Figure 9.1
Multiplex DNA amplification of DMD exons 8, 13, 19, 45, and 47. Lane 1: normal control; lane 2: DMD patient with exon 13 deletion; lane 3: DMD patient with exon 8 and 13 deletions.
Testing using MLPA has been utilized for both DMD deletion/duplication detection and carrier dosage determinations. The MLPA analysis has been shown to reproducibly generate each of the 79 DMD exons and their sizes should correspond to each probe pair. MLPA technology not only accurately detects deletions but also quantifies the copy number of each exon and therefore can be used for duplication and carrier determinations. Using capillary electrophoresis for analysis of the MLPA products, computer-aided scoring is a sensitive method to normalize the peak height or area of each PCR product to an endogenous two-copy control locus product. A heterozygous deletion should give a ratio of approximately 0.5 whereas an elevated copy number should give a reference value greater than 1.5. MLPA technology has been shown to exhibit a high degree of accuracy for the quantitative detection dystrophin exons, a high degree of reproducibility, and a capacity for testing a large number of samples simultaneously. One major limitation of MLPA technology is that DNA polymorphic sequence variants located under the probe-binding sites may interfere with probe hybridization and may result in false-positive results.
The identification of a deletion in a DMD patient not only confirms the diagnosis but also allows accurate carrier testing for other family members. Carrier status is determined by gene dosage analysis, which is used to assess whether a female at risk of being a carrier exhibits no reduction or 50 % reduction in band intensity for bands that are deleted in the affected male relative. A 50 % reduction (single-copy intensity) for the deleted band or bands indicates a deletion on one of her X chromosomes and confirms her carrier status. Southern blot, quantitative multiplex PCR, or MLPA can be used for gene dosage analysis. Dosage determinations permit direct carrier analysis and eliminate the inherent problems of linkage testing (recombinations, noninformative meioses, unavailability of family members, and spontaneous mutations). This is important since, unlike affected males, carrier females are generally asymptomatic, and creatine phosphokinase is elevated in only approximately two-thirds of known carriers.
As previously described, large genomic deletions and duplications have been identified in approximately two-thirds of the DMD/BMD population. The other mutations are due to smaller types of mutations within the dystrophin gene which require a sequence-based testing strategy. In most clinical molecular laboratories, these mutations have gone undetected because sequencing the entire gene is both expensive and labor intensive. With the advances in DNA sequencing technology, point mutation analysis of exons in the DMD gene is now feasible. The identification of these mutations is not only important for the confirmation of the diagnosis, but also important for the determination of carrier status. Due to the high mutation rate in the dystrophin gene, carrier testing based on indirect linkage results often is limited for extended family members of isolated cases of the disease. Knowledge of the exact causative mutation allows for the determination of the origin of the mutation in families with simplex cases of the disease.
Interpretation of Test Results
The analysis of gene mutations and protein determinations has greatly improved diagnosis, carrier detection, and prenatal counseling. Amplification techniques will confirm the clinical diagnosis in approximately 65 % of the DMD and BMD patients and should be performed as the initial molecular test due to the relatively common occurrence of deletions and duplications. If the patient tests negative by deletion/duplication analysis, gene sequencing of the coding regions and intron/exon boundaries should be performed. By gene sequencing, detection rates can be increased to greater than 90 %; however, even with sequencing, not all mutations are identified. Some of the undetected mutations may reside in the large dystrophin introns or in regulatory regions. Furthermore, genetic heterogeneity in the DMD phenotype may be another reason for less than 100 % sensitivity of testing. If there is any question of the diagnosis after negative results by molecular testing, western blot analysis of the dystrophin protein should be performed on a muscle biopsy specimen.
When gene dosage testing indicates that the mother does not have the deletion present in the affected child, she still has an uncertain risk of carrier status, due to the possibility of germline mosaicism [16]. Cases of germline mosaicism in DMD have been reported, in which a deletion is transmitted to more than one offspring by a mother who shows no evidence of the mutation in her somatic cells. Cases of germline mosaicism have important counseling implications. First and most obvious is the need to perform carrier studies on all female siblings of affected males, regardless of the outcome of testing for the mother. Furthermore, a negative deletion result in a mother does not rule out a recurrence risk for future pregnancies, and prenatal diagnosis still should be offered. The exact recurrence risk in germline female carriers is unknown because the risk is related to the percentage of the mutant clone in the mosaic mother. In these cases, the recurrence risk for subsequent pregnancies is significantly higher than a new mutation with a low recurrence risk. Mothers of apparently sporadic DMD cases have an estimated 15 % recurrence risk in future pregnancies.
Linkage analysis can provide valuable information but is limited by the possibility of recombination between the polymorphic marker and the unknown mutation, the presence of sporadic mutations, and unavailability of family members. The intragenic recombination rate over the entire length of the DMD gene is estimated to be as high as 12 %. The high recombinational error rate can be partially overcome by using microsatellite markers throughout the gene. Linkage analysis results often are extremely limited for extended family members of isolated cases of DMD/BMD, due to the possibility of the occurrence of a new mutation. Linkage analysis indicates only whether the female at risk inherited the same X chromosome as the affected male, not whether she is a carrier of a defective gene. Furthermore, since the gene mutation remains unidentified, a correct clinical diagnosis is essential. This is extremely important with patients presenting with the milder BMD phenotype, since this phenotype can overlap with other neuromuscular disorders.
Laboratory Issues
As a result of the discovery of the DMD gene and elucidation of the disease mutation spectrum, clinical diagnostic testing for DMD and BMD has significantly improved. Until an effective treatment is found to cure or arrest the progression of the disease, prevention of new cases through accurate diagnosis, and carrier and prenatal testing is of the utmost importance. Furthermore, molecular therapies (such as antisense oligonucleotides, antibiotics, or chimeric RNA/DNA) are being applied according to the specific dystrophin mutation. This requires a comprehensive mutation analysis and identification of all types of dystrophin mutations. Although the majority of laboratories offer testing for dystrophin deletions and duplications, sequencing of the entire gene becomes necessary for all patients who are negative for the deletion/duplication screen. Proficiency testing for both DMD deletion/duplication testing and carrier testing is currently offered through the College of American Pathologists (CAP), and proficiency specimens are sent to participants twice per year.
Myotonic Dystrophy
Molecular Basis of Disease
Myotonic dystrophy type I (DM1) is the most common inherited form of muscular dystrophy affecting adults, with an incidence of approximately 1 in 8,000 individuals. DM1 is an autosomal dominant, multisystem disorder characterized by progressive muscle weakness, myotonia, intellectual impairment, cataracts, and cardiac arrhythmias. The diagnosis can be problematic because of the wide range and severity of symptoms. Often, affected individuals have children before they are diagnosed. A severe congenital form of DM results in mental retardation, respiratory distress, hypotonia, and in many cases death due to respiratory complications shortly after birth. Those that survive the neonatal period initially follow a static course, eventually learning to walk but with significant mental retardation. The congenital form is most often observed in the offspring of women who are themselves affected, although the disease in a mother may not be diagnosed until after the birth of a congenitally affected child [17].
The myotonic dystrophy protein kinase gene (DMPK) at chromosome 19q13.3 is the only known gene associated with DM1. The DM1 mutation involves an expanded CTG trinucleotide repeat, located at the 3′ untranslated region (UTR) of the DMPK gene [18–20]. The number of CTG triplets varies in the normal population from 5–34 repeats, and within this range the alleles are stably transmitted. Individuals with 35–49 repeats (premutation alleles) do not have symptoms but their children are at an increased risk of inheriting larger repeats and having symptoms. When the number of repeated CTGs exceeds 50 (in some patients up to several thousands), the allele becomes unstable and results in the DM1 phenotype. There is a significant correlation between the number of CTG repeats and the age of onset and clinical severity. Mildly affected patients have 50–100 repeats, and these patients may only have cataracts. More classically affected patients have 100–1,000 repeats and congenital cases often have 1,000–6,000 repeats. Somatic mosaicism of the CTG repeat occurs and results in patients with similar repeat sizes having different severity of symptoms, which calls for caution in genetic counseling. The trinucleotide repeat region is mitotically and meiotically unstable, with a bias towards length increase in the next generation, accounting for the clinical phenomenon of “anticipation” (increasing severity in successive generations of the same family, with earlier age of onset). Although repeat expansions occur through both maternal and paternal transmissions, the larger repeat expansions observed in congenital cases are almost exclusively due to maternal transmissions.
The CTG repeat is located within the 3′ UTR of the DMPK gene that encodes a protein kinase, named myotonin protein kinase. Since protein kinases are involved in signal transduction pathways in all cells in the body, a defective protein kinase was considered as a mechanism in which a single-gene defect could result in the diverse symptoms characteristic of DM1; however, since the repeat is not in the protein-coding region of DMPK, the molecular mechanism by which the mutation exerts its dominant expression is difficult to explain. One theory is that the myotonin kinase mRNA with long CUG repeats, and not the protein, results in a gain-of-function RNA pathogenesis [21, 22]. Novel RNA-binding proteins that specifically bind to CUG repeats may be depleted by excessive CUG repeats in the DM1 transcripts. Muscleblind-like protein (MBNL1) and CUG-binding protein (CUGBP1) are two RNA-binding proteins proposed to be involved in the pathogenesis. The depletion of these CUG-binding proteins causes splicing alterations of the chloride channel gene (CLC1) and the insulin receptor genes (IR), resulting in myotonia and insulin resistance, phenotypes that are related to the clinical features of DM1.
Clinical Utility of Testing and Available Testing
The majority of clinically significant mutations can be identified by Southern blot analysis; however, PCR is essential for the detection of smaller CTG expansions (<100 repeats). Although the PCR test is less expensive and faster than Southern blot analysis, longer repeats often are not reliably amplified. Southern blot analysis can be performed using HindIII restriction digestion and the probe pMDY1 [19], which spans the repeat region (Fig. 9.2). The probe pMDYI detects a HindIII polymorphism with normal alleles of 8.5 and 9.5 kb, the frequencies of which are approximately 0.6 and 0.4, respectively. The polymorphism is due to a 1 kb insertion telomeric to the CTG repeat, and is almost in complete linkage disequilibrium with the CTG repeat mutation in most populations [20]. The mutation is found on the larger 9.5 kb allele, suggesting that there were a limited number of ancestral mutations that occurred on a chromosome having the 1 kb insertion. Alternatively, the larger allele may be predisposed to DM1 mutations. Typical increases in the range of 1–4 kb are observed on Southern blots in the DM1 population. Many of the larger expansions are detected as smears, indicating somatic cell heterogeneity of the expanded alleles. Background signal may interfere with the detection of larger expanded alleles since expanded alleles often appear as diffuse smears due to the somatic instability of the mutation. The efficiency of the detection of these somatically variable expansions is increased by use of a restriction enzyme with a recognition sequence which occurs relatively infrequently in the human genome, which will generate a larger restriction fragment containing the expansion. (EcoRI creates a large 9–10 kb fragment.) The larger fragment lengths reduce the smearing effect of the somatic variability of the unstable repeats. Decreasing the electrophoretic migration also improves the detection of larger somatic mosaic expansions. Molecular testing can identify individuals who are asymptomatic or exhibit equivocal symptoms, such as cataracts. These smaller expansions are detected using alternate restriction enzymes which reduce the normal size fragment, such as BamHI, or by PCR amplification across the repeat region. The most efficient approach for identifying expansions is a combination of different restriction enzymes and variation in electrophoretic duration. PCR amplification across the repeat region is used to exclude DM1 by demonstrating the presence of two different normal size alleles (8.5 and 9.5 kb). Since the heterozygosity frequency for the CTG repeats is approximately 75 % in the normal population, approximately 25 % of unaffected individuals will be homozygous for one normal allele. Therefore, the presence of a single PCR band does not confirm a diagnosis of DM1. All single bands require a Southern blot confirmation.
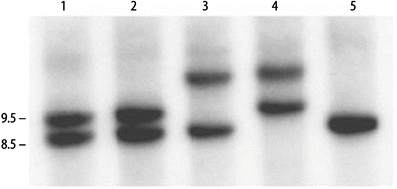
Figure 9.2
Autoradiogram of a Southern blot for myotonic dystrophy. Normal alleles are 8.5 and 9.5 kb. Lanes 1, 2, and 5: unaffected individuals; lanes 3 and 4: expanded and normal alleles in DM patients with repeat expansions
Several cases of reverse mutations have been reported in DM1, whereby there is a spontaneous correction of a deleterious expansion mutation upon transmission to an unaffected offspring. The mechanism for the DM1 reverse mutations remains unknown. A gene conversion mechanism, whereby the normal parental allele replaces the expanded allele, may best account for the reversion events in DM. The reversions may provide an explanation of the lack of penetrance observed in some DM families.
Interpretation of Test Results
PCR and/or Southern blot analysis for the DMPK CTG repeat region is used for symptomatic confirmatory diagnostic testing. Onset in adulthood typically presents with muscle weakness and myotonia. DMPK testing can be particularly useful in individuals for whom DM1 is part of a wider differential diagnosis, such as infants with muscular hypotonia. Prenatal problems, with congenital DM1, may be suspected when reduced fetal movements and polyhydramnios are observed on ultrasound. The testing also is helpful in identifying individuals who are asymptomatic or exhibit equivocal symptoms, such as cataracts. No new mutations have been described for DM1, which is consistent with the linkage disequilibrium data. To account for the maintenance of the mutation in the population, the theory developed that there is a high incidence of minimally expanded alleles in DM1 families which produce few symptoms and are stably transmitted over several generations [23]. For counseling purposes, testing can be used to identify the side of the family carrying the mutation, which is important for appropriate genetic counseling.
The triplet repeat size does correlate with muscular disability and is inversely related with the age of onset of the disease. However, there is a significant overlap of repeat size in patients with differing severity. When comparing unrelated affected individuals with small to moderate differences in repeat sizes, it is generally difficult to accurately predict the severity of the disease in each case. This is due to the overlap of triplet repeat size in patients with differing severity of the disease and the sometimes lack of a correlation between organ involvement and repeat size. As a result of the somatic heterogeneity observed in DM1, genotype/phenotype associations derived from leukocytes may not be as accurate as the measurement of the repeat size in the affected tissues (muscles, heart, others) [24]. Within a family, when a child has a significant increase in allele size compared to the parent, prediction of an earlier age of onset and more severe disease is more certain. Genetic counseling is very important not only for the affected patient, but also for other at-risk interested family members. DM1 testing can be used for prenatal diagnosis using either amniotic fluid cells or chorionic villus samples. Molecular testing has largely replaced muscle biopsy, muscle enzyme studies, and electromyography as the first diagnostic procedure.
Myotonic dystrophy type 2 (DM2, MIM 602668), previously termed proximal myotonic myopathy is due to a CCTG expansion located in intron 1 of the zinc finger protein 9 (ZNF9) gene on chromosome 3q21 [25]. Individuals affected with DM2 also have a complex clinical presentation that is similar to DM1, including myotonia, cardiac involvement, insulin insensitivity, and histological abnormalities in skeletal muscle. Patients with DM2 often can be distinguished from DM1 by more proximal muscle weakness and sparing of facial muscle involvement. Further distinguishing characteristic features of DM2 include muscle pain, the absence of congenital cases, and milder course of the disease without significant mental involvement.
Laboratory Issues
Many molecular pathology laboratories offer DNA testing for DM1. The majority of laboratories are using a combination of PCR and Southern blot testing. Although the PCR test is less expensive and faster than Southern blot, longer repeats are not reliably amplified. The availability of DNA testing has reduced the use of invasive (muscle biopsy) and noninvasive (electromyography) tests for the diagnosis of DM1. The combination of PCR and Southern blot analysis can detect all DM1 mutations. Proficiency testing is offered through the College of American Pathologists and proficiency specimens are sent to participants twice per year.
Spinal Muscular Atrophy
Molecular Basis of Disease
The autosomal recessive disorder proximal spinal muscular atrophy (SMA) is a severe neuromuscular disease characterized by degeneration of alpha motor neurons in the spinal cord, which results in progressive proximal muscle denervation and atrophy resulting in the symptoms of weakness and paralysis. SMA is the second-most common fatal autosomal recessive disorder after cystic fibrosis, with an estimated prevalence of 1 in 10,000 live births [26]. Childhood SMA is subdivided into clinical groups on the basis of age of onset and clinical course. Type I SMA (Werdnig-Hoffmann disease) is characterized by severe, generalized muscle weakness and hypotonia at birth or within the first 3 months after birth. Death from respiratory failure usually occurs within the first 2 years of life. Children affected with Type II SMA are able to sit, although they cannot stand or walk unaided, and survive beyond 4 years of age. Type III SMA (Kugelberg-Welander syndrome) is a milder form, with onset during infancy or youth, and patients may walk unaided. Type III SMA is further subdivided into two groups, type IIIa (onset before 3 years of age) and type IIIb (onset at age ≥3 years). Cases presenting with the first symptoms of the disease at the age of 20–30 years are classified as type IV, or proximal adult-type SMA. The described classification is based on age of onset and clinical course, although SMA demonstrates a continuous range of severity.
SMA is caused by mutations in the survival motor neuron (SMN1) gene which has nine exons [27]. Two almost identical SMN genes are present on 5q13: the telomeric SMN1 gene that is the SMA-causing gene, and the centromeric SMN2 gene. Exon 7 of the SMN1 gene is deleted in approximately 95 % of affected patients, while small, more subtle mutations have been identified in the majority of the remaining affected patients. The genotypic explanation for the phenotypic variability results from variability in the number of SMN2 gene copies, which influences the severity of the disease [28–30]. The number of SMN2 gene copies varies from 0–3 copies in the normal population, with approximately 10 % of unaffected individuals having no gene copies of SMN2. However, milder patients with type II or III SMA on average have more gene copies of SMN2 than do type I SMA patients. The extra SMN2 gene copies are thought to arise through gene conversions, whereby the SMN2 gene is copied either partially or totally into the telomeric SMN1 locus.
SMA is caused by low levels of SMN protein rather than by a complete absence of the protein. Complete loss of SMN protein function results in loss of viability of a fetus and in utero death. SMA is possible because SMN2 can partially replace the lost function of SMN1. Five base pair differences exist between SMN1 and SMN2 transcripts, and none of these differences change amino acids. The normal SMN1 gene produces predominately a full-length transcript, whereas the SMN2 gene produces predominately an alternate, exon-7-deleted product with only 10–20 % of transcripts being full-length. The inclusion of exon 7 in SMN1 transcripts and exclusion of this exon in the majority of SMN2 transcripts is caused by a single nucleotide difference at +6 in SMN exon 7, which alters splicing efficiency by disrupting an exonic splicing enhancer and results in absence of exon 7 in the majority of SMN2 transcripts. The SMN exon 7 region encodes a highly conserved tyrosine-glycine (Y-G) dodecapeptide motif which is crucial for the oligomerization and function of the SMN protein [31]. SMA arises because the levels of full-length SMN protein from the SMN1 gene are lost, and only partially rescued by the low number of full-length SMN protein from the SMN2 gene. The SMN2 gene cannot completely compensate for the lack of SMN protein; however, the small amounts of full-length transcript generated by SMN2 are able to prevent in utero lethality. Additional copies of the SMN2 gene result in greater amounts of full-length SMN protein and produce a milder type II or III phenotype.
SMN plays a role in small nuclear ribonucleoprotein (snRNP) biogenesis and function [32]. The SMN protein is required for pre-mRNA splicing. Immunofluorescence studies using a monoclonal antibody to the SMN protein demonstrate that the SMN protein is localized to novel nuclear structures called “gems,” which display similarity to and possibly interact with coiled bodies, which are thought to play a role in the processing and metabolism of snRNAs. SnRNPs and possibly other splicing components require regeneration from inactivated to activated functional forms. SMN functions in the reassembly and regeneration of these splicing components. Mutant SMN, such as that present in SMA patients, lacks the splicing-regeneration activity of wild-type SMN. SMA may be the result of a genetic defect in spliceosomal snRNP biogenesis in motor neurons. Consequently, the motor neurons of SMA patients may be impaired in their capacity to produce specific mRNAs and as a result become deficient in proteins that are necessary for the growth and function of these cells. Possibly, the altered splicing of a unique set of premessenger RNAs results in deficient proteins, which are necessary for motor neuron growth and survival. In addition to its role in spliceosomal ribonucleoprotein assembly, SMN may have other functions in motor neurons. A subset of SMN complexes are located in axons and growth cones of motor neurons and may be involved in some aspects of axonal transport and localized translation of specific mRNAs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


