Figure 61.1
Comparison of forward-phase and reverse-phase protein microarrays. (a) Forward-phase microarray. Capture antibodies are first immobilized on the slide surface. These immobilized antibodies are used to capture the antigens they recognize from a test sample. (b) Reverse-phase microarray. Complex samples are immobilized on the surface and targeted by antibodies overlaid on them
Forward-phase microarrays are the most frequently used formats allowing the simultaneous analysis of multiple parameters per sample. Specific proteins of interest are selectively trapped from complex protein mixtures (serum, plasma, cell lysate, or cell culture supernatant) by well-defined capture or “bait” molecules (usually antibodies but full-length functional proteins or protein active domains could be used) that are immobilized onto a solid surface. Each spot of the array contains one type of immobilized antibody or bait molecule. Each array is incubated with one sample and the bound analytes are visualized either by direct labeling of the analytes or via labeled secondary antibodies. Standard detection methods include fluorescence, chemiluminescence, and colorimetry. A planar microarray-based system can generate high-density protein microarrays to screen a large number of analytes in a single experiment. The concept of planar microarray-based systems has been successfully miniaturized and operationalized into a bead-based microarray system where each bead is coupled to a specific bait molecule. This bead-based microarray system provides an interesting alternative since it is more flexible, robust, and advanced with respect to automation to screen thousands of samples within a short time.
For reversed-phase microarrays, a multitude of different samples (tissues or cell lysates) are immobilized as spots in rows on a solid support such that the array contains numerous spots representing the proteome of different patient samples or cell lysates. Then, each microarray is incubated with one highly specific detection molecule or antibody, measuring a single analyte with direct comparison of the analyte across multiple samples. This approach allows sets of proteins to be detected in large collections of tissue or cell samples.
Clinical Applications of Protein Microarrays
To date, many commercial forward-phase microarrays are available to investigate different classes of proteins such as cytokines and chemokines, cancer biomarkers or molecules involved in signaling pathways. Interestingly, several clinical protein microarrays have been cleared by the US Food and Drug Administration (FDA) or CE-marked for use in the European Union. So far, the main applications of these protein microarrays are diagnosis of infectious diseases or immune diseases. For example, the AtheNA Muli-Lyte® Test System (Zeus Scientific, Raritan, NJ, USA) can be used for the diagnosis of the infection by Borrelia burgdorferi by multiplex sandwich immunoassay for the quantitative detection of distinct IgG antibody to Vlse-1 and distinct IgM antibody to pepC10. Another example is the Bio-Plex™ 2200 ANA Screen (Bio-Rad Laboratories, Hercules, CA) which detects autoantibodies in serum or plasma as an aid in the diagnosis of systemic rheumatic diseases such as systemic lupus erythematosus, mixed connective tissue disease, undifferentiated connective tissue disease, Sjogren’s syndrome, scleroderma, dermatomyositis, polymyositis, rheumatoid arthritis, CREST syndrome, and Raynaud’s disease.
Mass Spectrometry-Based Proteomic Analysis
Introduction to Mass Spectrometry
Instrumentation
Since the first description of a mass spectrometry (MS) experiment in the early 1900s by Thomson [1], MS-based approaches have come a long way from being narrowly applied in analytical chemistry to a key device for clinical testing. The description in the late 1980s of two different soft ionization methods named Matrix-Assisted Laser Desorption/Ionization (MALDI) [2, 3] and ElectroSpray Ionization (ESI) [4] (Fig. 61.2) significantly accelerated MS-based interrogation of large biologically important molecules. The principle of operation of MALDI-based proteomics involves the co-crystallization of a protein/peptide sample on a designated matrix medium (Fig. 61.2a). The vaporization and ionization of the protein/peptide sample is achieved by pulse-irradiation of the surface with a laser. This process generates ions that are accelerated into the mass analyzer for analysis, filtering, and detection. By comparison, the ESI process involves the transfer of ions from solution to gaseous phase at atmospheric pressure (Fig. 61.2b). The application of a very high voltage across a sample solution at very low flow rates in a capillary needle causes ions to accumulate at the narrow tip of the needle where they are then drawn out as the sample solution is pushed out of the needle creating charged droplets that are accelerated in an electric field. Via the Coulomb explosion, large charged droplets are sequentially blown apart to form smaller droplets upon reaching a certain limit leaving only the discrete peptide sample ions to enter the mass analyzer.
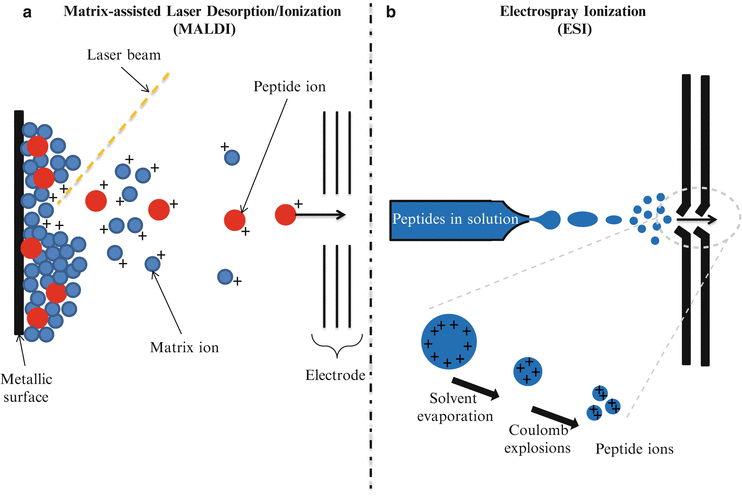
Figure 61.2
Principles of operation of Matrix-Assisted Laser Desorption Ionization (MALDI) and Electrospray Ionization (ESI). (a) MALDI. The protein/peptide sample is co-crystallized with a matrix, then the vaporization and ionization of the protein/peptide sample is achieved by a laser pulse-irradiation of the surface. The matrix material heavily absorbs UV laser light, leading to the ablation of the upper layer of the matrix. This process generates ions that are usually accelerated into a mass analyzer for analysis. (b) ESI. The liquid containing the analyte is dispersed by electrospray into a fine aerosol. Via Coulomb explosions, large charged droplets are sequentially blown apart to form smaller droplets. Upon reaching a certain size limit, only the discrete peptide sample ions enter the mass analyzer
After ionization the sample reaches the mass analyzer which separates ions by their mass-to-charge (m/z) ratios. Ion motion in the mass analyzer can be manipulated by electric or magnetic fields to direct ions to a detector which registers the number of ions at each individual m/z value. Different types of mass analyzers are available: time-of-flight (TOF), ion trap, and quadrupoles. These mass analyzers differ considerably in sensitivity, resolution, mass accuracy, and the possibility to obtain fragment peptide ions which results in mass spectra with high content of information (MS/MS spectra). The choice of the combination of the ion source with the mass analyzer and the detector depends on the specific application.
Classical Workflow for Mass Spectrometry Proteomics
Mass spectrometry-based proteomic analyses can be divided into top-down or bottom-up processes. In top-down proteomics, intact proteins or polypeptides are directly analyzed by mass spectrometry. On the other hand, bottom-up proteomics entails digestion of complex protein mixtures into peptides using a proteolytic enzyme (typically trypsin). Resulting peptides are separated by liquid chromatography (LC) and analyzed by tandem mass spectrometry (MS/MS). Proteins are identified by matching experimental spectra with those from theoretical spectra of translated genomic databases generated by in silico cleavage using specific enzymes (Fig. 61.3).
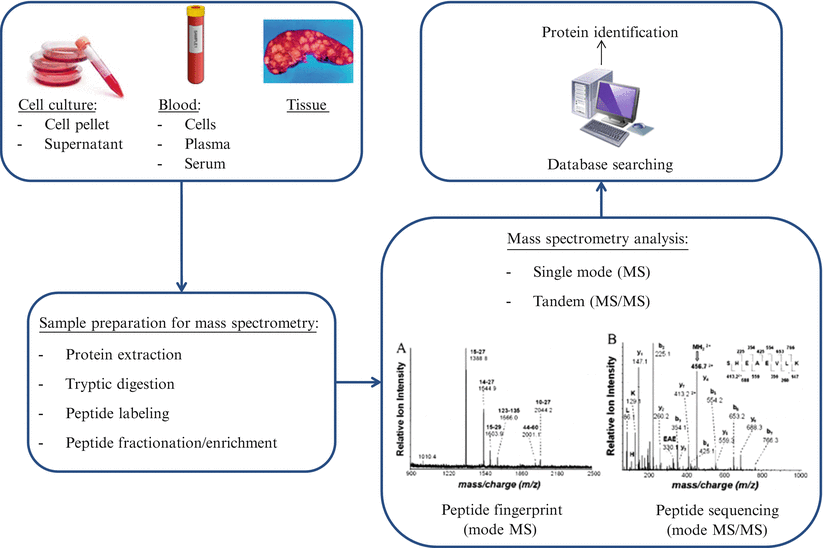
Figure 61.3
Workflow for mass spectrometry proteomics
Quantitative Mass Spectrometry-Based Proteomics
Quantitative measurement of protein concentrations is one of the key advancements in MS-based proteomics. Global quantitation of protein levels can be achieved by stable isotope labeling of proteins/peptides, use of heavy peptides as standards, and label-free quantitation (Fig. 61.4). Isotope labels can be introduced metabolically (i.e., in vivo), chemically, or enzymatically (i.e., in vitro). Since the metabolic labeling (stable isotope labeling with amino acids in cell culture, [SILAC]) requires maintaining cells in culture for several passages (usually six or seven passages) in the presence of stable isotopes of amino acids (heavy Arg, Lys, Leu, or Ile), its direct application to clinically relevant scenarios is less likely. The in vitro methods for labeling include isotope-coded affinity tagging (ICAT) and isobaric tags for relative and absolute quantitation (iTRAQ). These methods may be adaptable to clinical scenarios since cell viability is not required for labeling. In the original ICAT labeling method, cysteine residues were specifically derivatized with a reagent containing either zero or eight deuterium atoms as well as a biotin group for affinity purification of cysteine-derivatized peptides and subsequent mass spectrometry analysis. The iTRAQ method is another in vitro labeling reagent which targets the N-terminus amino acid of peptides with a varying mass. The AQUA (absolute quantification) quantitative approach uses isotope-labeled synthetic peptides as standards spiked at a known amount into a sample preparation. The quantitation is achieved by comparing the mass spectrometric signal of the synthetic peptide to the endogenous peptide in the sample. Label-free quantitation methods are gaining in popularity and are increasingly being employed as an alternative to label-based approaches. These techniques do not use isotopic labeling, but instead directly compare signal intensities across different mass spectrometry runs using either the signal intensity of peptide precursors or the number of fragment spectra identifying peptides of a given protein.
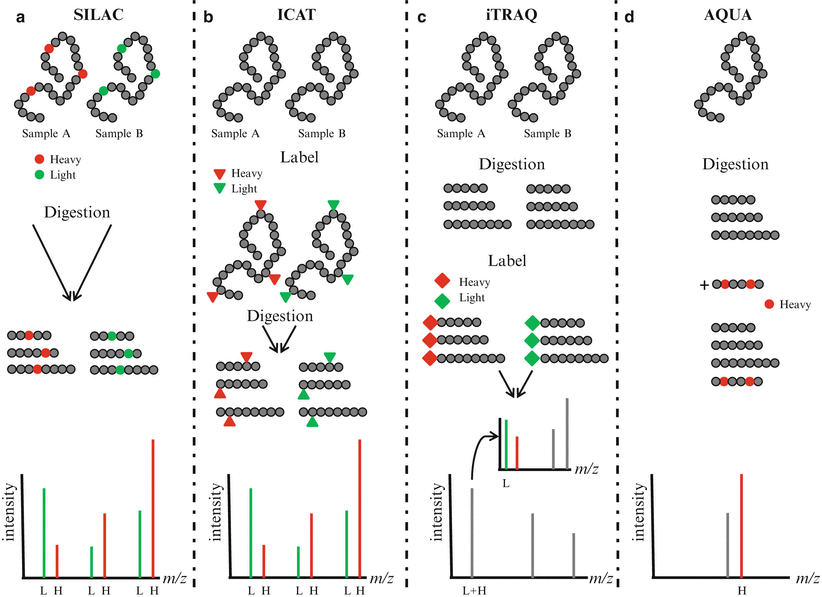
Figure 61.4
Quantitative proteomics. (a) In stable isotope labeling with amino acids in cell culture (SILAC), two cell cultures are differentially labeled with heavy amino acids containing stable isotopes (heavy, H) and normal amino acids (light, L). Lysates from labeled cells are digested, mixed, and analyzed by mass spectrometry. Differentially labeled peptides having the same amino acid sequence are detected in MS spectra and the relative abundance of the peptides can be compared. (b) In isotope-coded affinity tagging (ICAT), proteins are labeled at cysteine residues with reagent containing either 0 (light, L) or 8 (heavy, H) deuterium atoms. After digestion, labeled peptides are combined and analyzed by mass spectrometry. Peptides with the same amino acid sequence are detected in MS spectra and the relative abundance of the peptides can be compared. (c) In isobaric tags for relative and absolute quantitation (iTRAQ), peptides are labeled using isobaric amine-specific tandem tags. The iTRAQ reagent consists of a reporter region with 1 Da difference and a balance region that adjusts the molecular weight of the labeled parent ions. Each tag generates a unique reporter in MS/MS spectra and the relative abundance of peptides can be compared. (d) In absolute quantification (AQUA), an isotope-labeled peptide is spiked at a known amount into the sample preparation. The abundance of peptide can be compared with the abundance of the spiked peptide
Selected Reaction Monitoring (SRM)
Selected reaction monitoring (SRM), formerly referred to as multiple reaction monitoring mass spectrometry, utilizes two rounds of mass selection by quadrupole mass analyzers to discriminate specific ion peptides within a complex sample based on m/z ratios (Fig. 61.5) [5]. Following sample processing with trypsin and separation by nano-LC, ionized peptide precursors of predetermined interest are selected in the first quadrupole (Q1). After Q1 selection, only ions with the predefined m/z value are transmitted to the second quadrupole (Q2) where a collision-assisted dissociation causes peptide fragmentation. Detection and quantitation of the target analyte is achieved following the selective transmission in the third quadrupole (Q3) of a predefined product ion (a fragment of the precursor peptide). Ultimately, SRM allows the quantitation of a large set of target proteins in complex biological samples.
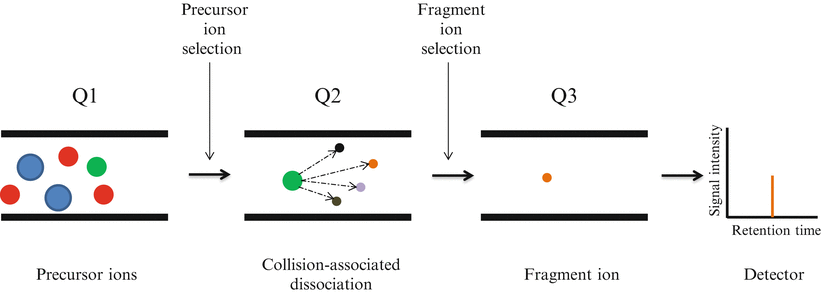
Figure 61.5




Selected Reaction Monitoring (SRM) analysis on triple quadrupole mass spectrometer. Several analytes are co-eluted from the chromatographic system. The ionized peptide precursors with a specific m/z are selected in the first quadrupole (Q1). Only ions with the predefined m/z value are transmitted to the second quadrupole (Q2) where a collision-assisted dissociation causes peptide fragmentation. Detection and quantitation of the target analyte is achieved following the selective transmission in the third quadrupole (Q3) of a predefined product ion
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
