Key point
The search for true bioequivalence is important. Given the variability of human drug response, to which we have referred several times, it is even more important that the product which enters the body is as standardised as possible. The combination of human variability and product variability is unthinkable.
16.2 Regulatory statements on generic products
A generic drug should be identical or bioequivalent to a brand-name drug in dosage form, safety, strength, route of administration, quality, performance characteristics and intended use. The US and European authorities adopt similar approaches.2 To gain approval, a generic drug must:
- contain the same active ingredients as the innovator drug (inactive ingredients may vary)
- be identical in strength, dosage form and route of administration
- have the same use indications
- be bioequivalent
- meet the same batch requirements for identity, strength, purity and quality
- be manufactured under the same strict standards of good manufacturing practice regulations required for innovator products.
The question of bioavailability has to be looked at in several ways (Fig. 16.1). Products can be bioequivalent yet not therapeutically equivalent, perhaps because the rate of absorption of the drug differs in the first 30 minutes or so. If bioavailability is measured by the area under the plasma concentration–time curve (AUC) over 24 or 48 hours, then these measures of bioavailability might not show up subtle differences.
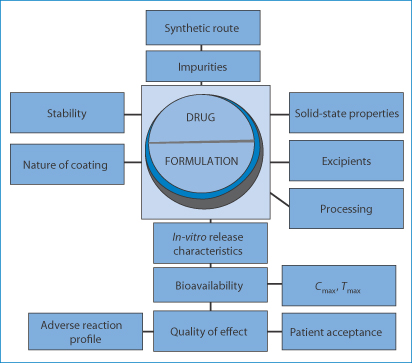
Figure 16.1 The range of issues in determining the equivalence or essential similarity of formulations.
16.2.1 Generics: a question of quality
Equivalence between medicinal products can be thought of at two levels:
1. chemical equivalence, which refers to dosage forms containing the same amount of the same drug in similar dose forms
2. therapeutic equivalence, which refers to medicines having not only the same bioavailability (as measured by the AUC), but the same clinical effects.
Figures 16.2 and 16.3 demonstrate the limits of bioequivalence and non-bioequivalence. Figure 16.3 shows how, while two generic products may be equivalent to the first brand product, the two generics may not be equivalent to each other, which may pose problems in practice.
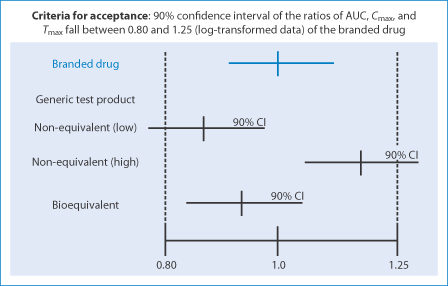
Figure 16.2 The issue of variability in branded and generic products with their 90% confidence limits and the definition of non-equivalence and equivalence in visual form.
Reproduced from Medscape.
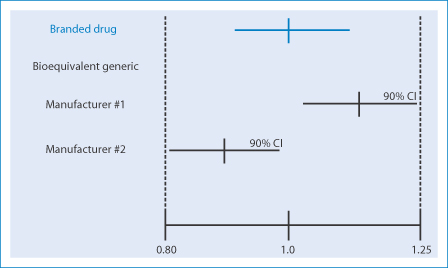
Figure 16.3 Visual demonstration of bioequivalence between generics and branded drugs, which demonstrates clearly, that while generic 1 can be equivalent to the brand, generic 2 may not be equivalent to generic 1. From Medscape.
Essential similarity of dose forms of the same drug focuses on the essential similarity of purity of the drug substance as well as similarity of release rates. Chemical equivalence is ensured by pharmaceutical processes and quality assurance and is one prerequisite for therapeutic equivalence. Limits are set for drug substance; for example, tetracycline hydrochloride contains not less than 96% and not more than 102% of the drug. As far as therapeutic equivalence is concerned, the product should have essentially the same safety profile as the comparator product. Regulations do not speak of identicality between products, but essential similarity. One reason is that drug substances can vary in purity within pharmacopoeial guidelines, and there are variations in the amount of drug within each unit of product, again within limits. On top of this there are interpatient and intrapatient variability. This does not excuse differences and indeed sometimes exacerbates differences. Many drugs are absorbed readily and raise no concerns when generic versions become available. It is with drugs that have proven bioavailability problems or that require special formulation techniques that the issue of similarity becomes real. Drugs with a narrow therapeutic index have to be considered with care. On the other hand, solutions and syrups do not need such scrutiny. Simple formulations of soluble drugs for injection are unlikely to cause concern. The only concern might be with drug purity. Pharmacopoeias of course provide limits on undesirable impurities in the drug substance. Toxic 4-epi-anhydrotetracycline is limited in tetracycline products. Even if the limit is very low (say 0.005%), there may be three products (or the same product in different batches) with levels of such an impurity at 0.0048%, 0.004% and 0.002%. Are these identical? Much depends on the nature and activity of the impurity.
For conventional tablets, solutions, simple injections and suspensions, generic versions can be used generally without qualms. However, once we enter the field of formulation modification, as with sustained-release products, the picture becomes more complex. One knows that towards the end of the patent life of conventional products, modified-release forms (or sometimes new polymorphs of the drug substance) are introduced to delay the introduction of generics and to retain brand loyalty. The relative potentials of oral dosage forms to exhibit bioavailability problems are listed in Table 16.1. It is obvious that the more complex formulations can rarely be identical in originators and branded preparations. Among the ‘complex’ formulations, one might include liposomes and microparticle and nanoparticulate systems.
Table 16.1 Relative potential of oral medicines to display bioavailability problems
High | Intermediate | Low |
Enteric-coated tablets | Suspensions | Solutions |
Sustained-release tablets | Chewable tablets |
|
Complex formulations | Capsules |
|
Slowly disintegrating tablets |
|
|
There are generic versions of some sustained-release formulations, but within a given class of sustained-release product of a given drug, such as theophylline, there will be a spectrum of release rates. The proliferation of sustained- and modified-release forms of branded products at the end of the patent life of a product results not necessarily from the prospect of improved clinical outcomes but for the very reason that generic versions are difficult to produce. Generic modified-release products themselves are therefore usually branded. Having said this, the benefit of having a range of formulations is recognised.3
16.2.2 Specific medical conditions and the use of generics
There is just the chance that in the treatment of some conditions differences in the performance of products are relevant. There are also some medical conditions where particular care has to be taken in titrating patients and maintaining therapeutic levels closely throughout treatment. Such is the case with anti-epileptic drugs (AEDs). While generic medicines have been an issue at least since the 1970s, problems with AEDs are still current,4,5 as we discuss below. There are reports on the re-emergence of psychotic symptoms after conversion (a study with n = 7) from a brand-name clozapine to a generic formulation.6 However, the problem with such reports is that they are often specific to a country; the results will depend on which brand and which generic are being studied. Papers do not always give full pharmaceutical information and hence their value is diminished.
The difficulty in extrapolating results across borders is exhibited by clozapine. It has been found that generic preparations of the drug licensed in the UK are bioequivalent with the branded Clozaril: as the author7 states: ‘There was no evidence of clinical deterioration or the need to use higher doses. Generic clozapine is not inferior to Clozaril.’ Some US reports suggest that there are problems in substituting the originators’ product with a generic. But another study in the USA of a generic clozapine (Mylan) and Clozaril (Novartis)8 concluded that they were therapeutically equivalent. Generic bupropion (I) extended-release (Budeprion, Impax Labs) was withdrawn for lack of bioequivalence with Wellbutrin XL, the branded product (Fig. 16.4) used for the treatment of depression.
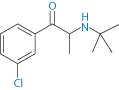
Structure I Bupropion
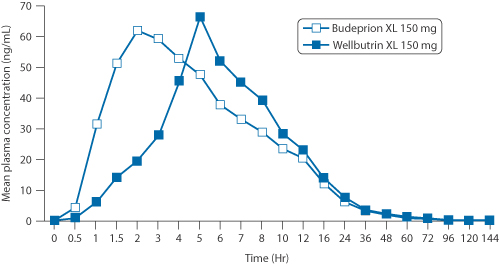
Figure 16.4 Mean plasma concentration of bupropion (Budeprion XL and Wellbutrin XL) as a function of time in 24 fasting healthy volunteers.
See details in Woodcock MD et al. Withdrawal of generic budeprion for nonbioequivalence. N Engl J Med 2012;367:2463–2465.
16.2.3 Antiepileptic drugs: clinical experience and the literature
There is, however, a relative shortage of literature comparing generic substitutions for AEDs. Most of the case reports, letters to the editor and some papers deal with three drugs, carbamazepine, phenytoin and valproate. These reports document breakthrough seizures or adverse events when switching from a branded AED to a generic version. Of around 300 US neurologists, 56% reported adverse events, and 68% reported breakthrough seizures in at least one patient when medication was switched from a branded to a generic AED. Burkhardt et al.9 identified eight adult patients whose seizures worsened after switching from branded phenytoin to generic phenytoin. The few blinded, controlled studies reported in the literature have evaluated relative pharmacokinetics of a brand versus generic formulations. Sometimes only one generic version was studied (note Fig. 16.2). No controlled studies have mirrored practice by evaluating safety, efficacy and compliance with the therapeutic regimen when multiple generic versions are used in succession.
The American Academy of Neurology has produced a number of recommendations regarding generic substitution for AEDs10,11:
- Such substitutions can be approved only if the safety and efficacy of treatment are not compromised.
- Specific pharmacokinetic information about each AED generic should be made available to physicians, who should avoid switching between formulations of AEDs.
- Labelling should identify specific manufacturers.
- Pharmacists should be required to inform patients and physicians when switching a product between manufacturers.
- Organisations that encourage or mandate substitution of AEDs should evaluate their responsibility for any problems arising from their policies.
Because changing from one formulation of an AED to another can usually be accomplished, and risks minimised, if physicians and patients monitor blood levels, seizures and toxicity, it is maintained in the USA that the individual and physician should be notified and should give their consent before a switch in medications is made, whether it involves either generic substitution for brand-name products, or generic-to-generic substitutions.
16.3 Reading and deconstructing the literature on bioequivalence
If pharmacokinetic data are available, one needs to ask whether the analytical methodology was up to the task. Was the study sound? The literature can be biased because studies supported by manufacturers of products under study (both generic and branded)8 might not be published if the results, say, show inequivalence in the former case or equivalence in the latter. In one case, with levothyroxine, it was estimated that analytical techniques commonly used were not sufficiently precise to ascertain equivalence. Figure 16.5 shows data on the levels of thyroxine in plasma after administration of doses from 400 to 600 μg, without taking into account baseline thyroxine concentrations.12 Such a method would not be able to distinguish between generics if it cannot distinguish between the doses over the range given. Figure 16.5b shows the data taking baseline levels into account. The latter method would be adequate.
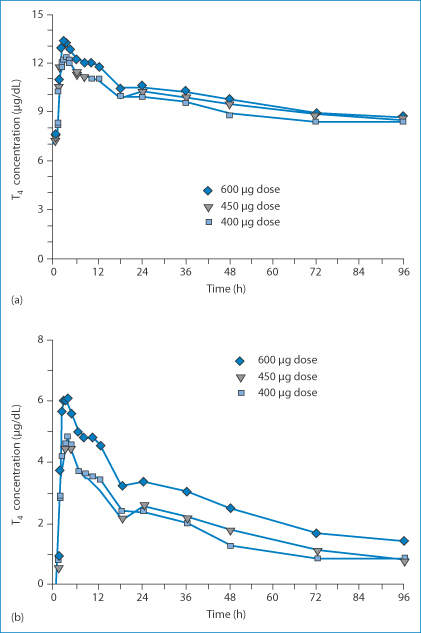
Figure 16.5 Thyroxine levels in plasma as a function of time. (a) Mean levothyroxine concentration time profiles on study day 1 following a single-dose administration of levothyroxine sodium, uncorrected for baseline levothyroxine concentrations. (b) Figures corrected for baseline levothyroxine concentrations.
Reproduced from Blakesley VA. Current methodology to assess bioequivalence of levothyroxine sodium products are inadequate. AAPS J 2005;7:E42–E46, with kind permission from Springer Science and Business Media.
The other issue is whether active and active metabolites are being measured, as with risperidone, whose active metabolite, 9-hydroxy-risperidone has a much longer half-life than the parent drug.
16.3.1 Antiretroviral drugs
It is always difficult to generalise from single studies on generic equivalence or non-equivalence. In comparisons of generic and branded anti-human immunodeficiency virus (HIV) products containing the three drugs stavudine, lamivudine and nevirapine made in HIV-infected adults,13 stavudine levels were found to be significantly lower using a generic formulation. A similar but larger study, also in infected adults, found that generic fixed-dose combinations of these drugs were efficacious and safe.14 Two generic fixed-dose combinations of these drugs for children (Pedimune Baby and Pedimune Junior, Cipla Pharmaceuticals) have been found to be similar to the branded products when tested in healthy adults.15 As with many products, the true measure of quality is not minor differences in peak plasma levels or AUCs but therapeutic outcome. Never more so than in developing countries. The stark facts are that, after the introduction of cheaper antiretroviral therapies in Southern India, the numbers using these products increased and death rates decreased from 25 to 5 deaths per 100 persons between 1997 and 2003.16
16.3.2 Bioequivalence of ophthalmic products
It is difficult to assess the bioequivalence of ophthalmic products. There have been issues, however,17 one being high rates of precipitation in a generic prednisone formulation when administered to the eye. Some products will employ different excipients, which might lead to subtle differences in behaviour of the preparation; a generic timolol gel-forming product possessed a different ‘feel’ from the branded product. This also apparently gave the impression to the patient that the generic formulation was less efficacious.
16.3.3 The case of sevoflurane
The apparent simplicity of the anaesthetic molecule sevoflurane (II) masks differences in synthetic methods and degradation that may be significant in deciding between products.
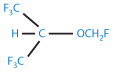
Structure II Sevoflurane
Sevoflurane is available in the USA from two manufacturers as Ultane (Abbott) and as a generic product, sevoflurane inhalation anaesthetic (Sevoness) (Baxter Healthcare Corp.). These products are rated therapeutically equivalent by the US Food and Drug Administration, but there are some differences. Ultane is made in a single-step synthetic process, whereas generic sevoflurane is manufactured using a three-step process, as described by Baker.18 In the UK sevoflurane is a non-proprietary product. As there is only one UK product, the question of differences will arise only if there are several manufacturers. In the USA, as Baker points out, Ultane contains >300 ppm water and generic sevoflurane contains ≤130 ppm water. Ultane is supplied in a plastic poly(ethylene naphthalate) polymer bottle, while generic sevoflurane is supplied in lacquer-lined aluminium bottles. Here then is an example that typifies some generic issues. The products have different manufacturing processes (Fig. 16.6) and hence different impurities, have different containers, and have potential differences in the rate or extent of sevoflurane degradation.
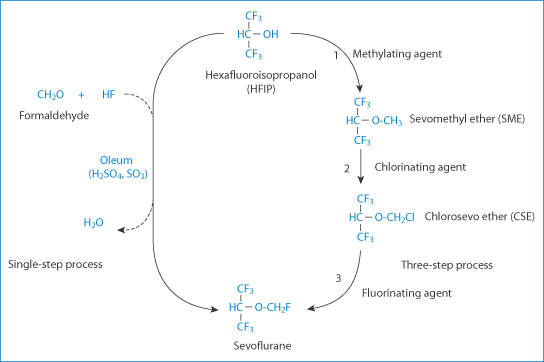
Figure 16.6 Pathways for the single-step and three-step syntheses of sevoflurane from the starting compound hexafluoroisopropanol. The potential impurity from the single-step method is formaldehyde, and from the three-step process is sevomethyl ether (SME) and chlorosevo ether (CSE).
Reproduced with permission from Reference 18.
The significance of the containers has been discussed18 and relates to the discovery that sevoflurane can be unstable in glass bottles in which the anaesthetic was originally supplied. Reports of a cloudy product with a strong odour appeared. Such batches were found to contain hydrofluoric acid in concentrations up to 863 ppm, as well as a pH below unity. This was linked to Lewis-acid defluorination of the drug. (Lewis acids are usually metal-containing compounds that accept electrons from Lewis bases and result in Lewis base degradation. There are many Lewis acids, such as metal halides and metal oxides, including aluminium oxide (Al2O3)). The Lewis acid in this case was identified as rust (iron oxide) on a valve on a bulk shipping container. The drug is especially susceptible to degradation because of the monofluoroalkyl ether group. The whole story can be read in Baker’s paper.18
The base-catalysed conversion of sevoflurane to ‘compound A’ is as shown in Fig. 16.7. Figure 16.8 shows a range of degradation pathways for the anaesthetic.19
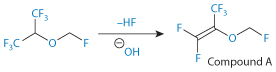
Figure 16.7 Base-catalysed conversion of sevoflurane to so-called compound A.
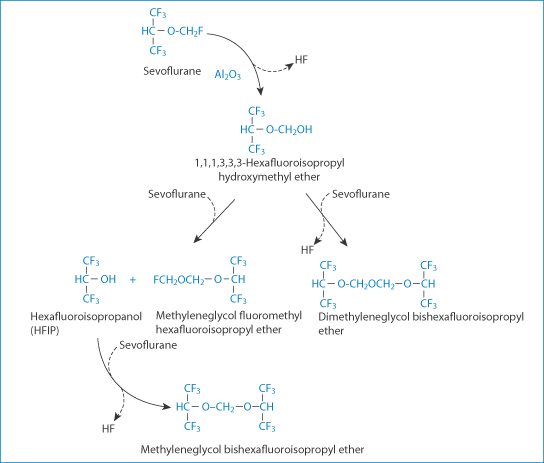
Figure 16.8 Potential pathways of degradation and the products of degradation of sevoflurane on reaction with alumina (Al2O3). The degradation products are detected at very low concentrations in low-flow, long-duration anaesthesia.
Reproduced from Bito H, Ikeda K. Long-duration, low-flow sevoflurane anesthesia using two carbon dioxide absorbents. Quantification of degradation products in the circuit. Anesthesiology 1994;81:340–345.
Carbon dioxide absorption enables the use of low-flow anaesthesia, and a decreased consumption of medical gases and halogenated anaesthetics, as well as reduced pollution. Chemical absorbents (soda lime and barium hydroxide lime (Baralyme)) may produce toxic compounds: carbon monoxide with all halogenated anaesthetics and ‘compound A’ with sevoflurane (Fig. 16.9). Simple measures against desiccation of the lime prevent carbon monoxide production. The toxicity of compound A, shown in the rat, has not been demonstrated in clinical anaesthesia. Recent improvements in manufacturing processes have decreased the powdering of lime. Moreover, filters inserted between the anaesthesia circuit and the patient abolish the risk for powder inhalation.
A Japanese product is marketed as Sevofrane (Maruishi Pharmaceutical Co., Osaka, Japan)20 and is available there along with Sevoness. Both products studied contain over 99.998% sevoflurane and fluoride concentrations were the same, at around 0.43 ppm.
The formation and toxicity of anaesthetic degradation products can be an issue.21 Soon after its introduction in 1935 there were reports of neuropathies following trichloroethylene anaesthesia. These were attributable to the formation of dichloroacetylene through base-catalysed elimination of HCl from the compound. The common cause of their formation is the reaction of the anaesthetic with the bases in adsorbents in the circuit. (These include sodium hydroxide (soda lime), barium hydroxide lime, potassium hydroxide (KOH)-free soda lime, calcium hydroxide and non-caustic lime. These have different reactivities: sodium hydroxide > soda lime > KOH-free soda lime > calcium hydroxide.) Understanding the nature of these reactions that affect trichloroethylene, desflurane (Fig. 16.9), norflurane and enflurane has helped towards the safe use of these agents.
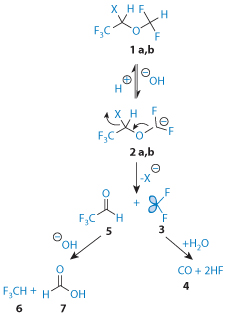
Figure 16.9 Base-catalysed conversion of desflurane and isoflurane (1a and 1b) to CO (4). Note also trifluroacetaldehyde (5) and trifluoromethane (6) and formic acid (7).
Although these examples are of a specialised use of these agents, nevertheless the potential formation of very low concentrations of degradants by whatever means can lead to adverse events or certainly to differences in the behaviour of products.
16.4 Biosimilars: protein drugs, monoclonal antibodies
The complexity of proteins as drugs is well known. Peptides and proteins are subject to a variety of chemical and physical instabilities in aqueous solution and at interfaces, and can also suffer during the stresses of some manufacturing processes.
16.4.1 Issues with biomolecular generics
Now that the protein therapeutic market has matured, generic versions of biologicals are becoming available. The source and mode of production of the protein therapeutic are important issues. With the so-called ‘follow-on’ protein drugs,22 compound or product identicality cannot always be demonstrated as it can with conventional molecules. Even with the latter there are often small regulated amounts of impurities, such as dimers, breakdown products and products of side-reactions. The impurities in proteins are often very similar to the parent compound and, although present in low concentrations, may be pharmacologically or immunologically active. Small changes in manufacturing processes may lead to final products that are not identical to the originator’s product. Aggregation, protein folding and glycosylation may be affected, any of which can lead to differences in pharmacokinetics, immunogenicity or, indeed, efficacy. The formulation may or may not influence outcomes. One example has been seen with recombinant human erythropoietin (Eprex). A change of stabiliser from albumin to polysorbate 80 has been thought to be the cause of the formation of antierythropoietin antibodies revealed in pure red cell aplasia,** but the issue is complex, as explained in reference 23.
16.4.2 Aggregation of biologicals
The processing of many biological agents is complex. Aggregation of monoclonal antibodies during manufacturing processes is a key problem because, if the correct form of the monoclonal antibodyis not retained, then biological activity will be compromised.24
Aggregation during processing includes the events in the following steps:
- cell culture
- purification
- agitation
- ultrafiltration
- pumping at higher shear rates
- buffering procedures
- exposure to pH values close to isoelectric points
- freezing and thawing
- final filling.
PEGylation of some proteins has been shown to reduce the extent of adsorption and surface-induced aggregation and to increase the reversibility of adsorption compared to native protein. Aspects of PEGylation are discussed in Chapter 13.
16.4.3 Generic biologicals (biosimilars)
A generic is, as stated above, a product that has been shown to be ‘essentially similar’ to the originator’s product. However, biological products (biologics, biologicals, biopharmaceuticals) are more complex than drugs that are small or relatively small organic molecules. Because of their labile nature and the fact that the products may be dependent on the mode of manufacture, it is important to remember that it is has been stated that a process ‘cannot be exactly duplicated by another manufacturer’.25
Why do these products have such specific problems? The molecules are large and often prepared in cell-based systems; they have complex tertiary and quaternary structures related to their activity; and they may be glycosylated. They are very sensitive to stresses such as temperature and even to shear forces that might be used in their production. DNA, for example, suffers from degradation due to shear forces. A practical pharmacy aspect is the use of International Non-proprietary Names (INNs) for such products. With conventional drugs an identical INN means a bioequivalent product and the same drug substance within pharmacopoeial or other limits. There are dangers if INNs are used for biogenerics that are subtly different.
Methods used to show that small-molecule therapeutics are nearly identical to each other are clearly not sufficient for biologicals. Bioequivalence is usually defined in terms of AUCs, but this is only part of the story with biological products. The nature of impurities is different. These impurities might indeed be analogues with a single amino acid difference, and be potent. If this is the case, this can lead to clinical problems. Impurities might be more difficult to detect in biological products if they are analogues of the main agent, so there is the risk of immunological and other side-effects. It is also possible that the formulation may be the cause of differences in protein products, as discussed above, as with recombinant human erythropoietin (Eprex).
Figure 16.10 summarises the issues that must be considered when dealing with biological generics.26 The most appropriate description for generic biologicals is that they are ‘biosimilar’.
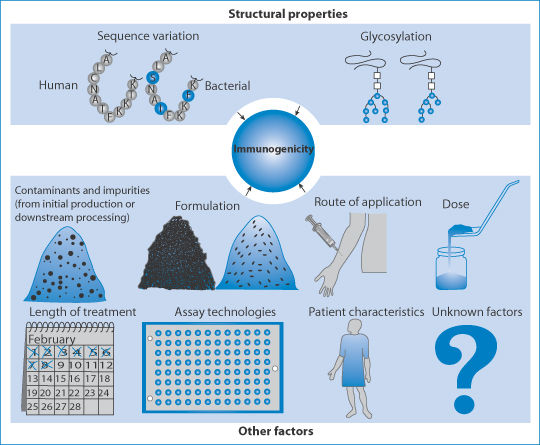
Figure 16.10 Areas of importance in determining the bioequivalence and the immunogenicity of biopharmaceuticals.
Reprinted by permission from Macmillan Publishers Ltd: Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov 2002;1:457–462. Copyright 2002.
The European Medicines Agency (EMA) has stated that the generic route is not appropriate for biologics, and the US Food and Drug Administration (FDA) stated in September 2006 that it ‘has not determined how interchangeability can be established for complex proteins’. Note, however, that for biologics such as insulin, the transfer of patients from one product to another of the same insulin type is commonplace, though carried out with circumspection.
16.4.4 Regulatory views on biosimilar biologicals
The FDA acknowledges that ‘biosimilars have not been demonstrated to be interchangeable through any scientific process’. A different naming scheme for these products might involve utilising a different level of granularity, which may be more detailed or less detailed. If the outcome of assigning the same INN to two products with highly similar ingredient(s) creates the implication that the two products are pharmacologically interchangeable and there were no scientific data to support that finding, then the FDA would have serious concerns about such an outcome, especially with more complicated proteins. At present, the FDA has not determined how interchangeability can be established for complex proteins.
This initiative reinforces the EMA communication on biosimilar medicines27 and recognises the uniqueness of these products. It states that they cannot be classified as ‘generics’ in the same way that chemical compounds may be, owing to the differences stemming from the variability of the active biotechnological substances and manufacturing processes. Further, the EMA document clarifies that ‘since biosimilar and biological reference medicines are similar but not identical, the decision to treat a patient with a reference or a biosimilar medicine should be taken following the opinion of a qualified healthcare professional’. This is effective advice against automatic substitution of one biological medicine over another that is ostensibly the same.
16.4.5 PEGylated proteins
Some PEGylated proteins are discussed in Chapter 13. Attachment of long-chain polyoxyethylene glycols (PEGs) to proteins is the basis of PEGylation. This can increase the circulation time of proteins with short half-lives and can reduce immunogenicity; however, there are cases28 where the process can reduce the functional activity of the protein by blocking access to receptors. These modified proteins are clearly not biosimilar to the parent proteins and are essentially new molecules.
16.5 Conclusions
For many medicinal products the initial brand and the subsequent generic products are therapeutically equivalent. There are of course effects of differences, such as the colour of capsules and tablets or the appearance of liquids, which can prejudice patients. There are drugs that have a narrow therapeutic index and that are perhaps poorly soluble, which perhaps suggests that they might not be used interchangeably, but these are few in number; experts are not agreed even with drugs such as the antiepileptic agents. There is a greater practical problem in that biostudies might have shown both generic 1 and generic 2 to be bioequivalent to the brand leader, but this does not necessarily mean that generic 1 is equivalent to generic 2. Pharmacists have thus to be aware of the source of generics.
With generic versions of biologicals or biosimilars the issues are somewhat more complex. Development in analytical methods and other tests might assist in elucidating the similarity or otherwise of this growing array of drugs in the future. With both conventional drugs and biological drugs, wherever a formulation has been devised to alter the rate of release or delivery of the active agent, it is not automatic that generic versions will produce identical results. To be able to make informed decisions, pharmacists and drugs and therapeutics committees need access to the facts about such products and in particular their biopharmaceutical profiles in patients or volunteers.
References
1. Hassali MA et al. Knowledge and perceptions of recent pharmacy graduates about generic medicines. Pharm Ed 2007;7;89–95.
2. US Food and Drug Administration, Centre for Drug Evaluation and Research, Office for Generic Drugs. Available online at: http://www.fda.gov/Drugs/ResourcesForYou/Consumers/BuyingUsingMedicineSafely/UnderstandingGenericDrugs/ucm144456.htm (updated 2009; accessed 23 September 2009).
3. Frijlink HW. Benefits of different drug formulations in psychopharmacology. Eur Neuropsychopharmacol 2003;13:S77–S84.
4. Wilner AN. Therapeutic equivalence of generic antiepileptic drugs: results of a survey. Epilepsy Behav 2004;5:995–998.
5. Crawford P et al. Are there potential problems with generic substitution of antiepileptic drugs? A review of issues. Seizure 2006;15:165–176.
6. Mofsen R, Balter J. Case reports of the re-emergence of psychotic symptoms after conversion from brand name clozapine to a generic formulation. Clin Ther 2001;23:1720–1731.
7. Paton C. Generic clozapine: outcomes after switching formulations. Br J Psychiatry 2006;189:184–185.
8. Healy DJ et al. Clinical equivalence of generic clozapine. Commun Ment Health J 2005;41:393–398.
9. Burkhardt RT et al. Lower phenytoin serum levels in persons switched from brand to generic phenytoin. Neurology 2004;63:1494–1496.
10. Assessment: generic substitution for antiepileptic medication. Report of the Therapeutics and Technology Assessment Sub-committee of the American Academy of Neurology. Neurology 1990;40:1641–1643.
11. Liouw K et al. Position statement on the coverage of anticonvulsant drugs for the treatment of epilepsy. Neurology 2007;68:1249–1250.
12. Blakesley VA. Current methodology to assess bioequivalence of levothyroxine sodium products are inadequate. AAPS J 2005;7:E42–E46.
13. Byakika-Kibwika P et al. Steady-state pharmacokinetic comparison of generic and branded formulations of stavudine, lamivudine and nevirapine in HIV-infected Ugandan adults. J Antimicrob Chemother 2008;62:1113–1117.
14. Laurent C et al. Effectiveness and safety of a generic fixed dose combination of neviparine, stavudine and lamivudine in HIV-1 infected adults in Cameroon: open label multicentre trial. Lancet 2004;364:29–34.
15. L’homme RFA et al. Pharmacokinetics of two generic fixed dose combinations for HIV infected children (Pedimune Baby and Pedimune Junior) are similar to the branded products in healthy adults. J Antimicrob Chemother 2007;59:92–96.
16. Kumarasamy N et al. The changing natural history of HIV disease: before and after the introduction of generic antiretroviral therapy in Southern India. Clin Infect Dis 2005;41:1525–1528.
17. Cantor LB. Generic ophthalmic medications: as good as Xerox? Medscape Ophthalmol 2008; 26 Nov. Available online at: http://cme.medscape.com/viewarticle/583866 (accessed 23 September 2009).
18. Baker MT. Sevoflurane: are there differences in products. Anesth Analg 2007;104:1447–1451.
19. Bito H, Ikeda K. Long-duration, low-flow sevoflurane anesthesia using two carbon dioxide absorbents. Quantification of degradation products in the circuit. Anesthesiology 1994;81:340–345.
20. Yamakage M et al. Analysis of the composition or ‘original’ and generic sevoflurane in routine use. Br J Anaesth 2007;99:819–823.
21. Anders MW. Formation and toxicity of anesthetic degradation products. Annu Rev Pharmacol Toxicol 2005;45:147–176.
22. US Food and Drug Administration. Follow-On Protein Products: Regulatory and Scientific Issues Related to Developing. Available online at: http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/ucm085854.htm (accessed 9 January 2015).
23. Locatelli F et al. Pure red-cell aplasia ‘epidemic’ – mystery completely revealed? Peritoneal Dialysis Int 2007;27:S303–S307.
24. Vazques-Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng 2011;108:1494–1508.
25. Bio (Biotechnology Industry Organization). See www.bio.org for various statements on biosimilars; 2015.
26. Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov 2002;1:457–462.
27. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) Guideline on Similar Biological Medicinal Products (CHMP/437/04). 2015. www.ema.europa.eu (accessed 6 April 2015).
28. Kubetzko S et al. Protein PEGylation decreases observed target association rates via a dual blocking mechanism. Mol Pharmacol 2005;68:1439–1454.
Further reading
Johnston A et al. Effectiveness, safety and cost of drug substitution in hypertension. Br J Clin Pharmacol 2010;70:320–344. N.B. Although the emphasis in this paper is on hypertension treatment, the review deals with wider therapeutic categories and many of the challenging aspects of the problem of substituting generic products for originator brand products.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


