Figure 38–1 Comparison of the cell envelopes of gram-positive and gram-negative bacteria. (A) The gram-positive bacterium has a thick cell wall but does not have an outer membrane. β-Lactamases are located in the outer portion of the cell wall. Penicillin-binding proteins are found in the cytoplasmic membrane. (B) The gram-negative bacterium has a thin cell wall. It also has an outer membrane that contains lipopolysaccharide and protein channels called porins.
Cytoplasmic and Outer Membranes
The cytoplasmic membrane is a trilaminar membrane. It contains various types of transport proteins, which facilitate the uptake of a wide variety of substrates used by bacteria, and it also contains several enzymes required for the synthesis of the cell wall. These enzymes, whose functions are described later in this chapter, are collectively known as penicillin-binding proteins (PBPs).
The outer membrane of gram-negative bacteria is also a trilaminar membrane. It contains species-specific forms of a complex lipopolysaccharide and various types of protein channels called porins. One portion of lipopolysaccharide (the lipid A portion) is the endotoxin responsible for gram-negative sepsis. This endotoxin activates immunologic mechanisms that lead to fever, platelet aggregation, increased vascular permeability, and other adverse effects on tissues. Porins allow ions and other small hydrophilic molecules to pass through the outer membrane, and they are responsible for the entry of several types of antibiotics. Acquired alterations in porin structure can lead to microbial resistance to antibiotics, such as is the case with resistance to imipenem.
The bacterial cytoplasmic membrane is the target of two peptide antibiotics, daptomycin and polymyxin. These drugs act directly on the cell membranes to increase membrane permeability and thereby cause the cytoplasmic contents to leak out of the cell. The properties and uses of these antibiotics are discussed in Chapter 40.
Cell Wall
The cell wall consists primarily of peptidoglycan, a polymer constructed from repeating disaccharide units of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc). Each disaccharide is attached to others through glycosidic bonds. Each molecule of MurNAc has a peptide containing two molecules of D-alanine and a pentaglycine side chain (Fig. 38–2). The strands of peptidoglycan in the cell wall are cross-linked by a transpeptidase reaction in which the glycine pentapeptide of one strand is attached to the penultimate D-alanine molecule of another strand. During this reaction, the terminal D-alanine is removed.
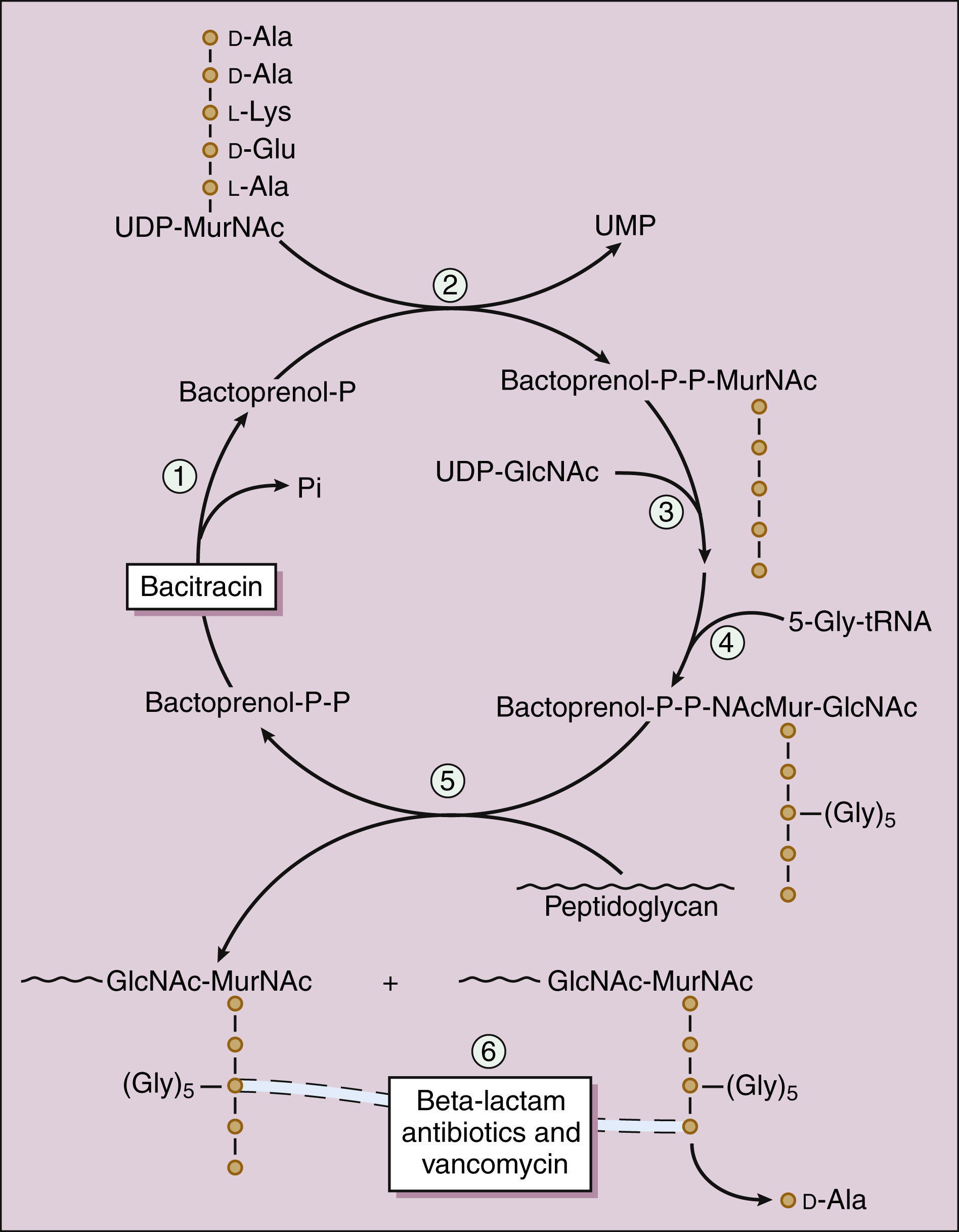
Figure 38–2 Sites of action of bacterial cell wall synthesis inhibitors. The bacterial cell wall consists primarily of peptidoglycan, a polymer constructed from repeating disaccharide units of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc). Numbers indicate the steps involved in the synthesis of the cell wall of Staphylococcus aureus. In step 1, bactoprenol pyrophosphate (bactoprenol-P-P) is dephosphorylated to regenerate the carrier molecule, bactoprenol phosphate (bactoprenol-P). In step 2, uridine diphosphate-MurNAc (UDP-MurNAc) is added. In steps 3 and 4, UDP-GlcNAc and a glycine pentapeptide (5-Gly-tRNA) are added. In step 5, the disaccharide peptide is transferred to the peptidoglycan growth point. In step 6, the cross-linking of peptidoglycan strands is catalyzed by transpeptidase, a type of penicillin-binding protein. In this reaction, a glycine of one strand forms a peptide bond with the penultimate D-alanine of an adjacent strand, and the terminal D-alanine is released. Bacitracin blocks step 1 and β-lactam antibiotics and vancomycin block step 6 by different mechanisms. Fosfomycin (not shown) inhibits enolpyruvyl transferase, the enzyme that catalyzes the condensation of UDP-GlcNAc with phosphoenolpyruvate to synthesize UDP-MurNAc.
The cell wall maintains the shape of the bacterium and protects it from osmotic lysis if it is placed in a hypotonic medium. Without a cell wall, the bacterium is unprotected. This is why inhibition of cell wall synthesis by antimicrobial drugs is usually bactericidal. Because a cell wall is not found in higher organisms, antimicrobial drugs can inhibit its formation without harming host cells. The cell wall is synthesized during bacterial replication, and drugs that inhibit cell wall synthesis are more active against rapidly dividing bacteria than they are against bacteria in the resting or stationary phase. For the same reason, the effectiveness of cell wall inhibitors is usually reduced by concurrent administration of bacteriostatic antibiotics that slow the growth of bacteria.
SITES OF DRUG ACTION
β-Lactam Drugs
The β-lactam antibiotics bind to a group of bacterial enzymes, the PBPs. These enzymes are anchored in the cytoplasmic membrane and extend into the periplasmic space. The PBP are responsible for the assembly, maintenance, and regulation of the peptidoglycan portion of the bacterial cell wall. Some of the PBP have transpeptidase activity, whereas others have carboxypeptidase and transglycosylase activity.
The β-lactam antibiotics form a covalent bond with PBPs and thereby inhibit the catalytic activity of these enzymes. Inhibition of some PBPs prevents elongation or cross-linking of peptidoglycan (see Fig. 38–2), whereas inhibition of other PBPs leads to the bacterium’s autolysis or to its change to a spheroplast or a filamentous form.
Each bacterial species has a set of unique PBPs to which particular β-lactam antibiotics bind with varying affinities. This partly accounts for the variation in the sensitivity of different organisms to β-lactam antibiotics.
Other Drugs
Bacitracin and fosfomycin inhibit cell wall peptidoglycan synthesis by blocking specific steps in the formation of the disaccharide precursor, MurNAc-GlcNAc. As shown in Figure 38–2, bacitracin inhibits the dephosphorylation of bactoprenol pyrophosphate, which is the carrier lipid required for regeneration of bactoprenol phosphate, the active carrier of the disaccharide precursor. Fosfomycin inhibits enolpyruvyl transferase, the enzyme that catalyzes the condensation of uridine diphosphate–GlcNAc (UDP-GlcNAc) with phosphoenolpyruvate to synthesize UDP-MurNAc. Vancomycin binds tightly to the D-alanyl-D-alanine portion of the peptidoglycan precursor and prevents bonding of the penultimate D-alanine to the pentaglycine peptide during cross-linking of peptidoglycan strands.
β-LACTAM ANTIBIOTICS
The β-lactam antibiotics include penicillins, cephalosporins, carbapenems, and a monobactam antibiotic.
Penicillins
Penicillins were the first antibiotics to be isolated from microorganisms and used to treat bacterial infections. Alexander Fleming is credited with the discovery of penicillin, but he was unable to isolate the substance in sufficient purity and quantity for clinical use. Later, E. B. Chain and H. W. Florey, working in England, obtained enough penicillin to establish its clinical effectiveness. The production of sufficient quantities of penicillin for widespread use around the world was made possible by advances in microbial fermentation technology in the United States.
Penicillins can be grouped according to their antimicrobial activity. Narrow-spectrum penicillins include penicillin G and penicillin V. Penicillinase-resistant penicillins include dicloxacillin and nafcillin. Extended-spectrum penicillins include amoxicillin, ampicillin, piperacillin, and ticarcillin.
CHEMISTRY
The penicillins consist of a β-lactam ring fused to a thiazolidine ring, with an R group that is unique for each antibiotic (Fig. 38–3). The natural penicillins isolated from strains of Penicillium were originally assigned letter designations because their chemical structures could not be identified at that time. Penicillin G and penicillin V are the only natural penicillins still used today, and they are classified as narrow-spectrum drugs. Semisynthetic penicillins are produced by substituting a different R group for the R group of a natural penicillin. The penicillinase-resistant penicillins have a large, bulky R group that protects them from hydrolysis by staphylococcal β-lactamase.
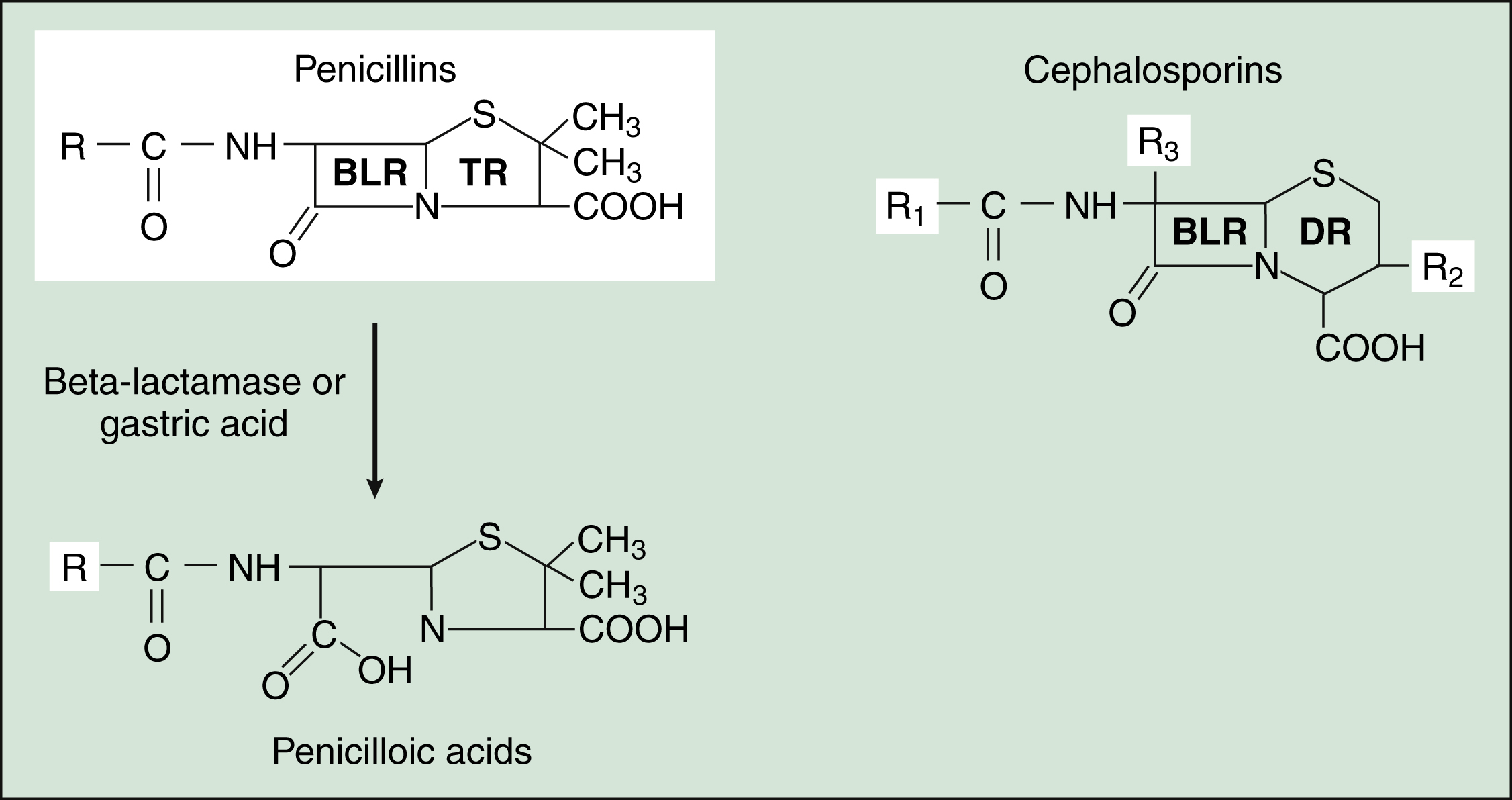
Figure 38–3 Structures of β-lactam drugs. Penicillins contain a β-lactam ring (BLR) and a thiazolidine ring (TR), whereas cephalosporins contain a BLR and a dihydrothiazine ring (DR). β-Lactamases hydrolyze the BLR and convert penicillins to penicilloic acids, which may react with body proteins to form antigens. β-Lactamases convert cephalosporins to inactive cephalosporanic acids (not shown).
PHARMACOKINETICS
The route of administration of penicillins depends on the stability of the drugs in gastric acid. Acid-stable penicillins, which include amoxicillin, dicloxacillin, and penicillin V, are effective when given orally. In contrast, acid-labile penicillins, which include piperacillin and ticarcillin, must be administered parenterally. Penicillin G has intermediate sensitivity to gastric acid and can be given orally in large doses, but penicillin V has better acid stability and oral bioavailability.
The penicillins are widely distributed to organs and tissues except the central nervous system. Because penicillins readily penetrate the cerebrospinal fluid when the meninges are inflamed (Fig. 38–4), they can be administered intravenously for the treatment of meningitis.
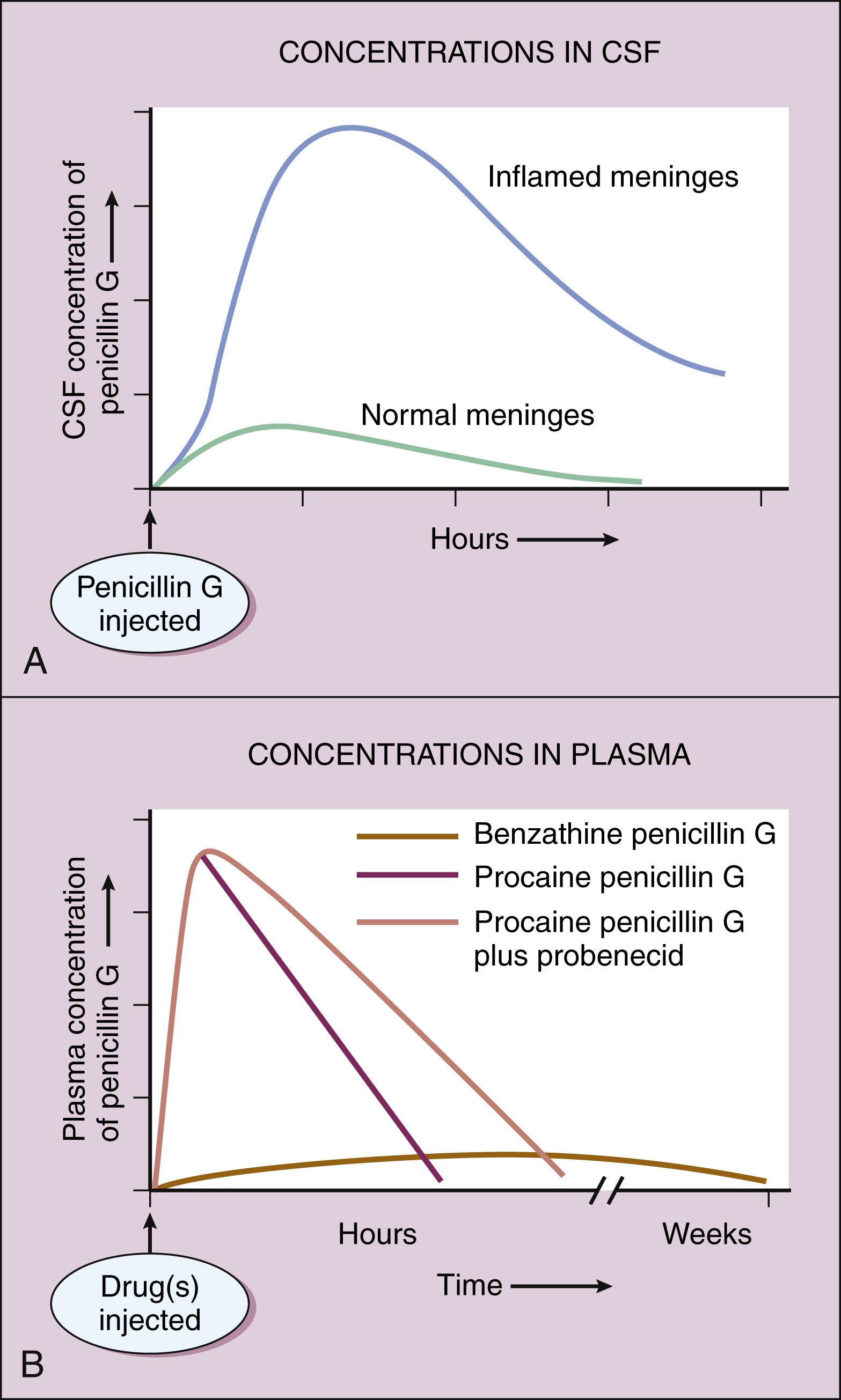
Figure 38–4 Pharmacokinetics of penicillin G preparations. (A) When penicillin G is administered to patients with normal meninges, the concentration in cerebrospinal fluid (CSF) remains low, but when it is administered to patients with meningitis, the CSF concentration is much higher because meningeal inflammation increases meningeal permeability to penicillin G. (B) Administration of benzathine penicillin G produces low plasma concentrations of the drug for several weeks. Administration of procaine penicillin G produces higher plasma concentrations for about 24 hours. Probenecid inhibits the renal excretion of penicillin G, prolongs its half-life, and increases its plasma concentrations.
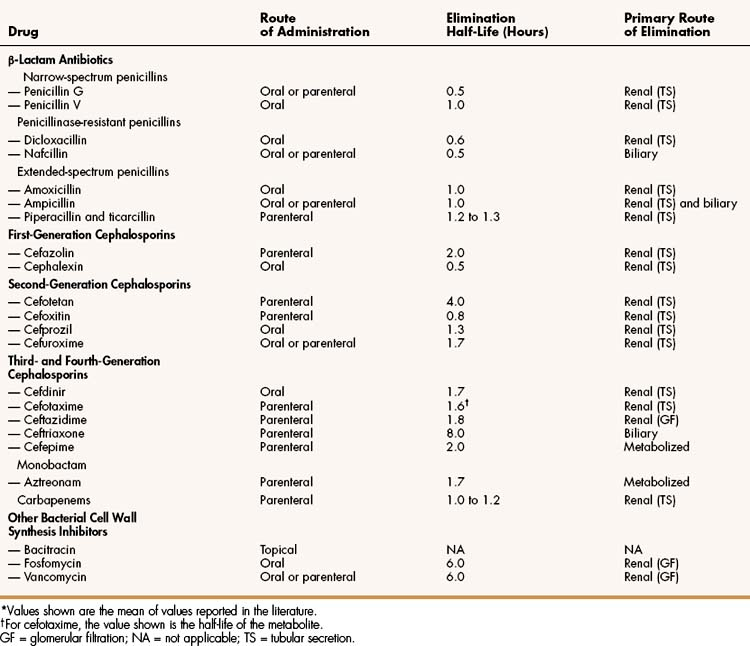
Most penicillin antibiotics are eliminated primarily by active renal tubular secretion and have short half-lives of about 0.5 to 1.3 hours (Table 38–1). A few penicillins (e.g., ampicillin and nafcillin) are excreted primarily in the bile. The renal tubular secretion of penicillins is inhibited by probenecid, a drug that competes with penicillins for the organic acid transporter located in the proximal tubule. Probenecid has been used to slow the excretion and prolong the half-life of penicillin G (see Fig. 38–4).
Penicillin G is available in two long-acting forms for intramuscular administration, procaine penicillin G and benzathine penicillin G. Penicillin G is slowly released from these two preparations for absorption into the circulation following an intramuscular injection. Benzathine penicillin G provides low plasma concentrations of the drug for several weeks, whereas procaine penicillin G produces higher plasma concentrations of penicillin for about 24 hours (see Fig. 38–4B).
SPECTRUM AND INDICATIONS
Table 38–2 outlines the spectrum and major clinical uses of penicillins.
TABLE 38–2 Major Clinical Uses of Selected Bacterial Cell Wall Synthesis Inhibitors
| Drug | Infections | Major Pathogens |
|---|---|---|
| Penicillins | ||
| Penicillin G | Syphilis | Treponema pallidum |
| Endocarditis | Viridans streptococci, enterococci | |
| Meningitis | Meningococci | |
| Pneumonia | Pneumococci | |
| Various | Streptococci | |
| Penicillin V | Pharyngitis | Streptococcus pyogenes |
| Nafcillin, oxacillin | Osteomyelitis, endocarditis, pneumonia, skin/soft tissue | Staphylococcus aureus |
| Amoxicillin ± clavulanate | Otitis, upper respiratory tract, pneumonia, skin/soft tissue, urinary tract | Pneumococci, streptococci, staphylococci, Haemophilus influenzae, Moraxella catarrhalis, E. coli, Pasteurella multocida |
| Ampicillin ± sulbactam | Meningitis | Listeria monocytogenes |
| Decubitus and diabetic foot ulcers | Gram-positive and anaerobic organisms | |
| Endocarditis | Streptococci, enterococci | |
| Lyme disease | Borrelia burgdorferi | |
| Piperacillin ± tazobactam | Intra-abdominal, skin/soft tissue, pneumonia, and other | Aerobic and anaerobic organisms, including Pseudomonas aeruginosa |
| Cephalosporins | ||
| Cephalexin | Skin and soft tissue infections | Streptococci, staphylococci |
| Cefazolin | Perioperative prophylaxis | Staphylococci, E. coli |
| Cefotetan | Intra-abdominal, gynecologic, biliary tract | Aerobic and anaerobic bacilli |
| Cefdinir, cefprozil, cefuroxime axetil | Respiratory tract, skin/soft tissue | Pneumococci, Haemophilus influenzae, Moraxella catarrhalis |
| Ceftriaxone | Gonorrhea,∗ urinary tract, otitis, meningitis, pneumonia, Lyme disease | Gonococci, pneumococci, meningococci, Borrelia burgdorferi, H. influenzae, other gram-negative bacilli |
| Ceftazidime | Urinary tract, pneumonia, others | Pseudomonas aeruginosa |
| Cefepime | Intra-abdominal, urinary tract, pneumonia, skin/soft tissue | Drug-resistant gram-negative bacilli, including Citrobacter and Enterobacter species |
| Carbapenems | Intra-abdominal, meningitis, febrile neutropenia | Aerobic, anaerobic, and drug-resistant gram-negative bacilli |
| Monobactam (aztreonam) | Urinary tract, gynecologic, intra-abdominal, skin, lungs, and others | Aerobic, gram-negative bacilli |
| Vancomycin | Bone and joint, skin and soft tissue, pneumonia, septicemia, endocarditis, and others | Methicillin-resistant staphylococci, enterococci, and others |
| Bacitracin | Skin and eye | Staphylococci, streptococci |
| Fosfomycin | Lower urinary tract | E. coli, Enterococcus faecalis, others |
∗ Also cefpodoxime as a single oral dose.
The narrow-spectrum penicillins, penicillins G and V, are used to treat infections caused by sensitive strains of streptococci (including pneumococci), meningococci, and spirochetes (e.g., Treponema pallidum). For example, penicillin G is used to treat group A streptococcal infections and to treat syphilis. Penicillin G is also active against Clostridium perfringens, the cause of gas gangrene, and other pathogens. Most staphylococci and gonococci and some strains of pneumococci are now resistant to penicillin G.
The penicillinase-resistant penicillins (e.g., dicloxacillin and nafcillin) were developed to treat penicillin-resistant strains of staphylococci that elaborate penicillinase (β-lactamase). These penicillins are not active against most other species of penicillinase-producing bacteria. Nafcillin is usually preferred when parenteral administration is required, whereas dicloxacillin can be given orally for less severe infections.
The penicillinase-resistant drugs are used to treat serious staphylococcal infections, such as acute endocarditis and osteomyelitis, as well as skin and soft tissue infections. Staphylococci that are resistant to these penicillins are often designated methicillin-resistant Staphylococcus aureus(MRSA) because methicillin was the original drug in this class. Methicillin is seldom used today, however, because of its greater toxicity and tendency to cause interstitial nephritis. Bacteria that are resistant to methicillin are also cross-resistant to nafcillin and all other penicillinase-resistant penicillins. Most strains of methicillin-resistant Staphylococcus aureus are also resistant to cephalosporins.
The extended-spectrum penicillins can be subdivided into the aminopenicillins (amoxicillin and ampicillin) and the antipseudomonal penicillins (piperacillin, ticarcillin, and others).
The aminopenicillins are active against many streptococci, some strains of enterococci, and a limited number of gram-negative bacilli. Amoxicillin and ampicillin are often combined with a β-lactamase inhibitor (described below), and drug products containing amoxicillin and clavulanate (Augmentin) or ampicillin and sulbactam (Unasyn) are widely used to treat infections caused by β-lactamase–producing bacteria.
Amoxicillin can be used alone to treat respiratory tract infections caused by sensitive bacteria, including otitis media, sinusitis, bronchitis, and community-acquired pneumonia. Many strains of Haemophilus influenzae and Moraxella catarrhalis, however, produce penicillinase, and infections caused by these organisms should be treated with amoxicillin-clavulanate. In addition, many strains of Streptococcus pneumoniae (pneumococci) have become increasingly resistant to amoxicillin, necessitating the use of larger doses to treat upper respiratory infections caused by this organism. For this reason, two formulations of amoxicillin-clavulanate have been developed to provide higher plasma levels of these drugs for longer periods of time. A sustained release tablet containing 1000 and 62.5 mg of amoxicillin and clavulanate, respectively, is given twice daily for treatment of adult respiratory tract infections caused by drug-resistant pneumococci, H. influenzae, or M. catarrhalis. A liquid suspension providing 90 and 6.4 mg/kg/day of amoxicillin-clavulanate is available to treat children with otitis media (Box 38–1) due to resistant strains of pneumococci, H. influenzae, or M. catarrhalis. For infections not caused by resistant organisms, the usual dose of amoxicillin is 30 to 45 mg/kg/day in two divided doses.
BOX 38–1 A CASE OF COUGH, NASAL CONGESTION, AND IRRITABILITY
CASE PRESENTATION
A previously healthy 18-month-old infant is brought to her pediatrician with cough, nasal congestion, and irritability. Examination reveals a temperature of 39° C and redness and bulging of the tympanic membrane under pneumatic otoscopy, suggesting acute otitis media. Because of the possibility of an infection caused by pneumococci with intermediate penicillin resistance, she is placed on amoxicillin at a dose of 90 mg/kg/day in three divided doses for 10 days. Her mother is instructed to contact the pediatrician if the infant does not respond to treatment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


