous administration of drugs percutaneously at a controlled rate should permit elimination of pulse entry into the systemic circulation, an effect that is often associated with side-effects.
- Absorption of medication can be rapidly terminated whenever therapy must be interrupted.
Not all drugs are suitable for transdermal delivery. The limitations of the route are indicated in Table 9.5, which compares the percutaneous absorption of a range of drugs, from aspirin (only 22% of which is absorbed, even after 120 hours) to chloramphenicol, of which only 2% is absorbed. The transdermal route is now routinely used for a range of drugs, including oestrogens, clonidine and nicotine. The four basic forms of patch systems are shown in Fig. 9.15.
Table 9.5 Percutaneous absorption of a range of drugs in humans
Drug | Percentage dose absorbed (120 h) |
Aspirin | 22 |
Chloramphenicol | 2 |
Hexachlorophene | 3 |
Salicylic acid | 23 |
Urea | 6 |
Caffeine | 48 |
Reproduced with permission from Beckett AH et al. Comparison of oral and percutaneous routes in man for the systemic administration of ephedrine. J Pharm Pharmacol 1972;24:65P.
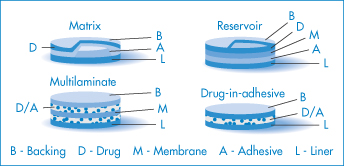
Figure 9.15 The four main types of transdermal patch: matrix, reservoir, multilaminate and drug-in-adhesive designs. The matrix and reservoir systems are cut away to show the drug.
Illustration courtesy of 3M.
Maximum flux from a saturated aqueous system of several drugs was estimated to be 300 μg cm−2 h−1 and from a mineral-oil system 250 μg cm−2 h−1. Using a ‘patch’ with a rate-limiting polymeric membrane, delivery is controlled to 40–50 μg cm−2 h−1; that is, it is presented to the skin at that rate. A 10 cm2 device will therefore deliver 0.4–0.5 mg h−1. Control of release can be exercised by altering the membrane’s properties and by changing the pH of the reservoir solution for a drug such as hyoscine (Fig. 9.16).
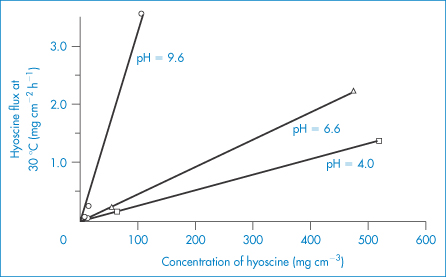
Figure 9.16 The effect of hyoscine concentration and pH on the flux of hyoscine across a membrane; increasing the pH increases the flux as ionisation is decreased.
Reproduced from Michaels AS et al. Drug permeation through human skin, theory and in vitro experimental measurement. A I Chem J 1975;21:985. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Iontophoresis
Iontophoresis is the process by which the migration of ionic drugs into tissues such as skin is enhanced by the use of an electric current, a technique that has found application favour in facilitating the delivery of peptides and proteins. A typical iontophoretic device is shown in Fig. 9.17.
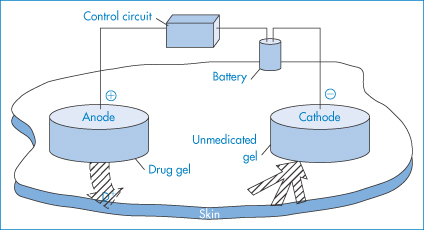
Figure 9.17 The major components of an iontophoretic drug delivery system. To optimise iontophoretic transdermal drug delivery, the drug should be charged. It should be applied under an electrode of the same polarity, and a counterelectrode opposite in charge to the drug must be placed at an indifferent site on the body. Electric current must be allowed to flow for an appropriate duration at a level minimising skin irritation. Iontophoresis can deliver neutral drugs by inducing solvent flow (electro-osmosis) that results from the migration of ionic species in the applied field. Drug candidates for iontophoretic transdermal delivery must usually be in a salt form with adequate aqueous solubility, be stable in an aqueous environment, and be sufficiently potent to allow therapeutic doses to be delivered at an acceptable current density.
Reproduced from Lattin GA et al. Electronic control of iontophoretic drug delivery. Ann N Y Acad Sci 1991;618:450. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Current can be applied in a continuous manner using either direct current (e.g. 0.1 mA cm−2) or pulsed (0.1 mA cm−2; 2000 Hz) to either solutions or gels of a drug. Enhancement of migration results from several possible sources, of which two are:
1. convective flow (electro-osmosis)
2. current-induced increases in skin permeability.
The flux of drug can be increased by increasing current density. With drug molecules (up to a molecular weight 1000 Da), the fraction of total current flowing that is carried by the drugs is small because of competition from smaller ions. Electro-osmosis probably plays a greater role here. Figure 9.18 shows the effect of pH, osmolality and current density on the transport of the nonapeptide buserelin through stratum corneum.
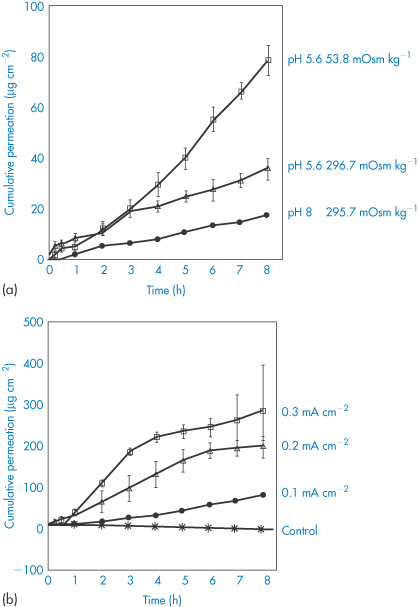
Figure 9.18 (a) The influence of donor pH and ionic strength of the donor medium on buserelin (a nonapeptide) permeation. (b) Continuous iontophoresis and release of buserelin through the stratum corneum, showing the effect of increasing the current.
Reproduced from Knoblauch P, Moll F. In vitro pulsatile and continuous transdermal delivery of buserelin by iontophoresis. J Control Release 1993;26:203. Copyright Elsevier 1993.
9.3.6 Ultrasound and transdermal penetration
Therapeutic ultrasound has been found2 first to expand and then collapse air bubbles in the stratum corneum (the process of cavitation) (Fig. 9.19). Cavitation tends to liquefy the solid fats and allows molecules such as insulin to pass through the skin at levels that in diabetic rats cause blood sugar lowering. The permeability of the skin was found to increase as the frequency of ultrasound decreased and no long-term damage was caused.
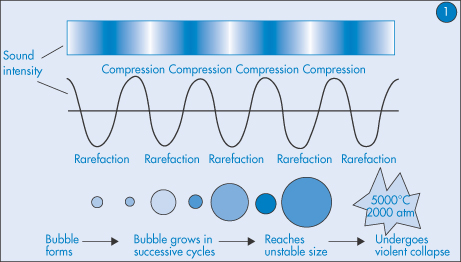
Figure 9.19 The process of cavitation leading to bubble collapse and change in fat structures and expulsion of drug.
9.3.7 Jet injectors
Invented in 1947 as a needle-free method of delivering drugs to the skin,3 jet injection techniques have received renewed attention recently. They perhaps have not lived up to recent promise, because of a certain unreliability of drug penetration, drug collection or pooling on the skin4 and rebound of particles from the skin surface. The systems are based on the high-velocity ejection of particles from the injector, through an orifice. Drug delivery is then due to either or both of skin ‘failure’ and flow through the skin.4 The theoretical maximum velocity (v) of the jet is related to the pressure (P) in the nozzle and the density (ρ) of the liquid by

but the velocity is affected by diameter of the orifice and is reduced by turbulence and friction.
Microneedles
When drug molecules have no natural tendency to penetrate the skin (hydrophilic molecules, proteins and oligopeptides and antigens), it is possible to take advantage of the development of microneedles fabricated, for example, from silicon, metal or polymers in solid or hydrogel form. There are five main categories of microneedle, as shown in Fig. 9.20:
1. solid
2. coated
3. dissolving
4. hollow
5. hydrogel-based systems.
These penetrate the stratum corneum, the viable epidermis and the dermis (Fig. 9.20). In the case of solid forms, their use is to make passages in these layers to allow a topical preparation to penetrate physically without invoking the processes discussed above. Other forms are coated with a drug so that the drug dissolves when inserted; dissolving systems contain the drug in the fabric of the needle and hollow needles allow drug from a reservoir to become available.*
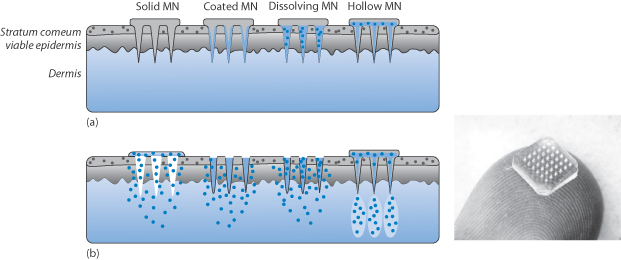
Figure 9.20 Diagram of the four types of microneedle (MN) showing (a) their structure: solid, coated. dissolving and hollow; and (b) their mode of behaviour once inserted into the skin. There are other configurations undergoing development. Right: photograph of a typical microneedle patch.
9.4 Topical treatment of the nails: transungual therapy
Treatment of fungal infections of the nail requires special topical approaches. It is a subject that has been the subject of several reviews. The same basic parameters hold for ensuring delivery via the nail as through the skin, but Murdan in her review5 concludes: ‘The permeability of the compact, highly keratinised nail plate to topically applied drugs is poor and drug uptake into the nail apparatus is extremely low. Topical therapy is worth pursuing, however, as local action is required in many nail disorders.’ Research aimed at enhancing ungual drug uptake following topical application may be divided into three approaches6: (1) understanding the physicochemical factors that influence drug permeation into the nail plate; (2) the use of chemical enhancers which cause alterations in the nail plate, thus assisting drug permeation; and (3) the use of drug-containing nail lacquers which are brushed on to nail plates and which act as a drug depot from which drug can be continuously released into the nail.
9.5 Medication of the eye and the eye as a route for systemic delivery
The eye is not, of course, a conduit for the administration of drugs to the body, although it has been explored for the systemic delivery of peptides and proteins such as insulin. It is considered here because absorption of drugs does occur from medication applied to the eye, producing sometimes toxic systemic effects, although often the desired local effect is on the eye or its component parts. We will consider the factors affecting drug absorption from the eye, and those properties of formulations that affect drug performance. A wide range of drug types are placed in the eye, including antimicrobials, antihistamines, decongestants, mydriatics, miotics and cycloplegic agents.
Drugs are usually applied to the eye in the form of drops or ointments for local action. The absorbing surface is the cornea. Drug absorbed by the conjunctiva enters the systemic circulation. It is useful to consider some of the properties of the absorbing surfaces and their environment.
|
|

9.5.1 The eye
The eye (Fig. 9.21) has two barrier systems: a blood–aqueous barrier and a blood–vitreous barrier. The former is composed of the ciliary epithelium, the epithelium of the posterior surface of the iris and blood vessels within the iris. Solutes and drugs enter the aqueous humour at the ciliary epithelium and at blood vessels. Many substances are transported out of the vitreous humour at the retinal surface. Solutes also leave the vitreous humour by diffusing to the aqueous humour of the posterior chamber.
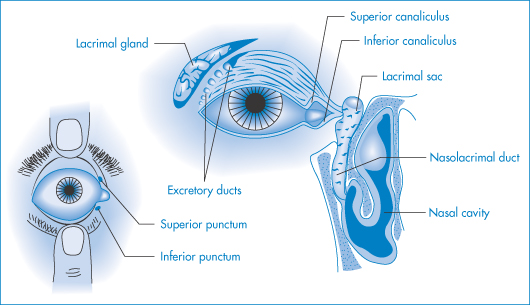
Figure 9.21 Diagrams of parts of the eye of importance in medication: the superior and inferior punctae are the drainage ports for solutions and tear fluids, and medicaments can drain via the canaliculi into the nasolacrimal duct and then to the nasal cavity, from whose surfaces absorption can occur.
Modified with permission from Robinson JR (ed.) Ophthalmic Drug Delivery Systems. Washington, DC: American Pharmaceutical Association; 1980.
Figure 9.21 is a diagrammatic representation of those parts of the eye involved in drug absorption. The cornea and the conjunctiva are covered with a thin fluid film, the tear film, which protects the cornea from dehydration and infection. Cleansed corneal epithelium is hydrophobic, physiological saline forming a contact angle of about 50° with it, and it has, in this clean condition, a critical surface tension of 28 mN m−1. The aqueous phase of tear fluid is spread by blinking.
Tears
Tears comprise inorganic electrolytes – sodium, potassium and some calcium ions, chloride and hydrogen carbonate counterions – as well as glucose. The macromolecular components include some albumin, globulins and lysozyme. Lipids that form a monolayer over the tear fluid surface derive from the Meibomian glands, which open on to the edges of the upper and lower lids. This secretion consists mainly of cholesteryl esters with low melting points (35°C) due to the predominance of double bonds and branched-chain structures.
Tear fluid
Tears are released from the Meibomian glands in the eye. Tears are important for lubrication of the eye. When we blink the tear film is replenished and re-spread, thus compensating for evaporation of the aqueous film and preventing the drying that would otherwise occur. In some patients, the supply of tear fluid, comprising a largely aqueous solution containing protein, lipids and enzymes, is impaired. Tears spread over the surface of the cornea, which is hydrophobic. The tears contain phospholipids, which act as a surfactant, lowering the surface tension of the fluid and allowing spreading. The thin lipid layer on the surface of the tear film (like most lipid monolayers) also prevents or slows down evaporation of the aqueous medium beneath. Dry eye7 (xerophthalmia; Sjögren’s syndrome) is caused when the tear film thins to such an extent that it ruptures, exposing the corneal surface to the air. Drying out of patches on the corneal surface follows. This can be painful and must be avoided. Evaporation over 5–10 min can actually eliminate the tear film completely.8
Artificial tear fluids
Artificial tear fluids may replace the natural tear fluid with varying degrees of similitude. Most aim for formulations that have an appropriate viscosity, but not necessarily identical rheological properties. Hydroxypropylmethylcellulose (HPMC) is a component of many commercial replacement or ‘artificial’ tear products, as are the water-soluble macromolecules carboxymethylcellulose, polyvinyl alcohol (PVA) and polyvinylpyrrolidone (PVP). Trehalose9 has also been studied as an agent for amelioration of the symptoms of dry eye. Trehalose is one of those molecules which, because of its hydrophilicity, increases the surface tension of water, but at 0.8 mol L–1 it has a viscosity of only 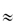 2 cP at 30°C. The aim is usually to reproduce the wetting and rheological properties of tear fluids, but in this case wetting is not enhanced. Increased concentrations, however, will have higher viscosities.
2 cP at 30°C. The aim is usually to reproduce the wetting and rheological properties of tear fluids, but in this case wetting is not enhanced. Increased concentrations, however, will have higher viscosities.
|
Tear fluid lies on the surface of the cornea and its importance lies in the possibility that components of formulations or drugs themselves can so alter the properties of the corneal surface, or interact with components of the tear fluid, that tear coverage of the eye is disrupted. When this occurs the so-called dry-eye syndrome (xerophthalmia) may arise, characterised by the premature break-up of the tear layer, resulting in dry spots on the corneal surface. |

9.5.2 Absorption of drugs applied to the eye
The cornea, which is the main barrier to absorption, consists of three parts: the epithelium, the stroma and the endothelium. Both the endothelium and the epithelium have a high lipid content and, as with most membranes, they are penetrated by drugs in their un-ionised lipid-soluble forms. The stroma lying between these two structures has a high water content, however, and thus drugs that have to negotiate the corneal barrier successfully must be both lipid-soluble and water-soluble to some extent (Fig. 9.22).
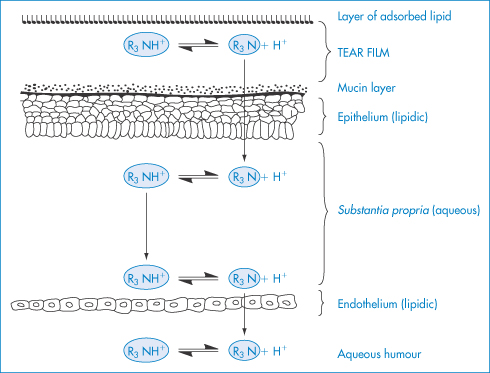
Figure 9.22 Diagrammatic representation of a tear film and the penetration of a base through the cornea; in this example R3N represents a drug such as homatropine.
Modified with permission from Moses RA. Adler’s Physiology of the Eye, 5th edn. St Louis: Mosby; 1970.
Tears have some buffering capacity so, as we noted before, the pH-partition hypothesis has to be applied with some circumspection. The acid-neutralising power of the tears when 0.1 cm3 of a 1% solution of a drug is instilled into the eye is approximately equivalent to 10 μL of a 0.01 mol dm−3 strong base. The pH for either maximum solubility (see Chapter 4) or maximum stability (see Chapter 3) of a drug may well be below the optimum in relation to acceptability and activity. Under these conditions it is possible to use a buffer with a low buffering capacity to maintain a low pH adequate to prevent change in pH due to alkalinity of glass or carbon dioxide ingress from the air. When such drops are instilled into the eye, the tears will participate in a fairly rapid return to normal pH.
|
In agreement with the pH-partition hypothesis, raising the pH from 5 to 8 results in a two- to threefold increase in the amount of pilocarpine reaching the anterior chamber. It is also found, however, that glycerol penetration increases to the same extent. The clue to why this should be so lies in the effect of the buffer solutions on lacrimation. Increased tear flow reduces the concentration of drug in the bathing fluid; loss of drug solution through the punctae and nasolacrimal ducts does not affect concentration, as the whole fluid drains away. |

Solution concentration is reduced only by diffusion of drug across the cornea or conjunctiva or by tear inflow. The pH 5 solution induced more tear flow than the pH 8 solution; thus the concentration gradient is reduced and transport of both ionised and non-ionic drugs is less at pH 5. At pH 11, absorption of pilocarpine is reduced because of the irritant effects of this solution on the eye.10 Mechanical irritation can produce the same effects and this can override the influence of other formulation factors.
Aqueous humour
As the examples above have shown, both water-soluble and lipid-soluble drugs can enter the aqueous humour. The pH-partition hypothesis thus accounts only imperfectly for different rates of entry into the aqueous humour. Sucrose and raffinose pass through the leaky ciliary epithelium and reach steady-state aqueous/plasma concentration ratios of 0.2 and 0.3, respectively. Lipid-soluble drugs, including chloramphenicol and some tetracyclines, can achieve higher concentrations as they can enter by both pathways. Penicillins, however, reach only low aqueous/plasma concentration ratios because they are removed from aqueous humour by absorption through the ciliary epithelium. Proteins are excluded, so that protein binding of ophthalmic drugs limits their absorption.
9.5.3 Influence of formulation
Pilocarpine activity has been compared in various formulations. Figure 9.23 shows some of the results on formulations, including results on ointments designed to prolong the contact of the drug with the cornea. One of the most difficult problems is to design vehicles that will retard drainage and prolong contact. Viscous polymer vehicles help to some extent but are not the complete answer. The rate of drainage of drops deceases as their viscosity increases and these factors contribute to an increased concentration of the drug in the precorneal film and aqueous humour.11 The magnitude of the concentration increase was small considering the 100-fold change in the viscosity, and it was concluded that the viscosity of the solution is not as important a factor in bioavailability as was previously thought.
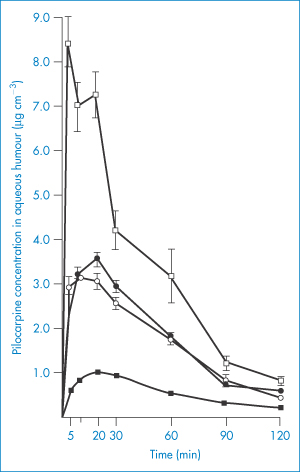
Figure 9.23 Aqueous humour levels of pilocarpine after dosing with 10−2 mol dm−3 ointment and solution in intact and abraded eyes. ●, 25 mg of ointment, intact eyes; ○, 25 mg of ointment, abraded eyes; ◼, 25 mm3 of solution, intact eyes; ◻, 25 mm3 of solution, abraded eyes; all points represent an average of 8–16 eyes.
Reproduced from Sieg JW, Robinson JR. Vehicle effects on ocular drug bioavailability II: Evaluation of pilocarpine. J Pharm Sci 1977;66:1222–1228. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
The results of incorporating pilocarpine (VI) (a relatively water-soluble drug) and fluorometholone (a lipophilic drug) into a water-in-oil ointment base can be compared in Fig. 9.24. Pilocarpine is thought to be released only when in contact with aqueous tear fluid, whereas the steroid, being soluble in the base, can diffuse through the base to replenish the surface concentrations and thus produce a sustained effect.
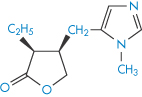
Structure VI Pilocarpine (pKa = 7.05)
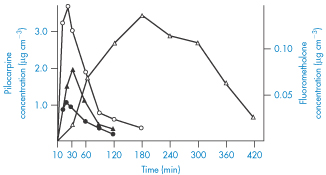
Figure 9.24 Aqueous humour levels after dosing with pilocarpine and fluorometholone in ointment and aqueous solution. ●, 10−2 mol dm−3 pilocarpine solution; ○, 10−2 mol dm−3 pilocarpine ointment; ▲, saturated fluorometholone solution;  , 0.1% fluorometholone ointment.
, 0.1% fluorometholone ointment.
Reproduced from Sieg JW, Robinson JR. Vehicle effects on ocular drug bioavailability II: Evaluation of pilocarpine. J Pharm Sci 1977;66:1222–1228. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Polymeric vehicles
Hydrophilic polymeric vehicles, such as PVA and HPMC, are used in ophthalmic formulations.12 PVA increases the effectiveness of the drug substance, most likely by slowing the drainage rate of the eye drops. The viscosity of Newtonian fluids should be greater than 4.4 cP to influence drainage times, with a maximum effect at 100 cP. HPMC has been found to enhance the activity and reduce the effective dose of neomycin sulfate required to prevent infection of corneas of experimental animals (Table 9.6).
Table 9.6 Calculated effective dose (ED50) of neomycin sulfate required to prevent infection in 50% of rabbit corneas when incorporated into various vehicles
Vehicle | Derived values (mg base per cm3) | |
| ED50 | 95% confidence limits |
HPMC, 0.5% | 0.50 | 0.403–0.620 |
PVA, 1.4% | 1.00 | 0.750–1.330 |
PVP, 1.4% | 1.10 | 0.89–1.35 |
Distilled water | 1.03 | 0.84–1.27 |
Reproduced from Bach FC et al. The influence of vehicles on neomycin sulfate prevention of experimental ocular infection in rabbits. Am J Ophthalmol 1970;69:659. Copyright Elsevier 1970.
|
Eye drops are often formulated to be isotonic with tear fluid, but deviations from tonicity do not cause major problems, although hypertonicity may cause stinging of the eye and hypotonicity may increase the permeability of the cornea. Some ingredients may increase the permeability of the cornea. Surfactants are known to interact with membranes to increase the permeability: benzalkonium chloride has surfactant properties and may well have some effect on corneal permeability, although its primary purpose is as a bacteriostat and bactericide. Chlorhexidine acetate and cetrimide, both of which are surface-active, are also used. |

Log P
The influence of log P of beta-blockers (Table 9.7) on their permeation coefficients into conjunctiva and cornea is shown in Fig. 9.25. In humans, the conjunctiva has 17 times the surface area of the cornea and therefore it absorbs significant amounts of drug. Work has centred on increasing corneal permeability or retention of the drug (or product) in the conjunctiva sac. Appropriate doses of drug can achieve a degree of corneal or conjunctival selectivity, represented in Fig. 9.25c by the ratio C/J. Increasing lipophilicity increases the C/J ratio.
Table 9.7 Log P values of beta-blockers (in increasing order of lipophilicity)
Compound |
| Log P |
Sotalol |
| −0.62 |
Atenolol |
| 0.16 |
Nadolol |
| 0.93 |
Pindolol |
| 1.75 |
Acebutolol |
| 1.77 |
Metoprolol |
| 1.88 |
Timolol |
| 1.91 |
Oxprenolol |
| 2.07 |
Levobunolol |
| 2.40 |
Labetalol |
| 2.55 |
Alprenolol |
| 2.61 |
Propranolol |
| 3.21 |
Betaxolol |
| 3.44 |
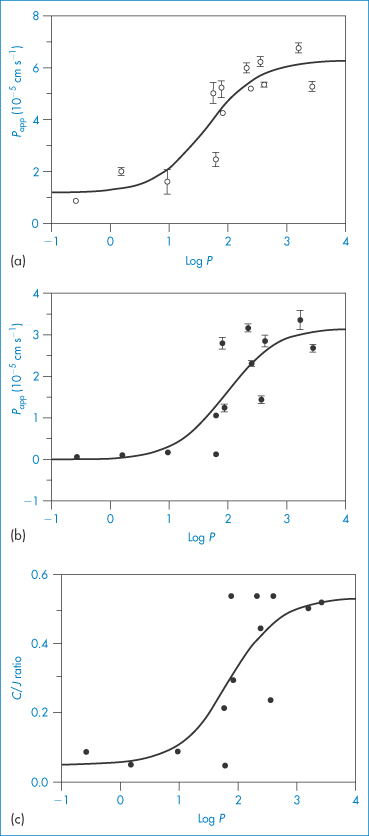
Figure 9.25 Influence of drug lipophilicity (log P) on the permeability coefficients (Papp) of beta blockers across (a) the conjunctiva and (b) the cornea of the pigmented rabbit. Plot (c) shows the influence of log P on the ratio of the corneal (C) and conjunctival (J) permeability coefficients.
Reproduced with permission from Wong W et al. Lipophilicity influence on conjunctival drug penetration in the pigmented rabbit: a comparison with corneal penetration. Curr Eye Res 1991;6:571.
The prodrug approach
Attempts have been made to improve the performance of drugs used in the eye. One approach has been to modify the drug substance to increase its ability to penetrate the corneal barrier. In Fig. 9.26, the results of application of 0.5% epinephrine (adrenaline) (VII) and 0.16% of the dipivoyl derivative of epinephrine (dipivefrine) (VIII) are shown. The more hydrophobic derivative is absorbed to a greater extent and is then hydrolysed to the active parent molecule in the aqueous humour.
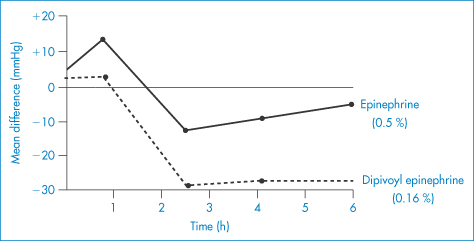
Figure 9.26 The action of 0.5% epinephrine (VI) and 0.16% dipivoyl epinephrine (VII) on intraocular pressure.
Reproduced with permission from McClure DA. In: Higuchi T, Stella V (eds) Prodrugs as Novel Drug Delivery Systems. Washington, DC: American Chemical Society; 1975. Copyright 1975 American Chemical Society.
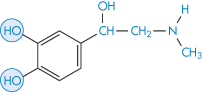
Structure VII Epinephrine (adrenaline)
Derivatives of pilocarpine such as the hexadecanoyloxymethylene chloride (IX) similarly show the advantages of the epinephrine prodrug.
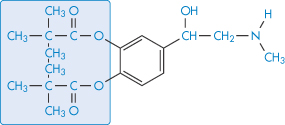
Structure VIII Dipivoyl epinephrine (dipivefrine; compare with Structure VII)
Structure IX Pilocarpine derivatives
Reservoir systems
Eye drops and eye ointments have their limitations, thus other means of delivering drugs to the eye have been developed. These include reservoirs such as contact lenses and the Ocusert device. Figure 9.27 traces the fate of drugs delivered in systems to the eye and some of the issues that must be take into account in their use.
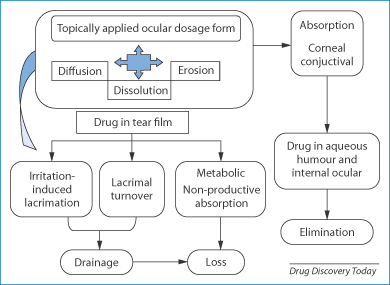
Figure 9.27 Illustration of the systems that release a drug by diffusion, erosion or dissolution, entering the tear film and being absorbed by the conjunctival tissue, and showing the means of loss of drug by drainage and elimination. One other relevant factor is that devices can induce lacrimation, thus increasing tear flow and diluting released drug.
Reproduced from Gupta H, Aqil M. Contact lenses in ocular therapeutics. Drug Discov Today 2012;17:522–527. Copyright Elsevier 2012.
Soft contact lenses
Since the advent of soft contact lenses, attempts have been made to use these as drug reservoirs13; a drug such as pilocarpine is imbibed from solutions into the polymer matrix and when this is placed in the eye the drug leaches out, generally over a period of up to 24 hours. Levels of prednisolone applied in a copolymer of 2-hydroxyethyl methacrylate and N-vinyl-2-pyrrolidone were compared with levels in aqueous humour, cornea and iris after applications of prednisolone in solution. When the drug was incorporated into the lens, aqueous and corneal levels of the drug were two- to threefold higher 4 hours after insertion. Figure 9.28 shows data for the delivery of the prostaglandin latanoprost, illustrating the levels achieved over 30 days from a contact lens. Issues discussed in Chapter 7 should be refreshed to appreciate the mechanism of drug release from polymeric systems and the means of rationalising their physicochemical behaviour. The results in the figure show prolonged action but marked differences in concentration of the drug over the 30-day duration.
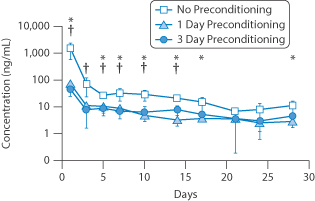
Figure 9.28 The effect of changes in lens pretreatment to produce different levels of latanoprost for the treatment of glaucoma over a 1-month period. Data from rabbit eyes. For comparison, the average concentration over 24 hours was about 12 mg mL–1 using topical drops.
Reproduced from Ciolino JB et al. In vivo performance of a drug-eluting contact lens to treat glaucoma for a month. Biomaterials 2014;35:432–439. Copyright Elsevier 2014.
Ocusert
The Ocusert device (Fig. 9.29) releases controlled amounts of pilocarpine over a period of 7 days and is designed for the treatment of glaucoma. It comprises a drug reservoir in which pilocarpine is embedded in an alginic acid matrix, and this is bounded by two rate-controlling membranes of polymer (vinyl acetate–ethylene copolymer). It is inserted into the lower conjunctival sac of the eye. The rate-controlling membranes are subject to strict quality control during manufacture and their thicknesses adjusted to give the appropriate flux of drug, either 20 or 40 μg h−1. The difference between the two available systems is effected by altering the amount of di-(2-ethylhexyl)phthalate in the polymer; increasing the concentration of this plasticiser increases the permeability to pilocarpine.
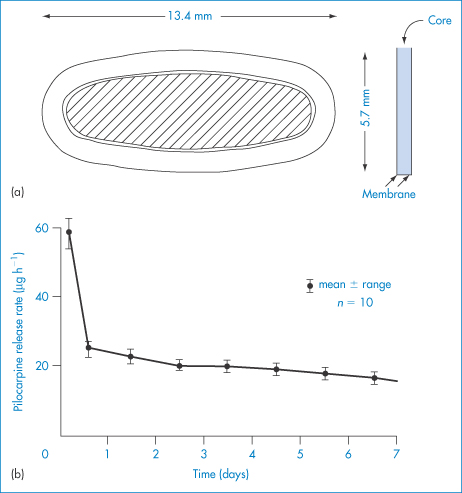
Figure 9.29 (a) Dimensions and structure of the Ocusert device. (b) A release rate profile over 1 week.
Reproduced from Snell JW, Baker RW. Diffusional systems for controlled release of drugs to the eye. Ann Ophthalmol 1974;6:1037.
9.5.4 Systemic effects from eye drops
Most of the dose applied to the eye in the form of drops reaches the systemic circulation and typically less than 5% acts on ocular tissues.
|
Atropine toxicity resulting from the use of eye drops to dilate the pupil has been reported,14 as has a rise in blood pressure in premature infants following the use of 10% phenylephrine eye drops in preparation for ophthalmoscopy.15 The mucosa in the eye, nose and mouth of infants is much thinner and more permeable to a drug than is, say, the skin. Drugs placed in the eye, nose or mouth, moreover, may bypass the metabolic transformations which may inactivate the drug given orally. Reduction in exercise tachycardia in normal volunteers is induced following administration of 0.5% timolol eye drops, showing the significant effects of beta-blockers. |

|
|

9.6 The ear (the aural route)
Medications are administered to the ear only for local treatment. Drops and other vehicles administered to the ear will occupy the external auditory meatus, which is separated from the middle ear by the tympanic membrane (Fig. 9.30).
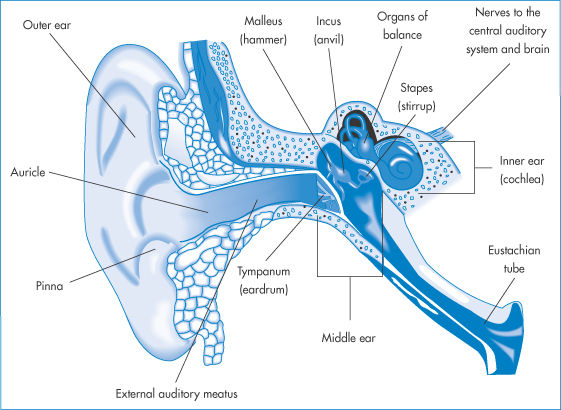
Figure 9.30 The ear and its associated structures; medications would normally enter only the external auditory meatus.
Various factors affect drug absorption from the ear or action in it. The sebaceous and apocrine glands of the mucosa secrete an oily fluid that, when mixed with exfoliated cells of the stratum corneum, forms the cerumen or wax composed of, inter alia, fats, fatty acids, protein, pigment, glycoprotein and water. The acidic environment of the ear skin surface (around pH 6), sometimes referred to as the acid mantle of the ear, is thought to be a defence against invading microorganisms.
Various ceruminolytic agents achieve their action by partially dissolving the wax. Several commercial ear drops contain surfactants such as poloxamers or sodium dioctyl sulfosuccinate (docusate sodium) to assist in the process. In otitis media, infection of the Eustachian tube is involved; antibiotic treatment is indicated along with oral analgesics. There is little evidence that topical analgesics give faster relief, suggesting poor absorption. However, cefmetazole sodium, a cephamycin antibiotic, passes through the membrane into the inner ear of the guinea pig. The concentration of the drug in the inner ear fluid indicated that a larger amount of the drug reached the inner ear by this means than when it was administered intraperitoneally.
9.7 Absorption from the vagina
The vagina cannot be considered to be a route for the systemic administration of drugs, although oestrogens for systemic delivery have been applied intravaginally. Certain medicaments are, however, absorbed when applied to the vaginal epithelium as it is permeable to a wide range of substances, including steroids, prostaglandins, iodine and some antibiotics.16 Econazole and miconazole are both also appreciably absorbed.
The epithelial layer of the vagina (Fig. 9.31a) comprises the lamina propria and a surface epithelium of non-cornified, stratified squamous cells (Fig 9.31b). The thickness of the epithelium increases after puberty and then again after menopause. The surface area is increased by folds in the epithelium and by micro-ridges. The pH in the vagina decreases after puberty, varying between pH 4 and 5 depending on the point in the menstrual cycle and also on the location within the vagina, the pH being higher near the cervix. There is little fluid in the vagina. The absorbing surface is under constant change, therefore absorption will be variable. While the presence of mucus is likely to retard absorption, there is unlikely to be other material in the vagina which will inhibit absorption. The uterine and pudendal arteries are the main sources of blood to the vagina; the venous plexus that surrounds the vagina empties into the internal ileac veins. Lymph vessels drain the vagina, and vaginal capillaries are found in close proximity to the basal epithelial layer.
Figure 9.31 (a) Anatomy of the urethra and vagina; drug delivery systems for both are available. (b) For other than local vaginal treatment, transport through the absorbing membrane structure (squamous epithelium (SE) lamina propria (LP) and muscularis (not shown)) is important in securing systemic levels of drug; inadvertent absorption after local therapy is, of course, possible.
9.7.1 Vaginal delivery systems
Conventional vaginal delivery systems include vaginal tablets, foams, gels, suspensions and pessaries. Vaginal rings (Fig. 9.32) have been developed to deliver contraceptive steroids. These commonly comprise an inert silicone elastomer ring that is covered with an elastomer layer containing the drug. In some systems a refinement has been to add a third, rate-modifying layer to the external surface of the ring, as shown. Some systems contain both an oestrogen and progestogen.
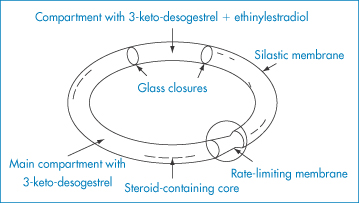
Figure 9.32 A combined contraceptive vaginal ring for the slow release of 3-keto-desogestrel (etonogestrel) and ethinylestradiol.
Reproduced from Sam AP. Controlled release contraceptive devices: a status report. J Control Release 1992;22:35. Copyright Elsevier 1992.
Hydrogel-based vaginal pessaries to deliver prostaglandin E2 (to assist in ripening of the cervix prior to labour), progesterone and bleomycin have been developed.
Tablets for vaginal use have included hydroxypropylcellulose or sodium carboxymethylcellulose and poly(acrylic acid) (such as Carbopol 934) as excipients. Micropatches in the size range 10–100 μm diameter prepared from albumin, collagen or dextrose will gel on contact with vaginal mucosal surfaces and adhere, prolonging contact between the delivery system and the absorbing surface.17
9.8 Inhalation therapy
The respiratory system provides a route of entry into the body for a variety of airborne substances but is also a route of medication. The large contact area of its surfaces extends to more than 30 m2. The surfaces have been described18 as ‘gossamer-thin membranes’ that separate the lung air from the blood, which courses through some 2000 km of capillaries in the lungs. There is consequently an ‘exquisite degree of intimacy between the lung tissue and blood and the atmospheric environment’. The route is thus used for rapid relief of asthmatic conditions, where both local and systemic effects are required, for chronic therapy, and for the administration of peptides and proteins.
Orally administered corticosteroids are effective in the treatment of chronic bronchial asthma. The inhalation route has been widely used in attempts to avoid systemic side-effects, such as adrenal suppression, but evidence suggests that inhaled steroids are absorbed systemically to a significant extent. The respiratory tract epithelium has permeability characteristics similar to those of the classical biological membrane, so lipid-soluble compounds are absorbed more rapidly than lipid-insoluble molecules. Cortisone, hydrocortisone and dexamethasone are absorbed rapidly by a non-saturable diffusion process from the lung; the half-time of absorption is of the order of 1–1.7 minutes. Quaternary ammonium compounds, hippurates and mannitol have absorption half-times, in contrast, of between 45 and 70 minutes.
Relative to the gastrointestinal mucosa the pulmonary epithelium possesses a high permeability to water-soluble molecules, which is an advantage with drugs such as sodium cromoglicate (X), a bischromone with two carboxylic acid groups and a pKa of approximately 1.9. The drug is well absorbed from the lungs with a clearance rate of about 1 hour, even though the molecule is completely ionised at physiological pH. The free acid is very insoluble in both polar and non-polar solvents and has virtually no lipid solubility. Because of this, and the insolubility of the un-ionised form, very little of an oral dose of sodium cromoglicate is absorbed. Powder swallowed after inhalation therefore contributes little to the systemic dose and is subsequently excreted in the urine and bile.
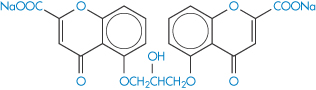
Structure X Sodium cromoglicate (DSCG; sodium cromoglycate; cromolyn sodium)
Drugs administered by inhalation are mostly intended to have a direct effect on the lungs. However, the efficiency of inhalation therapy is often not high because of the difficulty in targeting particles to the sites of maximal absorption. Only about 8% of the inhaled dose of sodium cromoglicate administered from a Spinhaler device reaches the alveoli.
Crude inhalers have been used for medicinal purposes for at least two centuries. Solutions of volatile aromatic substances with a mild irritant action, inhaled as vapour rising from hot aqueous solutions, have been used for many years. Particles from the older nebulisers would settle without reaching the patient’s face (a 100 μm particle of unit density settles in still air at a velocity of 8 cm s−1). The duration of existence of a suspension of particles of size around 10 μm is so brief that the upper limit of aerosols of therapeutic interest is well below this size. Special devices are required to generate these.
9.8.1 Physical factors affecting deposition of aerosols
Figure 9.33 shows the order of maximum size of particles that can penetrate to various parts of the respiratory tract, from trachea to alveoli. The major processes that influence deposition of drug particles in the respiratory tract are, as illustrated in Fig. 9.34,
- the interception of particles
- their impaction on epithelial surfaces
- their gravitational settling
- electrostatic attraction
- Brownian diffusion.
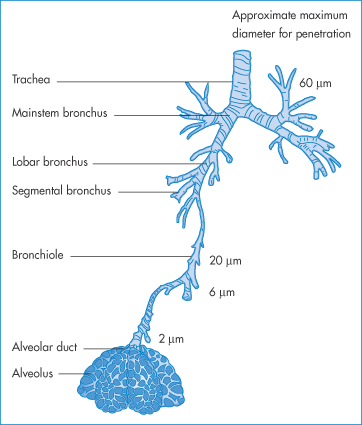
Figure 9.33 Deposition of particles in various anatomical regions of the respiratory tract from bronchus to alveoli according to particle size.
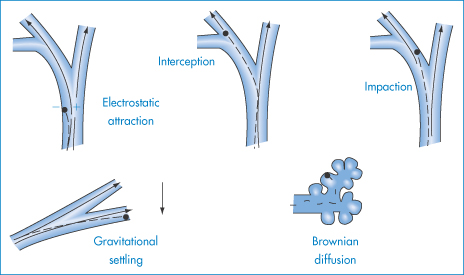
Figure 9.34 Representation of the major processes of particle deposition in the respiratory tract, as discussed in the text.
The passage of an aerosol particle from the alveolar sac into the blood stream is illustrated diagrammatically in Fig. 9.35.
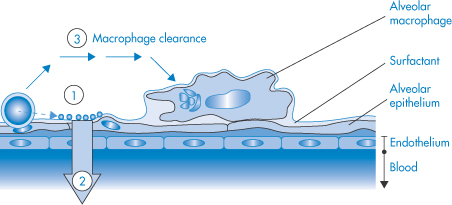
Figure 9.35 Diagram of the fate of an aerosol particle in a surfactant-coated alveolar sac. The particle dissolves after landing (1) and is taken up into the alveolar epithelial cell layer (2), transported by transcytotic vesicles into the pulmonary lymph and then into the blood via the capillary endothelium. Alveolar macrophages remove residues.
Reproduced from Ruge CA, Kirch J, Lehr CM. Lancet Resp Med 2013;1:402–413.
When a particle contacts the walls of the airways it is removed from the airstream; this process can occur during inspiration or expiration of a single breath, or later if the particle has been transferred to unexhaled lung air. Deposition increases with duration of breath holding and depth of breathing, hence the instruction to patients to breathe deeply when using their inhalers.
As flowing air moves in and out, inertial forces within the nasopharyngeal chamber and at the points of branching of the airways, or wherever the direction of flow changes, result in the collision of particles with surfaces. Along the narrower airways, particles are removed by gravity. Very fine particles may be exhaled.
The next sections present a more detailed account of deposition in the airways. There is no unified theory that predicts exactly what is occurring, but each subset of events is governed by an equation that demonstrates the key parameters of the aerosol. This section is included for completeness to illustrate the physicochemical basis of therapy of the lung.
Physical diameter and aerodynamic diameter
The aerodynamic diameter of a particle, da, is related to the particle diameter (d) and density (ρ) by the equation

To overcome problems of powder flow and agglomeration, porous particles (i.e. particles with a low density) have been developed. Equation (9.14) shows that a particle of 10 μm diameter with a density of 0.1 g cm−3 has an aerodynamic diameter of around 3 μm.
Gravitational settling
Fine particles falling through the air under the force of gravity do so at a constant velocity such that the resistance of the air balances the mass of the particle. The following equation relates particle diameter, d, and density, ρ, to terminal velocity, μt:

where g is the gravitational constant and η is the viscosity of the air. For air, η = 1.9 × 10−7 N m−2 s and therefore
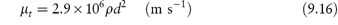
Table 9.8 shows the terminal velocities for particles of a range of diameters from 20 to 0.1 μm, reinforcing the importance of small size in preventing removal of particles before entry into the lower reaches of the respiratory tract. In still air, a cloud of powder particles of about 20 μm diameter takes a few seconds to settle, whereas powder of around 1 μm diameter takes approximately 60 seconds.
Table 9.8 Terminal velocity (μt) of spherical particles of unit density in air
Diameter (μm) | μt (cm s−1) |
20 | 1.2 |
10 | 2.9 × 10−1 |
4 | 5 × 10−2 |
1 | 3.5 × 10−3 |
0.6 | 1.4 × 10−3 |
0.1 | 8.6 × 10−5 |
Reproduced from Hatch TF, Gross P. Pulmonary Deposition and Retention of Inhaled Aerosols. New York: Academic Press; 1964.
Sedimentation
As the particles of drug move with the air in laminar flow in the airways, they fall under the force of gravity a distance equal to μtt, where t is the time of travel. If the tube (or part of the bronchial tree) in which they move is of radius R and is inclined at an angle ψ to the horizontal, the maximum distance of fall will no longer be 2R but 2R/cos ψ. The ratio, r, of the distance of fall to the maximum distance for deposition to be achieved is thus

The probability of deposition by sedimentation is proportional to this ratio; the closer μt is to 2R/cos ψ, the greater the likelihood of deposition by this mechanism. If the particles are evenly distributed over the cross-section of an airstream, it is theoretically possible to calculate the probability of deposition in tubular airways and in a spherical alveolus. Individual tubes are of course randomly positioned with reference to the horizontal and therefore an average value of ψ is used. Airflow is also not always laminar, and orderly deposition will not always occur, but in spite of these problems the following calculations of percentage sedimentation (S) may usefully be quoted. If S = 55% when d = 2 μm and the deposition diameter is 1.0 μm, then for 1 μm particles S = 29%, and for 0.5 μm particles S = 10%. Sedimentation, of course, reduces in importance as the drug particle size decreases.
Diffusion
The effectiveness of deposition by diffusion increases as particle size is reduced, which contrasts with the above. There must therefore be a particle diameter for which both processes have a combined minimum value; this occurs with particles approximately 0.5 μm in diameter. Particles of this size have the minimum probability of deposition in the upper respiratory tract.
Inertial impaction
When, during breathing, the airflow suddenly changes direction, a drug particle will continue in its original direction of flow owing to its inertia. In this way the particle may impact on the channel wall. In curved tubes the particle in an airstream that experiences a sudden bend suffers a similar fate, and the effective stopping distance at right angles to the direction of travel, hs, is given by

where μ is the velocity of the airstream with particles approaching a bend of angle θ. The term μ sin θ is therefore a component of initial particle velocity at right angles to the direction of airflow. The probability of inertial deposition, I, is proportional to the ratio of stopping distance, hs, to the radius, R, of the airway; that is

from a calculation similar to that discussed above for sedimentation. Calculated inertial deposition shows a dependence on particle size as follows: 10 μm particles 50%, 7 μm particles 33%, leading to 20% for 5 μm particles, and 1% for 1 μm particles.
9.8.2 Experimental observations
Although the complexity of the respiratory system prevents a precise mathematical approach to the problem, many clinical studies clearly demonstrate the importance of particle size, as one would anticipate from the preceding sections.
Particles of hygroscopic materials are removed more effectively than are non-hygroscopic particles, because of the growth of these particles by uptake of water from the moist air in the respiratory system. Apart from its importance in determining the efficiency of an aerosol in reaching the alveoli, particle size may be critical in determining response because of the influence of particle size on rate of solution.
The effect of particle size on the fate of particles inhaled from an aerosol is shown in Fig. 9.36.
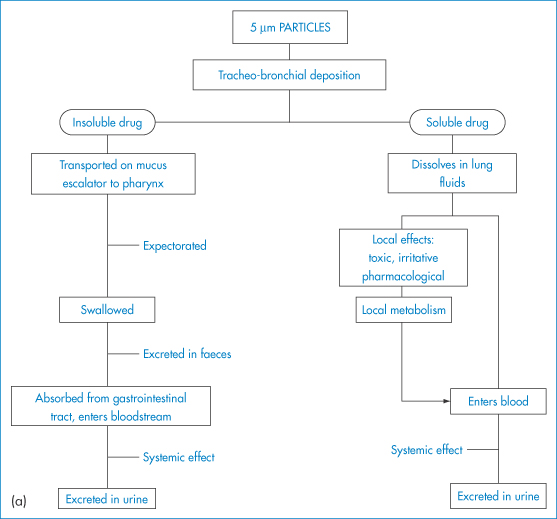
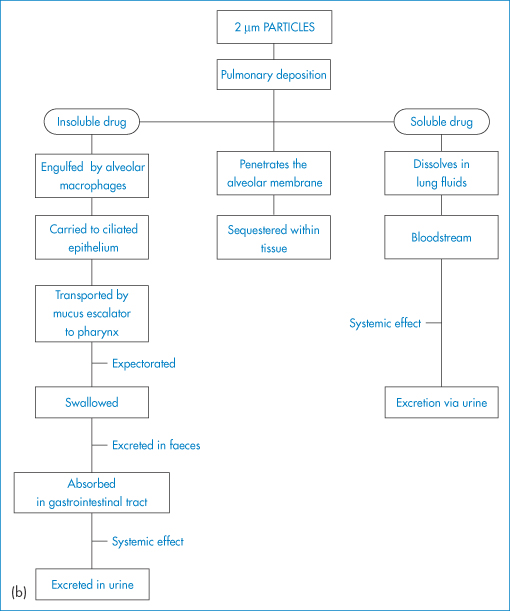
Figure 9.36 The fate of particles of (a) 5 μm and (b) 2 μm in diameter deposited in alveoli.
Reproduced from Clark DG. Proc Eur Soc Study Drug Toxic 1974;15:252.
When used by patients, the Spinhaler delivers about 25% by weight of sodium cromoglicate, which is normally dispersed as particles below 6 μm in diameter, about 5% being less than 2 μm diameter. The mass medium diameter (and geometric standard deviation) of the sodium cromoglicate particle batches used were, respectively, 2 ± 1.2 and 11.7 ± 1.1 μm.
|
There is no doubt that the biological effect of small particle material is dramatically greater than that of coarser material, hence the importance of storage conditions of drug cartridge capsules to prevent aggregation of the drug particles. Although the Spinhaler is designed to break up aggregates, its efficiency will be reduced if moisture uptake is increased by storage in humid conditions. If aerosols of drug with large particles (11 μm diameter) are administered, up to 66% of the dose will end up in the mouth. |

An alternative dry-powder aerosol device is illustrated in Fig. 9.37 and the mechanism of dispersion of powdered drug in a Ventodisk or Becodisk system is shown in Fig. 9.38.
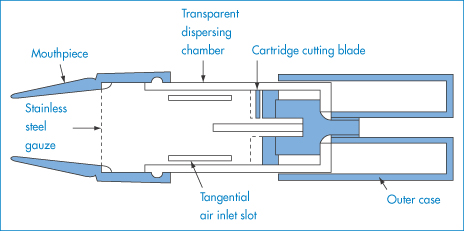
Figure 9.37 Longitudinal view of the Rotahaler.
Reproduced from Hallworth GW. An improved design of powder inhaler.Br J Clin Pharmacol 1977;4:689. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
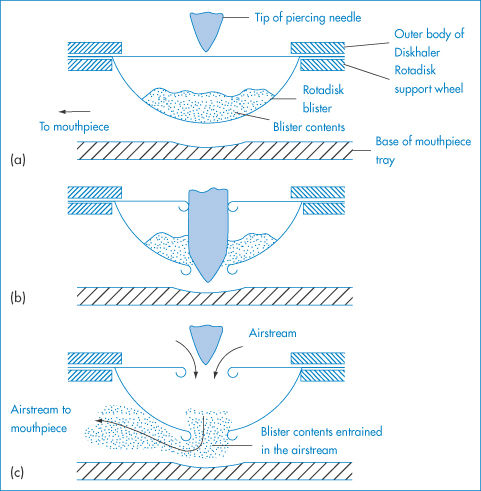
Figure 9.38 Dispersion of Ventodisk or Becodisk contents by breath actuation: a cross-section of a Diskhaler is shown with a disc located on the support wheel. (a) A disc is located beneath an aperture in the body of the Diskhaler through which a piercing needle enters. (b) The needle penetrates the upper and lower surfaces of the blister. (c) The patient inhales through the device and pierced blister, entraining the blister contents into the airstream.
Reproduced with permission from Farr SJ et al. In: Florence AT, Salole EG (eds) Routes of Drug Administration. London: Wright; 1990.
Types of pressurised aerosol
In two-phase systems the propellant forms a separate liquid phase, whereas in the single-phase form the liquid propellant is the liquid phase containing the drug in solution or in suspension in the liquefied propellant gas. The vapour pressure of metered-dose inhalers determines the aerosol droplet size, which, as discussed above, has an important influence on the efficiency of deposition in the lungs. The requirement of the Montreal Protocol in 1989 for the replacement of chlorofluorocarbon (CFC) propellants in pressurised metered-dose inhalers with hydrofluoroalkanes (HFAs) because of the ozone-depleting properties of CFCs led to a substantial review of the formulation of these devices as a consequence of major differences in physical and chemical properties of these propellants.19 The properties of the two most widely used HFAs, HFA 227 and HFA 134a, are summarised in Table 9.9.
Table 9.9 Physicochemical properties of hydrofluoroalkanes
Propellant | Molecular formula | Molecular weight | Boiling point (°C) at 1.013 bar (1 atm) | Gauge vapour pressure (psig) at 20°C |
HFA 134a | C2H2F4 | 102.0 | −26.5 | 68.4 |
HFA 227 | C3HF7 | 170.0 | −17.3 | 56.0 |
Data from reference 19.
The vapour pressure above an aerosol system in which there is an equilibrium between the liquefied propellant and its vapour can be calculated using Raoult’s law,

where pi is the partial vapour pressure of a component i in the vapour phase, xi is the mole fraction of that component in solution and 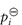 is the vapour pressure of the pure component. Its application to this type of aerosol system is illustrated in Example 9.1.
is the vapour pressure of the pure component. Its application to this type of aerosol system is illustrated in Example 9.1.
|
Assuming ideal behaviour, calculate the vapour pressure at 20°C above an aerosol mixture consisting of 30% w/w of HFA 134a (tetrafluoroethane, molecular weight 102 Da) with a vapour pressure of 68.4 psig and 70% w/w of HFA 227 (heptafluoropropane, molecular weight 170 Da) with a vapour pressure of 56.0 psig. Convert the pressure into pascals using the relationship between the various units of pressure shown in Box 9.1. Answer For the two propellants HFA 134a and HFA 227 with respective vapour pressures
where p134 and p227 are the partial pressures of components HFA 134a and HFA 227 respectively, and x134 and x227 are the mole fractions of these components in the liquid phase.
From Dalton’s law of partial pressures, the total vapour pressure P is the sum of the partial pressures of the component gases, assuming ideal behaviour. Thus,
and hence
Converting pressures into pascals (Pa) using
we obtain
|

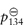
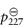


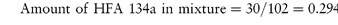
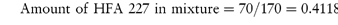
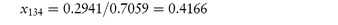
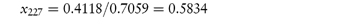

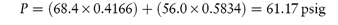
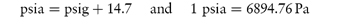

Box 9.1 Pressure units |
In practice, pressure is expressed in terms of a wide range of units. The SI unit is the pascal (Pa), where 1 Pa = 1 N m-2. Pressure should usually be converted to this unit before substitution into equations. The relationship of other commonly used pressure units to the pascal is as follows: 1 bar = 105 Pa 1 mmHg = 1 torr = 133.32 Pa 1 atmos = 1.013 × 105 Pa 1 psi* = 6894.76 Pa Standard atmospheric pressure is 760 mmHg = 760 torr = 1.013 bar = 1.013 × 105 Pa. *It is common to report vapour pressure of propellants as pounds per square inch gauge, psig. Gauge pressure uses the actual atmospheric pressure as the zero point for measurement and hence atmospheric pressure (14.7 psi at sea level) must be added to measurements quoted in psig to obtain the absolute pressure in pounds per square inch, psia, i.e. |
Binary mixtures of hydrofluoroalkanes show behaviour that approaches ideality.20 Figure 9.39a shows the vapour pressure–composition plots for a mixture of the propellants HFA 134a (tetrafluoroethane) and HFA 227 (heptafluoropropane); the linearity of the plots indicates that Raoult’s law is obeyed over the temperature range examined. It is frequently necessary to include a cosolvent such as alcohol in the aerosol formulation to enhance the solvent power of the propellant blend.21 Figure 9.39b shows large positive deviations from Raoult’s law due to interactions between the components of the formulation. In practical terms such behaviour is beneficial as it enables substantial addition of ethanol without reduction in vapour pressure and aerosol performance.
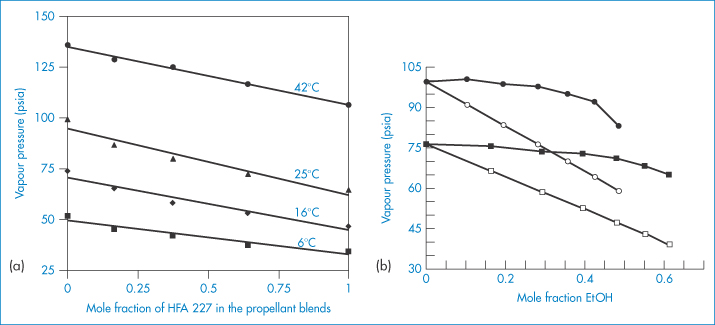
Figure 9.39 (a) Plots of vapour pressure versus mole fraction of HFA 227 for propellant systems composed of HFA 134a and HFA 227 at 6, 16, 25 and 42°C showing ideality of mixing (Raoult’s law obeyed). (b) Plots of vapour pressure of HFA 134a (circles) and HFA 227 (squares) versus mole fraction of ethanol at 21.5°C. Solid symbols represent experimental data; open symbols represent theoretical values calculated assuming ideal (Raoult’s law) behaviour.
(a) From Williams RO, Lie I. Int J Pharm 1998; 166:99–103.
(b) From Vervaet C, Byron PR. Int J Pharm 1999; 186:13–30.
Optimisation of vapour pressure, drug stability, solubility and spray patterns takes place during the design stage. Other ingredients of the formulation can include surfactants to act as solubilisers, stabilisers or lubricants to ease the passage of the particles when emitted from the valve.
The Autohaler has been devised as a breath-activated pressurised inhaler system because of the difficulty experienced by some patients in coordinating manual operation of an aerosol with inhalation. The Autohaler is activated by the negative pressure created during the inhalation phase of respiration and is specifically designed to respond to shallow inhalation in those with restricted pulmonary capacity.
Nebulisers22
Modern nebulisers for domestic and hospital use generate aerosols continuously for chronic therapy of respiratory disorders. A Venturi-type system is shown in Fig. 9.40 and an ultrasonic device in Fig. 9.40b. The particle size distribution and hence efficiency of such systems vary with the design and sometimes with the mode of use. Hence adequate monitoring of particle size is important. In vitro analysis of the particle size distribution of aerosols and nebulisers is discussed in Chapter 15.
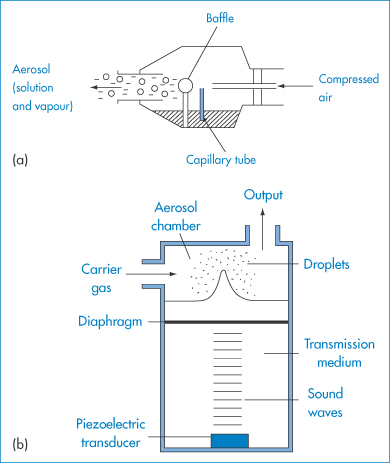
Figure 9.40 Schematic diagrams of (a) a Venturi-type nebuliser and (b) an ultrasonic nebuliser.
Modified with permission from Egan DF. Fundamentals of Respiratory Therapy, 3rd edn. New York: Mosby; 1977.
Other mechanisms of generating solid or liquid aerosols are available, such as vibrating mesh devices where the material to be inhaled is directed via the mesh, as shown simply in Fig. 9.41.
Figure 9.41 Vibrating mesh technology in which a mesh/membrane with 1000–7000 laser-drilled holes vibrates at the exit of a liquid reservoir, forcing a mist of very fine droplets through the holes. For further discussion, see www.ers-education.org (Vicellio L. Breathe 2006; 2:253–260).
|
|

9.9 The nasal route
Two main classes of medicinal agents are applied by the nasal route:
1. drugs for the alleviation of nasal symptoms
2. drugs that are inactivated in the gastrointestinal tract following oral administration and where the route is an alternative to injection, such as for peptides and proteins.
Intranasal beclometasone dipropionate in a dose as low as 200 μg daily is a useful addition to the therapy of perennial rhinitis. Considerable attention is being paid to the delivery by the nasal route of peptides and proteins such as insulin, luteinising hormone-releasing hormone (LHRH) analogues such as nafarelin, as well as vasopressin, thyrotropin-releasing hormone analogues and adrenocorticotrophic hormone.
Figure 9.42 shows the structures involved in delivery by the nasal route. Formulations have to be efficiently delivered to the epithelial surfaces: what physical factors affect the utility of this route? Factors such as droplet or particle size that affect deposition in the respiratory tract are involved if administration is by aerosol, but formulations may also be applied directly to the nasal mucosa. The physiological condition of the nose, its vascularity and mucus flow rate are therefore of importance. So too is the formulation used – the volume, concentration, viscosity, pH and tonicity of the applied medicament can affect activity.23 As the condition of the nasal passages changes with changes in the environment, temperature and humidity, it is clearly not an ideal route for absorption of drugs or vaccines, but may be the only feasible route for some agents. As with all routes, however, absorption decreases with the increasing molecular weight of the active, as seen in Fig. 9.43 for a series of model molecules.
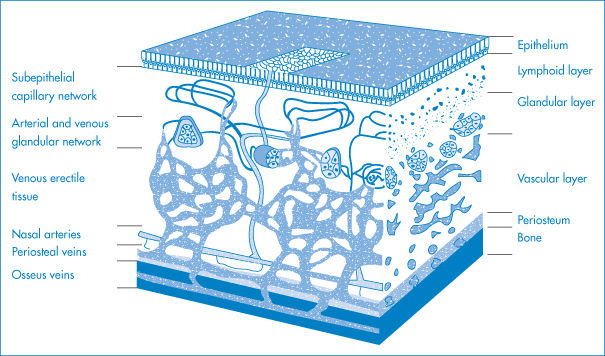
Figure 9.42 The vascular network of the nasal mucous membrane in the inferior turbinate.
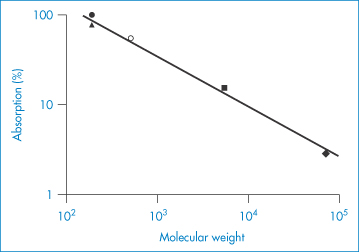
Figure 9.43 Correlation between the percentage dose absorbed after nasal administration and molecular weight: ●, 4-oxo-4H-1-benzopyran-2-carboxylic acid (chromocarb); ▲, p– aminohippuric acid; ○, sodium cromoglicate; ■, inulin; ♦, dextran; r = −0.996.
Reproduced from Fisher AN et al. The effect of molecular size on the nasal absorption of water-soluble compounds in the albino rat. J Pharm Pharmacol 1987;39:357. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
The air passages through the nasal cavity begin at the nares (nostrils) and terminate at the choanae (posterior nares). Immediately above the nares are the vestibules, lined by skin that bears relatively coarse hairs and sebaceous glands in its lower portion. The hairs curve radially downward, providing an effective barrier to the entry of relatively large particles. The division of the nasal cavity exposes the air to maximal surface area. As in the other parts of the respiratory tree, sudden changes in the direction of airflow cause impingement of large particles through inertial forces. The respiratory portion of the nasal passage is covered by a mucous membrane that has a mucous blanket secreted in part by the goblet cells. The ciliary streaming here is directed posteriorly so that the nasal mucus is transported towards the pharynx. Figure 9.44 shows the fractional deposition of inhaled particles in the nasal chamber as a function of their particle size. A diameter of not less than 10 μm minimises the loss of drug to the lung. In the external nares, removal of particles occurs on nasal hairs; further up inertial deposition takes place; and in the more tortuous upper passages deposition is assumed to be by inertia and sedimentation.
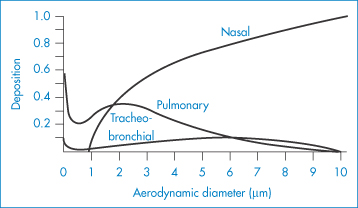
Figure 9.44 Regional deposition of inhaled particulate matter as a function of particle size; nose breathing at 15 respirations per minute and 730 cm3 tidal volume (the ‘pulmonary compartment’ refers to deposition beyond the terminal bronchiole).
Reproduced with permission from Muir CDF. Clinical Aspects of Inhaled Particles. London: Heinemann; 1972.
Comparison of the nasal route with other routes has been made in some instances. Desmopressin (1-desamino-8-D-arginine vasopressin), administered as 20 μg dose, elicits a response equivalent to approximately 2 μg administered by IV injection. A greater dose of virus is required to obtain an equivalent response to a nasal vaccine than when administered by other routes. The role of the intranasal route for the delivery of peptides and proteins has been widely researched (see Chapter 13); as can be inferred from above, the bioavailability of desmopressin acetate ranges from 9% to 20% in humans, buserelin 6% and salmon calcitonin 3% when compared with the IM route. Penetration enhancers can lead to increases in uptake, but insulin’s bioavailability in human subjects ranges from only 3% to 13% depending on the study involved.
In the treatment of nasal symptoms the patient adjusts the dose so that, perhaps, the theoretical bases of droplet and particle retention are less vital. Although formulation of the nasal drops, or sprays from plastic squeeze-bottles must obviously influence the efficiency of medication, little work has in the past been carried out relating formulation to the effect of intranasal medicines. Microsphere delivery systems have received some attention, however, with special interest being directed to bioadhesive microsphere carriers.
9.10 Rectal absorption of drugs
Drugs administered by the rectal route in suppositories are placed in intimate contact with the rectal mucosa, which behaves as a normal lipoidal barrier. The pH in the rectal cavity lies between 7.2 and 7.4, but the rectal fluids have little buffering capacity. As with topical medication, the formulation of the suppository can have marked effects on the activity of the drug. Factors such as retention of the suppository for a sufficient duration of time in the rectal cavity also influence the outcome of therapy; the size and shape of the suppository and its melting point may also determine bioavailability.
The once traditional suppository base cocoa butter (theobroma oil) is a variable natural product that undergoes a polymorphic transition on heating. It is primarily a triglyceride. Four polymorphic forms exist: γ, m.p. 18.9°C; α, m.p. 23°C; β′, m.p. 28°C; and the stable β form, m.p. 34.5°C. Heating above 38°C converts the fat to a metastable mixture solidifying at 15–17°C instead of 25°C, and this subsequently melts at 24°C instead of at 31–35°C. Reconversion to the stable β-form takes 1–4 days depending on storage conditions.
Modern bases include polyoxyethylene glycols of molecular weight 1000–6000 Da and semisynthetic vegetable fats. The appropriate bases must be selected carefully for each substance. The important features of excipient materials are melting point, speed of crystallisation and emulsifying capacity. If the medicament dissolves in the base, it is likely that the melting point of the base will be lowered, so that a base with a melting point higher than 36–37°C has to be chosen. If the drug substance has a high density, it is preferable that the base crystallises rapidly during production of the suppositories to prevent settling of the drug. Preservatives, hardening agents, emulsifiers, colouring agents and materials which modify the viscosity of the base after melting are common formulation additives.
9.10.1 The rectal cavity
The rectum is the terminal 15–19 cm of the large intestine. The mucous membrane of the rectal ampulla, with which suppositories and other rectal medications come into contact, is made up of a layer of cylindrical epithelial cells, without villi.
Figure 9.45 shows the blood supply to the rectal area. The main artery to the rectum is the superior rectal (haemorrhoidal) artery. Veins of the inferior part of the submucous plexus become the rectal veins, which drain to the internal pudendal veins. Drug absorption takes place through this venous network. Superior haemorrhoidal veins connect with the portal vein and thus transport drugs absorbed in the upper part of the rectal cavity to the liver; the inferior veins enter into the inferior vena cava and thus bypass the liver. The particular venous route the drug takes is affected by the extent to which the suppository migrates in its original or molten form further up the gastrointestinal tract, and this may be variable. The rectal route therefore does not necessarily, or even reproducibly, avoid the liver.
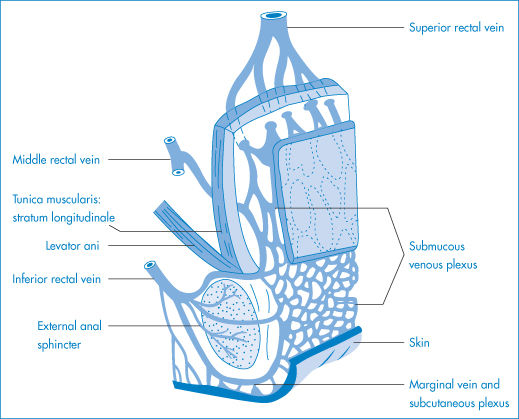
Figure 9.45 Blood supply to the rectum and anus. The significance of the location of the superior and the inferior rectal veins is discussed in the text.
Reproduced with permission from Tondury G. Topographical Anatomy. Stuttgart: Thieme; 1959.
A schematic representation (Fig. 9.46) shows the processes occurring following insertion of a suppository into the rectum. Cocoa butter suppositories usually liquefy within a few minutes, but the drug is not necessarily released from solution or suspension, as the fat in this case is not emulsified or absorbed. Surfactants may be required to aid dispersal of the fat and thus when this base is used the physicochemical properties of the drug are important. The rate-limiting step in drug absorption for suppositories made from a fatty base is the partitioning of the dissolved drug from the molten base, not the rate of solution of the drug in the body fluids.
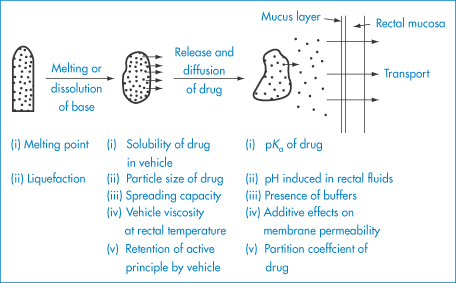
Figure 9.46 Schematic representation of rectal absorption of an active principle from a suppository, and the factors at each stage likely to affect the bioavailability of the drug.
Modified from Jaminet F. In: Guillot BR, Lombard AP (eds) The Suppository. Paris: Maloine; 1973.
The influence of the aqueous solubility on in vitro release from fat-based suppositories is shown in Fig. 9.47, the results being collated from the study of 35 drugs grouped into classes I–V in decreasing order of water solubility. The results may be explained as follows. The water-soluble active substances will be insoluble in the fatty base, while the less water-soluble material will tend to be soluble in the base, and will thus diffuse from the base more slowly. Water-soluble drugs are better absorbed from a fatty excipient than from a water-soluble one, and ethyl nicotinate, for example, which is lipid-soluble, is absorbed faster from a water-soluble excipient.
Type | Solubility in water |
I | 1 in 1 |
II | 1 in 10 |
III | 1 in 10 to 1 in 100 |
IV | 1 in 100 to 1 in 1000 |
V | 1 in 1000 to 1 in 10 000 |
Figure 9.47 Release of drugs of varying solubilities from fat-based suppositories of equal active agent content.
Modified from Voigt R, Falk G. Pharmazie 1968;23:709.
In a study of the various physical properties of suppositories, the most important parameter for the bioavailability of paracetamol was found to be rheological properties at 37°C. The relationship between the excretion of paracetamol (APAP; N-acetyl-p-aminophenol) and the rheology of the excipient drug suspension is shown in Fig. 9.48. The greater the limiting shear stress, τ, of the system, the lower the bioavailability of the drug.
Figure 9.48 Variation of the excretion of APAP (paracetamol) in the urine 2 hours after rectal administration of a formulation as a function of the limiting shear stress, τ, of the excipient–drug mixture at 37°C.
Modified with permission from Moës A. J Pharm Belg 1974;29:319.
Apparatus for studying the many variables in suppository formulation has been designed to measure rates of release in vitro. Both a circulating dissolution apparatus and a dialysis device utilising an aqueous and a non-aqueous phase have been described, and are discussed in Chapter 15. As the suppository base is heated before moulding, certain effects can be noted that are unique to this type of medication. Testosterone dissolves when hot in the semisynthetic excipient Witepsol H, to give, on cooling, crystals of about 2–3 μm in diameter. After dissolution in theobroma oil, the drug does not crystallise on cooling but remains dissolved as a solid solution. In the former case, high absorption rates are obtained, while in the latter poor absorption is achieved.
Absorption from the rectum depends on the concentration of drug in absorbable form in the rectal cavity and, if the base is not emulsified, on the contact area between molten excipient and rectal mucosa. Addition of surfactants may increase the ability of the molten mass to spread and tends to increase the extent of absorption. Significant increases in absorption can be obtained with polyoxyethylene sorbitan monostearate (polysorbate 60), sodium lauryl sulfate and cetyltriethylammonium (cetrimonium) bromide. Surfactants increase wetting and spreading and may increase the permeability of the rectal mucosal membrane.
A study of the effect of polyoxyethylene glycols on the rectal absorption of un-ionised sulfafurazole demonstrated the importance of the affinity of the drug substance for base and for rectal mucosa. As the amount of Macrogol 4000 (polyoxyethylene glycol 4000) was increased, the partition coefficient (lipid/vehicle) fell and absorption decreased correspondingly, because the affinity of the drug for the suppository base increased and its tendency to partition to the rectal lipids decreased. In a similar way, incorporation of water-soluble drug into a water-in-oil emulsified suppository base tends to decrease bioavailability as the drug is transferred in dispersed aqueous droplets in the molten base.
|
The hygroscopicity of some hydrophilic bases such as the polyoxyethylene glycols (each oxygen can interact with up to 4 water molecules under certain circumstances) results in the abstraction of water from the rectal mucosa. This causes a stinging sensation and discomfort and probably affects the passage of drugs across the rectal mucosa. The hygroscopicity of the glycols decreases as the molecular weight increases. The problem may be overcome by the incorporation of water into the base, although the presence of water may affect drug stability. Glycerogelatin bases are also hygroscopic. |

Deliberate incorporation of water into a formulation gives rise to the possibility that, on storage, water will be lost by evaporation and drug may crystallise as a result. Reactions between components of the formulation are more likely to occur in the presence of moisture than in its absence.
9.10.2 Incompatibility between base and drug
Various incompatibilities have been noted with polyoxyethylene glycol bases. Phenolic substances complex with glycol, probably by hydrogen bonding between the phenolic hydroxyl group and the glycol ether oxygens. Polyoxyethylene glycol bases are incompatible with tannic acid, ichthammol, aspirin, benzocaine, clioquinol and sulfonamides. High concentrations of salicylic acid alter the consistency of the bases to a more fluid state.
Glycerogelatin bases are prepared by heating together glycerin, gelatin and water. Although primarily used per se as an intestinal evacuant, the glycerogelatin base may be used to deliver drugs to the body. For this purpose the United States Pharmacopeia XVIII specified two types of gelatin to avoid incompatibilities. Type A is acidic and cationic, with an isoelectric point between pH 7 and 9; type B is less acidic and anionic, with an isoelectric point between pH 4.7 and 5. Use of untreated gelatin renders the base incompatible with acidic and basic drugs.
9.11 Drug administration to brain, spinal cord and tissues
Administration of drugs in solution by intrathecal catheter provides an opportunity to deliver drugs to the brain and spinal cord. Relatively hydrophilic drugs such as methotrexate (log P = −0.5) that do not cross the blood–brain barrier in significant amounts have been infused intrathecally to treat meningeal leukaemia, and baclofen (log P = −1.0) to treat spinal cord spasticity. High lumbar cerebrospinal fluid concentrations are achieved as a result. Figure 9.49 shows the anatomy of the epidural space and routes of drug transport. The spinal cerebrospinal fluid has a small volume (70 cm3) and a relatively slow clearance (20–40 cm3 h−1) of hydrophilic drugs.
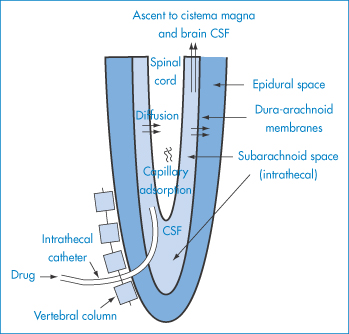
Figure 9.49 Anatomical structures and pathways of drug movement important in intrathecal drug administration; the lower part (sacral, lumbar) of the spinal cord is shown in this diagram.
Reproduced from Kroin JS. Intrathecal drug administration. Present use and future trends. Clin Pharmacokinet 1992;22:319, with kind permission from Springer Science and Business Media.
The intrathecal route is more invasive than IV, IM or SC routes. Both percutaneously implanted catheters and subcutaneously implantable pumps have been used to reduce the risk of infection on repeated puncture.
Diffusion of the drug following administration intrathecally is critical in determining activity. The cerebrospinal fluid pharmacokinetics of three drugs (morphine, log P = 0.15; clonidine, log P = 0.85; and baclofen, log P = −1.0) were found to be similar, leading to the suggestion that bulk flow mechanisms may be the dominant factor in determining distribution.
9.11.1 Brain implants
The brain offers many challenges in terms of treatment systemically because of the power of the blood–brain barrier to resist entry of certain drugs. It might be assumed that direct implantation of delivery systems into the brain, e.g. to treat brain tumours, might always be effective. However, the physical and chemical phenomena that control the transport of drugs in the brain tissue are complex and not fully understood.** The physicochemical issue is not necessarily the release of drug from any reservoir (e.g. polymer wafers) but the subsequent diffusion and spread of the drug into the surrounding tumour or other tissues.
A large variety of processes can be involved in the transport of the drug once it is released from the dosage form to its target site(s), including:
- diffusion within the extracellular space
- reversible and irreversible binding to the extracellular matrix (which comprises long-chain macromolecules)
- degradation of the dose form (implant or wafer) (e.g. by enzymes or hydrolysis)
- passive or active uptake into central nervous system cells (by diffusion or receptor-mediated internalisation)
- release from endolysosomes into the cytosol
- diffusion and convection within the cytosol of the cells
- uptake into the cell nuclei (where appropriate)
- bulk flow within the extracellular space
- direction-dependent drug transport (anisotropy), because the brain is not one homogeneous mass
and, of course, elimination into the blood stream.
Figure 9.50 shows the arrangement of Gliadel implants in brain tissue and the pathways of diffusion and interaction of a drug released from an implant in the brain.
Figure 9.50 (a, top) Implants of the polyanhydride-based Gliadel wafers containing carmustine (BCNU); drug release is controlled both by diffusion of the drug and erosion of the polymer matrix. (b, bottom) The pathways of diffusion and interaction of a drug released from implants. The results of the complex pathways in the brain tissues are shown by the data in Fig. 9.51.
(a) Reproduced with permission from Lesniak MS, Brem H. Targeted therapy for brain tumours. Nature Rev Drug Discov 2004;3:499–508.
The poor diffusion characteristics of one agent, human nerve growth factor, in the brain obviates the use of implants, as shown clearly in Fig. 9.51. This would require more direct intervention and application of the drug used as a prospective drug for the treatment of Alzheimer’s disease.
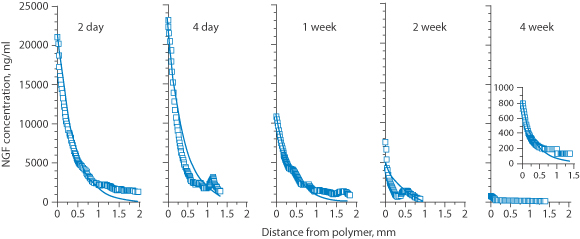
Figure 9.51 Concentration profiles of human nerve growth factor (NGF) in the vicinity of a polymer implant over 28 days. The solid lines represent the calculation of diffusion/elimination from days 2 and 4 and steady-state conditions thereafter. Note the small distance where levels are significant, some 1–2 mm from the implant surface.
Reproduced from Krewson CE, Saltzman WM. Transport and elimination of recombinant human NGF during long-term delivery to the brain. Brain Res 1996;727:169–181. Copyright Elsevier 1996.
9.11.2 Cardiovascular drug-eluting stents
A stent is a mesh ‘tube’ inserted into any natural passage such as a ureter or an artery to prevent or counteract a flow constriction. Cardiovascular stents are wire mesh tubular devices that are employed to enlarge diseased arteries. Early versions of cardiovascular stents caused problems of provoking clot formation and restenosis, a renewed narrowing of the vessel. A relatively new form of drug delivery has appeared in the form of ‘coated stents’ in which polymer coatings on the mesh are used to deliver drugs such as sirolimus (a drug that can prevent restenosis) directly to tissues (Fig. 9.52).
Figure 9.52 (a) A stent is mounted on a balloon catheter and advanced to the narrowed portion of the heart artery. (b) The balloon is inflated and the stent is expanded, opening the narrowed section of the artery. (c) The balloon is deflated and removed; the stent is embedded into the wall of the artery and stays in position.
Reproduced with permission from Maisel WH, Laskey WK. Cardiology patient page. Drug-eluting stents. Circulation 2007;115:e426–e427.
Some designs of stent use several polymers in several layers to control drug release, as in Fig. 9.53.
Figure 9.53 A diagrammatic representation of a polymer-coated stent (the Infinitum stent). The stent structure is coated with three different layers of polymers that have different functions from the outermost coat, which contains no drug but protects the underlying structures.
Some of the factors of importance in the design of new polymers for slow release of drugs on stents are shown in Fig. 9.54.
Figure 9.54 The range of properties of importance in the design of new polymers for slow release of drugs on stents.
Redrawn from Udipi K et al. J Biomed Mater Res A 2007; 85A:1064–1071.
Summary
The routes discussed in this chapter are sometimes simpler, and perhaps sometimes more complex than the oral route. In several of those we have covered there is a more intimate contact between the formulation and the absorbing surface – as in the topical and rectal routes – so the formulation and the thermodynamic activity of the drug in the vehicle become more directly important. In essence:
- Each route has its own special characteristics. Administration of the same dose of drug by different routes usually produces significantly different pharmacokinetics.
- The optimal lipophilicity of a drug to traverse the barrier membrane depends on the nature of the membrane.
- Some barriers as in the eye are complex, having the characteristics of typical lipid barriers, interspersed with more aqueous hurdles.
- In some cases, as with IM injections, the nature of the tissue into which the drug is injected, whether fatty or aqueous, is key to the process of transferring drug into the blood.
- No one equation can yet predict the pharmacokinetic profile of a drug delivered by a particular route in a particular formulation, but the equations presented in this chapter help to show the effect of key parameters.
- The overriding importance of lipophilicity is clear when drug is absorbed in molecular form.
- When drug is delivered as a suspension (as in an aerosol), the paramount importance of particle size in first getting the drug to the site of action is clear; once it has reached that site, its rate of solution and its lipophilicity are again important.
- There is no escaping the fact that the diffusion coefficient and flux of the active entity in the tissues concerned in each route of drug delivery have a paramount effect on the success or otherwise of a route. The body’s many biological and physical barriers present what we sometimes refer to as the complex realities of drug delivery and targeting, a topic to which we return in later chapters.
References
1. Balch CM et al. Continuous regional chemotherapy for metastatic colorectal cancer using a totally implantable infusion pump. A feasibility study in 50 patients. Am J Surg 1983;145:285–290.
2. Mitragotri S et al. Ultrasound-mediated transdermal protein delivery. Science 1995;269:850–853.
3. Figge FHJ, Barnett DJ. Anatomic evaluation of a jet injection instrument designed to minimise pain and inconvenience of parenteral therapy. Am Pract 1948;3:197–206.
4. Shramm J, Mitragotri S. Transdermal drug delivery by jet injectors: energetics of jet formation and penetration. Pharm Res 2002;19:1673–1679.
5. Murdan S. Drug delivery to the nail following topical application. Int J Pharm 2002;236:1–26.
6. Elkeeb R et al. Transungual drug delivery: current status. Int J Pharm 2010;384:1–8.
7. Tabbara KF, Sharara N. Dry eye syndrome. Drugs Today 1998;34:447.
8. Holly FJ. Formation and rupture of the tear film. Exp Eye Res 1973;15515–525.
9. Matsuo T et al. Trehalose eye drops in the treatment of dry eye syndrome. Ophthalmology 2002;109:2024–2029.
10. Sieg JW, Robinson JR. Vehicle effects on ocular drug bioavailability II: Evaluation of pilocarpine. J Pharm Sci 1977;66:1222–1228.
11. Chrai SS, Robinson JR. Binding of sulfisoxazole to protein fractions of tears. J Pharm Sci 1976;65:437–439.
12. Snell JW.. Ophthalmic drug delivery systems. Drug Deliv Res 1985;6:245–261.
13. Hull DS et al. Ocular penetration of prednisolone and the hydrophilic contact lens. Arch Ophthalmol 1974;92:413–416.
14. German E, Siddiqui N. Atropine toxicity from eyedrops. N Engl J Med 1970;282:689.
15. Borromeo-McGrail V et al. Systemic hypertension following ocular administration of 10% phenylephrine in the neonate. Paediatrics 1973;51:1032–1036.
16. Benziger DP, Edelson J. Absorption from the vagina. Drug Metab Ther 1983;141:37–68.
17. Kunth K et al. Hydrogel delivery systems for vaginal and oral applications. Formulations and biological considerations. Adv Drug Deliv Rev 1993;11:137–167.
18. Cox JSG et al. Disodium cromoglycate (Intal). Adv Drug Res 1970;5:115–196.
19. McDonald KJ, Martin GP. Transition to CFC-free metered dose inhalers-into the new millennium. Int J Pharm 2000;201:89–107.
20. Williams RO, Lie L. Influence of formulation additives on the vapor pressure of hydrofluoroalkane propellants. Int J Pharm 1998;166:99–103.
21. Vervaet C, Byron P. Drug–surfactant–propellant interactions in HFA-formulations. Int J Pharm 1999;186:13–30.
22. O’Callaghan C, Barry PW. The science of nebulised drug delivery. Thorax 1997;52(Suppl. 2):S31–S44.
23. Constantino HR et al. Intranasal delivery: physicochemical and therapeutics aspects. Int J Pharm 2007;337:1–14.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


