Characteristic
Hepatitis B virus
Hepatitis C virus
Type of virus
Hepadnavirus
Hepacivirus
Viral genome
3.2 kb, relaxed circular, partially double-stranded DNA
9.5 kb, positive-sense, single-stranded RNA
Routes of transmission
Parenteral
Parenteral
Sexual
Injection drug use
Injection drug use
Blood products before 1990
Perinatal
Perinatal (infrequent)
Sexual (infrequent)
Frequency of acute icteric disease
Common in adults
Uncommon
Uncommon in children
Frequency of evolution to chronic disease
Infrequent (<10 %) in adults
Frequent (>80 %)
Common in children
Estimated number of acute infections/year in the USA
185,000
38,000
Estimated number of chronically infected persons in the USA
1,250,000
2,700,000
Estimated number of chronically infected persons in the world
350,000,000
170,000,000
Treatment
Interferon pegylated interferon-α
Pegylated interferon-α with:
Nucleoside/nucleotide analogs
Ribavirin and Protease inhibitors or RNA polymerase inhibitors or NS5A inhibitors RNA polymerase inhibitor and Ribavirin alone
Prophylaxis
Recombinant Hepatitis B vaccine
None
Immune globulin
Hepatitis B Virus
Description of the Pathogen
The HBV genome is a 3.2 kilobase relaxed circular, partially double-stranded DNA molecule. It has four partially overlapping open reading frames encoding the viral envelope (pre-S and S), nucleocapsid (precore and core), polymerase, and X proteins. After binding to hepatocytes, the virion is taken up into the cell by endocytosis and uncoated. The partially double-stranded DNA genome is converted to a covalently closed circular DNA (cccDNA) in the cell nucleus. The cccDNA is used as a template for transcription of the pregenomic RNA (pgRNA) and messenger RNA in the cell nucleus. The pgRNA moves into the host cell cytoplasm and serves as the template for translation of the HBV reverse transcriptase (RT) as well as the core protein by the cellular translational proteins. Concurrently, the HBV RT reverse transcribes the pgRNA to a new circular DNA molecule. Early in the replication cycle, some of the newly synthesized genomes will circulate back to the nucleus to maintain and increase the pool of cccDNA [3].
Although HBV is a DNA virus, it replicates by a RT that lacks proofreading activity and, as a result, is prone to errors. The overlapping open reading frames of the genome limit the types of mutations that can be tolerated. However, variations in HBV sequences have been detected in almost all regions of the genome. Consequently, HBV exists as quasispecies, and different patients may be infected with different strains and genotypes.
Seven phylogenetic genotypes (A through H) of HBV have been identified, most of which have distinct geographic distribution. Genotypes are defined by intergroup divergence of greater than 8 % in the complete genome nucleotide sequence. All known genotypes have been found in the USA with the prevalence of A, B, C, D, and E–G being 35 %, 22 %, 31 %, 10 %, and 2 %, respectively [4]. Recent data suggest that HBV genotype plays an important role in the progression of HBV-related liver disease as well as response to interferon alpha (IFN-α) and pegylated IFN-α; however, HBV genotyping is not necessary in routine clinical practice [5].
Clinical Utility
Serologic assays with high levels of sensitivity, specificity, and reproducibility have been developed to detect HBV antigens and their respective antibodies. This complicated system of serologic markers is used for diagnosis of HBV infection and to define the phase of infection, degree of infectivity, prognosis, and the patient’s immune status. The presence of HBV DNA in the serum is a marker of viral replication in the liver and has replaced hepatitis B e antigen (HBeAg) as the most sensitive marker of viral replication. HBeAg is the extracellular form of the hepatitis B core protein. Molecular assays to quantify blood levels HBV DNA are useful for the initial evaluation of HBV infections, monitoring of patients with chronic infections, and assessing the efficacy of antiviral treatment [3, 5]. In addition, US blood donors are routinely screened for HBV DNA by qualitative tests to detect donors in the early stage of infection [6]. Antiviral resistance mutations are detected by molecular methods that identify known mutations associated with drug resistance.
The initial evaluation of patients found to have HBsAg in serum should include routine liver tests and a variety of virologic tests including HBV DNA tests [5]. Chronic HBV infection is a disease of variable course, and establishing baseline laboratory values at the time of diagnosis is important clinically for the tracking of disease progression over time and to evaluate candidates for liver biopsy. Monitoring disease activity in chronically HBV-infected patients is best done by measuring aminotransferase (ALT) levels at regular intervals in HBeAg-positive patients. However, serial HBV DNA testing is recommended in HBeAg-negative patients. The determination of serum HBV DNA levels (viral load) is important in the pretreatment evaluation and monitoring of therapeutic response in patients with chronic infection [5]. Currently, therapy for chronic HBV infection does not eradicate the virus and has limited long-term efficacy. The decision to treat should be based on ALT elevations, the presence of HBeAg or HBV DNA viral load of >2,000 IU/ml or both, the presence of moderate disease activity and fibrosis on liver biopsy, and virologic testing to exclude concurrent infections with hepatitis D virus (HDV), HCV, and human immunodeficiency virus (HIV). The goals of treatment of chronic hepatitis B are to achieve sustained suppression of HBV replication and to prevent further progression of liver disease. Parameters used to indicate treatment response include normalization of serum ALT, decrease in serum HBV DNA level, and loss of HBeAg with or without detection of anti-HBeAg. Currently, eight US Federal Drug Administration (FDA)-approved therapies are available for treatment of chronic HBV infection: IFN-α, pegylated IFN-α2a, four nucleoside analogs (NS; lamivudine, telbivudine, entecavir, and emtricitabine) and two nucleotide analogs (NT; adefovir and tenofovir). Several factors predict a favorable response to IFN treatment with the most important being high ALT and low serum HBV DNA viral load, which are indirect markers of immune clearance.
Therapy usually does not eradicate the virus because of the difficulty of eliminating the covalently closed circular form of the HBV genome from the liver and the existence of extrahepatic reservoirs of HBV. Endpoints of treatment have traditionally been clearance of HBeAg, development of anti-HBe antibodies, and undetectable serum HBV DNA using insensitive hybridization assays with detection limits of approximately 106 genome copies/ml. Achieving these endpoints usually is accompanied by resolution of liver disease as evidenced by normalization of the ALT level and decreased histologic activity on liver biopsy. The response usually is sustained at long-term follow-up. Nevertheless, most responders continue to have detectable HBV DNA when sensitive nucleic acid amplification tests are used. Responses to antiviral therapy are categorized as biochemical, virologic, or histologic and as on-therapy, or sustained off-therapy (Table 46.2) [5].
Table 46.2
Definitions of response and time of assessments for antiviral therapy for chronic hepatitis B virus (HBV) infection
Response definitions | |
Biochemical response | Decrease in serum ALT to within normal range |
Virologic response | Decrease in serum HBV DNA to undetectable levels by nucleic acid amplification assay, and loss of HBeAg in patients who were initially HBeAg positive |
Primary non-response (NS and NT analogs only) | Decrease in serum HBV DNA by <2 log10 IU/ml after at least 24 weeks of therapy |
Virologic relapse | Increase in serum HBV DNA of ≥1 log10 IU/ml after discontinuation of treatment by at least two tests performed >4 weeks apart |
Histologic response | Decrease in histology activity index by at least 2 points and no worsening of fibrosis score compared to pretreatment biopsy |
Complete response | Fulfill criteria of biochemical and virologic response and loss of HBsAg |
Time of assessment definitions | |
On-therapy | During therapy |
Maintained | Persists throughout the course of treatment |
Off-therapy | At the end of a defined course of therapy |
Sustained-6 | 6 months after discontinuation of therapy |
Sustained-12 | 12 months after discontinuation of therapy |
Several variations in the nucleotide sequence of HBV have important clinical consequences. An important mutation in the S gene is a glycine-to-arginine substitution at codon 145 (G145R) in the conserved “a” determinant, which causes decreased affinity of the HBsAg for anti-HBs antibodies [7]. HBV with this mutation has been found in children of HBsAg-positive mothers who develop HBV infection despite vaccination and an adequate anti-HBs antibody response after vaccination, as well as in liver transplant recipients who have recurrent infection despite administration of HBV immune globulin [8, 9]. These immune escape mutants have raised concern about vaccine efficacy and serologically silent infections. The G145R mutation has been reported in many countries and is responsible for 2–40 % of vaccine failures. Although there is diminished binding to anti-HBs antibodies, the vast majority of S mutants can be readily detected with the current generation of HBsAg tests. Thus, an initial concern that widespread use of HBV immune globulin and vaccination would result in HBV mutants that would escape detection in the HBsAg test was unfounded.
Mutations in the basal core promoter and the precore genes affect the synthesis of HBeAg and commonly arise under immune pressure [10]. The most common basal core promoter mutation has a dual change of A to T at nucleotide (nt) 1,762 (T1762) and G to A at nt 1,764 (A1764) that diminishes the amount of mRNA and hence HBeAg secretion [11]. The predominant precore mutation is a G to A change at nt 1,896 (A1896), which leads to premature termination of the precore protein at codon 28, thus preventing the production of HBeAg [12]. The A1896 mutation is infrequent in North America and Western Europe but is geographically widespread. This geographic variability in frequency is related to the predominant genotypes in a geographic region because the mutation is found only in genotypes B, C, D, and E.
The A1896 mutation was first reported in patients with chronic active hepatitis or fulminant hepatitis. However, the A1896 mutation also can be present in asymptomatic carriers and viruses with this mutation replicate no more efficiently than wild-type HBV. Thus, the pathophysiologic significance of this mutation is unclear [13]. However, the clinical picture of persistent HBV replication and active liver disease in HBeAg-negative patients appears to be increasingly prevalent, and in some regions the A1896-mutant virus may be more prevalent than wild-type virus.
Therapy for chronic hepatitis B requires long courses of treatment with NS or NT analogs. A major concern with long-term therapy is the development of antiviral resistance. The rate at which resistant mutants are selected is related to pretreatment serum HBV DNA viral load, rapidity of viral suppression, duration of treatment, and prior antiviral exposure. The incidence of genotypic resistance also varies with the sensitivity of the methods used to detect resistance mutations and the patient population tested. The definitions of terms used to describe resistance to NS and NT analogs are summarized in Table 46.3 [5].
Table 46.3
Definitions of terms used to describe antiviral resistance of HBV
Term | Definition |
|---|---|
Virologic breakthrough | Increase in serum HBV DNA >1 log10 above nadir after achieving a virologic response during continued treatment |
Viral rebound | Increase in serum HBV DNA >20,000 IU/ml or above pretreatment level after achieving a virologic response during continued treatment |
Biochemical breakthrough | Increase in ALT above upper limit of normal after achieving normalization during continued treatment |
Genotypic resistance | Detection of mutations that have been shown in vitro to confer resistance to drug being administered |
Phenotypic resistance | In vitro confirmation that a mutation decreases the susceptibility to the drug as demonstrated by an increase in the inhibitory concentration |
Typically, when a patient experiences a virologic breakthrough, HBV resistance genotyping should be performed. The standardized nomenclature of HBV antiviral resistance mutations is shown Table 46.4 [3, 14, 15]. No HBV mutations are associated with resistance to IFN-α or pegylated IFN-α2a.
Table 46.4
Antiviral agents and the HBV mutations associated with resistance
Antiviral agent | Description of agent | Mutations associated with resistance |
|---|---|---|
Lamivudine | Nucleoside analog (cytidine) | (L180M + M204V/I/S), A181V/T, S202G/I |
Telbivudine | Nucleoside analog (dTTP) | M204I, A181T/V |
Entecavir | Nucleoside analog (2-deoxyguanosine) | T184S/C/G/A/I/L/F/M, S202G/C/I, M250V/I/L |
Emtricitabine | Nucleoside analog (cytidine) | M204V/I |
Adefovir | Nucleotide analog (dATP) | A181V/T, N236T |
Tenofovir | Nucleotide analog (dATP) | A194T, N236T, A181V/T |
Available Assays
HBV DNA Detection and Quantification
The commercially available tests for quantification of HBV DNA in serum and plasma are listed in Table 46.5. These tests employ either branched DNA or real-time PCR for amplification and differ in their limits of detection and dynamic ranges. The COBAS Taqman (Roche Diagnostics, Indianapolis, IN) and the real-time (Abbott Laboratories, Abbott Park, IL) HBV tests are approved for in vitro diagnostic use by the US FDA. The others are available as research use only (RUO) kits or analyte specific reagents (ASRs). All of the quantitative HBV DNA test formats have been used in monitoring the status of HBV infection before and after treatment.
Table 46.5
Commercial assays for quantification of HBV DNA
Assay (manufacturer) | Method | Dynamic range |
|---|---|---|
COBAS Taqman HBV test (Roche Diagnostics, Indianapolis, IN)a | Real-time PCR | 1.7 × 102–8.5 × 108 copies/mlb |
Real-time HBV (Abbott Laboratories, Abbott Park, IL)a | Real-time PCR | 1.0 × 101–1 × 109 IU/ml |
Versant HBV DNA (Siemens Corp., Washington, DC) | Branched DNA | 3.3 × 103–1 × 108 copies/ml |
Artus HBV PCR (Qiagen, Valencia, CA) | Real-time PCR | 2 × 102–1 × 108 IU/ml |
Affigene HBV Trender (Cepheid, Sunnyvale, CA) | Real-time PCR | 1.7 × 102–1.7 × 108 IU/ml |
Qualitative assays for detection of HBV DNA in blood donations are produced by Gen-Probe Inc. (San Mateo, CA) (using transcription-mediated amplification) and Roche Diagnostics (using PCR) with limits of detection of 11 IU/ml and 15 IU/ml, respectively. However, the value of nucleic acid testing compared to serology for HBV screening of blood donors remains controversial and has not been universally adopted [6].
HBV Genotyping
Two commercial HBV genotyping systems are available as RUO kits. Innogenetics Inc. (Alpharetta, CA) offers three different line probe assays for HBV phylogenetic genotyping, detection of precore mutations, and detection of all relevant lamivudine, emtricitabine, telbivudine, adefovir, and entecavir resistance mutations as well as known compensatory mutations [16, 17]. All assays use PCR to amplify portions of the relevant genes to produce a biotinylated product. The PCR products are denatured and hybridized to a series of informative probes immobilized on a nitrocellulose strip. The hybrids are visualized on the strip after addition of streptavidin–alkaline phosphatase and colorimetric substrate. The mutations are identified by the colored patterns of PCR product hybridization to the probes. The line probe assays typically have better sensitivity for detection of sequence variants than direct Sanger sequencing.
Interpretation of Test Results
A major dilemma in the interpretation of serum HBV viral load results is what values should be used to define treatment indications and response. Because HBV DNA may persist in individuals who have serological recovery from acute infection, low levels may not be associated with progressive liver disease and viral clearance is an unrealistic treatment endpoint. An arbitrary value of 20,000 IU/ml (>105 copies/ml) was chosen as a diagnostic criterion for chronic hepatitis B at the 2000 NIH conference [18]. However, progressive liver disease can be found in individuals with lower levels. In addition, wide fluctuations in HBV viral load from undetectable to >2 million IU/ml are observed in some patients with chronic hepatitis B [19, 20]. Consequently, serial monitoring is more important than reliance on a single cutoff value in determining prognosis and the need for treatment.
The first manifestation of antiviral resistance is an increase in HBV viral load >1 log10 from nadir during treatment in a patient who had an initial virologic response. Virologic breakthroughs often are related to therapy noncompliance, so compliance should be assessed before testing for genotypic resistance. HBV viral load levels tend to be low initially because antiviral-resistant mutants have decreased replication fitness. However, compensatory mutations that can restore replication fitness often emerge during continued therapy leading to progressive increases in HBV viral load followed by biochemical breakthrough. Antiviral resistant mutations can be detected months to years prior to biochemical breakthrough. Thus early detection of these mutations can prevent hepatitis flares and liver decompensation. Cross-resistance mutations do occur that limit future treatment options and multidrug-resistant mutants in patients who have received sequential monotherapies have been described [21].
Laboratory Issues
A World Health Organization (WHO) international HBV standard was created in 2001 in response to the recognized need to standardize HBV DNA quantification assays [22]. The WHO HBV standard is a high-titer, genotype A virus preparation (code 97/746) which was assigned a potency of 106 IU/ml. A second standard (code 97/750) was established in 2006 and is quantitatively equivalent to the first standard. The standard established that 1 IU was equivalent to 5.4 genome equivalents by testing the pooled material with a range of commercially available and laboratory-developed tests. However, despite the availability of HBV DNA standards, the various quantitative assays usually have different conversion factors for copies to IU/ml, which may reflect their different amplification and detection chemistries. Laboratories should report HBV viral load test results in IU/ml as both log10 transformed and arithmetic values.
As with any viral load test, some of the variability in quantification may be attributable to sample processing. Either serum or plasma can be used for most tests. Whole blood should be centrifuged within 6 h of collection and the serum or plasma removed from the cells. Short-term storage of serum or plasma is safe at refrigerator temperatures (4 °C). Freezing at temperatures ≤20 °C is recommended for long-term storage of samples.
The standardized nomenclature for reporting of HBV antiviral resistance mutations shown Table 46.4 should be used when resistance genotyping is performed. The inability of genotyping assays to detect minor populations of circulating HBV is a significant technical issue. In general, direct sequencing is limited to resolution of populations which are ≥20 % of the viral population. HBV is included in the hepatitis viral load proficiency testing surveys available from the College of American Pathologists.
Future Directions
Molecular assays for quantification and genotyping of HBV will become more fully integrated in the testing strategies for the diagnosis and management of patients with chronic hepatitis B. Clinical practice guidelines now reflect the importance of HBV viral load and genotyping tests in improving patient outcomes and this is likely to create more demand for these tests. The current generation of real-time PCR viral load assays meets the clinical needs and development and adoption of the WHO international standard has improved the agreement between results of different assays. Improved HBV treatment will depend on development of better laboratory tools for assessing genotypic resistance. Genotyping beyond individual mutation detection to a more comprehensive genomic analysis to predict fitness and other polymutational phenotypes, and detection of minor resistant populations will be possible with next-generation sequencing methods [22, 24].
Hepatitis C Virus
Description of the Pathogen
HCV is an RNA virus with a positive-sense, single-stranded genome of approximately 9,500 nt encoding a single polyprotein of about 3,000 amino acids. The long open reading frame is flanked at each end by short untranslated regions (UTR). The genome structure is most similar to viruses of the family Flaviviridae, which includes many of the arthropod-borne viruses. As in other flaviviruses, the three N-terminal proteins of HCV (core, envelope 1 [E1], and envelope 2 [E2]) are probably structural and the four C-terminal proteins (nonstructural 2, 3, 4, and 5) are thought to function in viral replication. HCV is classified within the family Flaviviridae in its own genus, Hepacivirus.
The 5′ UTR is a highly conserved region of 341 nt and has a complex secondary structure. It contains an internal ribosome entry site and is important in the translation of the long open reading frame. The 3′ UTR contains a short region that varies in sequence and length, followed by a polypyrimidine stretch of variable length, and finally a highly conserved sequence of 98 nt, which constitutes the terminus of the genome. The function of the 3′ UTR is not known but is thought to be essential for viral replication.
The E1 and E2 regions of HCV are the most variable regions within the genome at both the nucleotide and amino acid levels. Two regions in E2, called hypervariable regions 1 and 2 (HVR1 and HRV2, respectively), show extreme sequence variability, which is thought to result from selective pressure by antiviral antibodies. E2 also contains the binding site for CD81, one of the putative cell receptors or coreceptors for HCV.
The nonstructural regions 2 (NS2) and 3 (NS3) contain a zinc-dependent autoprotease that cleaves the polyprotein at the NS2-NS3 junction. The aminoterminal portion of the NS3 protein also is a serine protease that cleaves the polyprotein at several sites. The carboxyterminal portion of the NS3 protein has helicase activity, which is important for HCV replication. The NS4A protein is a cofactor for NS3 serine protease. The NS5B region encodes the RNA-dependent RNA polymerase, which replicates the viral genome. A region in NS5A has been linked to IFN-α response and therefore is called the IFN-α-sensitivity determining region (ISDR).
HCV Genotypes
The first complete HCV genome sequence was reported by Choo et al. in 1991 [25]. As additional genome sequences from isolates from different parts of the world were determined and compared, it was evident that HCV exists as distinct genotypes with as much as 35 % sequence diversity over the whole viral genome [26]. Much of the early literature on genotyping is confusing because investigators developed and used their own classification schemes. However, a consensus nomenclature system was developed in 1994. In this system, the genotypes are numbered using Arabic numerals in order of their discovery, and the more closely related strains within some types are designated as subtypes with lowercase letters. The complex of genetic variants found within an individual isolate is termed the “quasispecies.” The quasispecies results from the accumulation of mutations that occur during viral replication in the host.
The genotype and subtype assignments and nomenclature rules for HCV have recently been updated [27]. Seven major genotypes and 67 subtype of HCV are now recognized with another 20 provisional subtypes. HCV strains belonging to different genotypes differ at 30–35 % of nucleotides and those that belong to the same subtype differ at <15 % of nucleotides at the genome level.
HCV genotypes 1, 2, and 3 are found throughout the world, but there are clear differences in their distribution [28]. HCV subtypes 1a, 1b, 2a, 2b, 2c, and 3a are responsible for more than 90 % of infections in North and South America, Europe, and Japan. In the USA, type 1 accounts for approximately 70 % of the infections with equal distribution between subtypes 1a and 1b. In Japan, subtype 1b causes more than 70 % of HCV infections. Although subtypes 2a and 2b have wide distributions in North America, Europe, and Japan, subtype 2c is widespread in a region of northern Italy. HCV subtype 3a is common among intravenous drug users in the USA and Europe. The other subtypes of genotype 3 are common in Nepal, Bangladesh, India, and Pakistan. Genotype 4 is prevalent in North Africa and the Middle East, and genotypes 5 and 6 are limited to South Africa and Hong Kong, respectively [29]. Subtype 4a constitutes the majority of infections in Egypt, and this and other subtypes of genotype 4 are found in Zaire and Gabon. Subtype 5a is predominant in South Africa, where some reports indicate that it is responsible for more than 50 % of infections. Subtype 6a infections are common in Hong Kong. To date, only 2 genotype 7 infections has been reported in patients from Central Africa [30].
The retrospective nature of most of the genotype studies has not allowed determination of the role of genotype as a risk factor for disease progression nor separation of genotype from other known risk factors for severe disease, such as older age at infection, male gender, alcohol consumption, and concurrent viral infection. However, in two prospective studies, viral genotype did not correlate with disease progression [31, 32].
Clinical Utility
HCV RNA Detection and Quantification
Detection of HCV RNA in serum or plasma by nucleic acid amplification methods is important for confirming the diagnosis of HCV, distinguishing active from resolved infection, assessing the virologic response to therapy, and screening the blood supply. These tests are incorporated into diagnostic algorithms for hepatitis C proposed by the CDC [33], American Association for the Study of Liver Diseases [34], and National Academy of Clinical Biochemistry [35].
The detection of HCV RNA in the plasma or serum is the earliest marker of infection, appearing 1–2 weeks after infection and weeks before elevation of liver enzyme levels or the appearance of anti-HCV antibodies. Approximately 80 % of individuals infected with HCV will be chronically infected with the virus. In antibody-positive individuals, HCV RNA tests can distinguish active from resolved infections. In patients with a high pretest probability of infection, a positive serologic screening test is usually confirmed with a test for HCV RNA rather than the recombinant immunoblot assay (RIBA). This strategy is cost-effective and more informative than using the RIBA to confirm positive antibody screening tests in a diagnostic setting [36]. However, with the discontinuation of the HCV RIBA by the manufacturer in 2012, all reactive HCV antibody screening tests should be followed by US FDA-approved HCV RNA testing [37].
HCV RNA testing also is helpful for the diagnosis of infection in infants born to HCV-infected mothers due to persistence of maternal antibody, and in immunocompromised or debilitated patients who may have blunted serologic responses. An HCV RNA test also should be used for patients suspected of having an acute infection and in patients with hepatitis of no identifiable cause.
HCV RNA tests are the most reliable means of identifying patients with active HCV infection. A negative HCV RNA test in a serologically positive individual may indicate that the infection has resolved or that the viremia is intermittent. Up to 15 % of chronically infected individuals have intermittent viremia and, as a result, a single negative HCV RNA determination may not be sufficient to exclude active infection when the index of clinical suspicion is high [38]. In these individuals a second specimen should be collected and tested.
The use of anti-HCV antibody tests to screen the blood supply has dramatically reduced the risk of transfusion-associated HCV infection in developed countries. The risk in the USA from blood that is negative for anti-HCV antibodies is less than 1 in 103,000 transfused units [39]. To drive the risk of infection from transfusion even lower, blood donor pools currently are tested for the presence of HCV RNA [40]. The serologic screening tests for HCV have a 70-day window period of seronegativity, and antigen detection tests are not yet available for blood product screening. HCV RNA testing is estimated to reduce the detection window by 25 days and reduce the number of transfused infectious units from 116 to 32 per year [41].
Assays for the detection and quantification of HCV core antigen in serum have recently been commercially developed but are not yet US FDA-cleared for diagnostic use (Ortho Clinical Diagnostics, Rochester, NY; Bio-Rad, Hercules, CA; and Abbott Laboratories, Abbott Park, IL) [42–46]. These tests significantly shorten the serologically silent window period using seroconversion panels, and their performance correlates closely with RNA detection tests in blood donors. However, the analytical sensitivity is less than most RNA tests, at approximately 10,000 IU/ml. The analytical sensitivity of the core antigen test is too high to be used in the monitoring of late events during and after treatment. Antigen detection may represent a cost-effective alternative to HCV RNA testing to distinguish active from resolved infections in resource-poor settings.
HCV viral load testing is useful in pretreatment evaluations of patients being considered for therapy, since a viral load of less 600,000 IU/ml is one of several predictors of achieving a sustained virologic response [47, 48]. Other factors associated with achieving a sustained response to therapy include the absence of cirrhosis, age < 40 years, female gender, white race, viral genotype 2 or 3, and presence of C or T at position rs12979860 near the gene for lambda IFN 3 (IL28B) [49, 50].
HCV viral load does not predict disease progression and is not associated with severity of liver disease [51]. This is in sharp contrast to HIV-1, in which the viral load is the principal factor determining the rate of disease progression. Monitoring HCV viral load in untreated patients is not warranted and should be discouraged. Until recently, the standard therapy for patients with chronic HCV infection was pegylated IFN-α in combination with ribavirin administered for either 48 weeks for HCV genotype 1, 4, 5, and 6 infections, or for 24 weeks for HCV genotype 2 and 3 infections. Sustained virologic response (SVR) rates were attained in 40–50 % of patients with genotype 1 and in 80 % or more of those with genotype 2 and 3 infections. SVR is defined as the absence of detectable HCV RNA in plasma or serum as determined with a test that has a limit of detection of ≤50 IU/ml and is considered a virologic cure.
The first direct acting antivirals (DAA) for treatment of hepatitis C were approved by the US FDA in 2011. Both are NS3/4A serine protease inhibitors, boceprevir (BOC) (Merck, Kenilworth, NJ) and telaprevir (TVR) (Vertex, Boston, MA). These DAA agents are used in combination with pegylated IFN-α and ribavirin. Triple therapy for genotype 1 infections has led to approximately a 30 % increase in SVR over the previous standard of care therapy in all patient subgroups. Limited phase 2 testing has shown that TVR also has activity against genotype 2 infections but not against genotype 3 infections. BOC appears to have activity against both genotypes 2 and 3 infections. However, neither drug should be used to treat patients with genotype 2 and 3 infections [52].
The algorithms for monitoring HCV viral load in patients receiving triple therapy and the stopping rules are shown in Fig. 46.1 [52]. The important time points for response-guided therapy are at 8, 12, and 24 weeks for BOC and 4, 12, and 24 weeks for TVR. Treatment with all three drugs should be stopped if HCV RNA level is >100 IU/ml at week 12 or detectable at week 24 for BOC triple therapy and if HCV RNA level is >1,000 IU/ml at weeks 4 or 12 and/or detectable at week 24 with TVR.
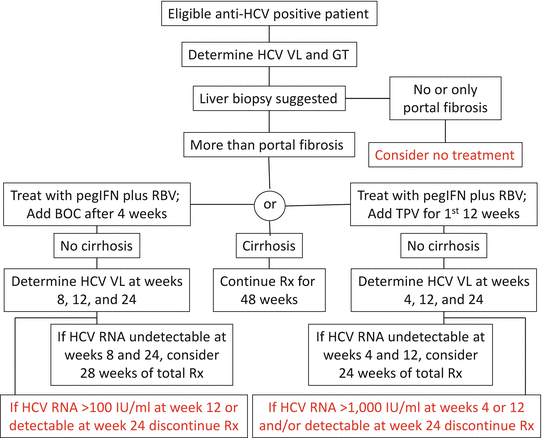
Figure 46.1
Triple therapy algorithms for c 1 infections. Stopping rules in red (Adapted from Ref. 52). BOC boceprevir, GT genotype, IU international units, pegIFN pegylated interferon-α, RBV ribavirin, Rx treatment, TVP telaprevir, VL viral load
The goal of treatment is a SVR, defined as no detectable HCV RNA in serum or plasma by a highly sensitive assay (limit of detection ≤10–15 IU/ml) 6 months after the end of treatment. Patients who achieve a SVR have little or no chance of virologic relapse of their disease.
In 2013, two more potent DAAs were approved by the US FDA: sofosbuvir (Gilead Sciences, Inc., Foster City, CA), a NS5B polymerase inhibitor [53], and simeprevir (Johnson & Johnson, New Brunswick, NJ), a second generation protease inhibitor [54]. Sofosbuvir was approved in combination with pegylated IFN-α and ribavirin for treatment of genotypes 1 and 4 and in combination with ribavirin alone for genotypes 2 and 3. Simeprevir was approved by the US FDA for treatment of genotype 1 infections in combination with pegylated IFN-α and ribavirin, but only for patients with genotype 1 who have not failed therapy with first generation protease inhibitors.
Monitoring of on-treatment viral load levels does not affect management decisions with a sosobuvir-based regimen since treatment failure is almost exclusively due to relapse [53]. However, given the expense of the drugs and the potential risk of viral resistance with inappropriate use, viral load testing at week 4 and end of treatment at either week 12 or 24 depending on the regimen seems prudent.
Viral load levels should be determined at weeks 4, 12, and 24 to assess treatment response and for stopping rules in patients treated with simeprevir, pegylated IFN-α, and ribavirin. Discontinuation is warranted for patients who are unlikely to achieve a SVR based on the on-treatment virologic response. If HCV RNA level is >25 IU/ml at week 4, the entire regimen should be discontinued. If the HCV RNA level is >25 IU/ml at week 12 or 24 after the simeprevir has been completed, the pegylated IFN-α and ribavirin should be discontinued [54].
Numerous other DAAs have been developed and are currently in clinical trials. These include NS3/4A protease inhibitors, NS5B polymerase inhibitors, and inhibitors of host cell proteins required for HCV replication. The most current recommendations for all aspects of HCV treatment can be found at http://www.hcvguidelines.org/.
HCV Genotyping
Sequence analysis of variable regions of the HCV genome has been used to investigate outbreaks of infection and to study modes of transmission. Two large outbreaks of infection associated with contaminated lots of anti-rhesus D immunoglobulin (anti-D) in Ireland and Germany were investigated using molecular typing [55, 56]. In both studies, sequence analysis showed that the HCV infecting the women was the same as that found in the implicated batches of anti-D. In another report, sequencing part of the NS3 region provided evidence of patient-to-patient transmission during colonoscopy [57]. Sequence analysis also has been used as part of investigations of HCV infections associated with blood transfusions. In addition, molecular genotyping has been used to study vertical and sexual transmission of HCV [58–61].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


