Figure 30–1 Pathogenesis of rheumatoid arthritis and sites of action of selected antirheumatic drugs. In this simplified view of the immune system, antigen-presenting cells phagocytose antigens and present them to T cells, thereby activating the T cells. Activated T cells produce cytokines (IL-2, IL-1, TNF-α, and others) that stimulate the production of B and T cells. B cells produce plasma cells that form antibodies. Helper T cells activate macrophages and cytotoxic T cells. Together, T cells, macrophages, and cytotoxic T cells produce cytotoxic cytokines and prostaglandins that cause joint inflammation, synovial proliferation, and bone and cartilage destruction. Glucocorticoids inhibit T-cell activation and interleukin-2 (IL-2) production by regulation of gene transcription. Glucocorticoids and nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit the formation of prostaglandins. Methotrexate inhibits the proliferation and activity of T cells and B cells. Etanercept and infliximab inactivate a cytokine called tumor necrosis factor-α (TNF-α). Immunoglobulin G-rheumatoid factor (IgG-RF) complexes are detected in the blood of patients with rheumatoid arthritis.
In patients with RA, NSAIDs are used to relieve pain and inflammation, and DMARDs are used to suppress the underlying disease process and slow the progression of joint destruction. The sites of action of selected antirheumatic drugs are depicted in Figure 30–1 and discussed below.
OSTEOARTHRITIS
Osteoarthritis, also called degenerative joint disease, is the most common joint disease in the world. It affects about 10% of persons over 60 years of age, and radiographic evidence of osteoarthritis can be found in most persons over 65. However, the disease is not simply associated with the aging process. Other factors that increase the risk for osteoarthritis include obesity, osteoporosis, smoking, heredity, repetitive use of joints through work or leisure activities, and joint trauma.
Osteoarthritis primarily affects weight-bearing joints and causes deformity, limitation of motion, and progressive disability. The cartilage undergoes thickening, inflammation, splitting, and thinning. Eventually, the cartilaginous layer is completely destroyed, leading to erosion and microfractures in the underlying bone. The major symptoms of osteoarthritis are pain, stiffness, and muscle weakness around affected joints.
Nonpharmacologic measures for treating osteoarthritis include joint protection and splinting, physiotherapy, orthotic prostheses to support the feet, and joint replacement surgery. Pharmacologic measures include NSAIDs, local glucocorticoid injections, and experimental chondroprotective drugs (e.g., chondroitin sulfate and glucosamine). Recently, sodium hyaluronate (Supartz) was approved for intra-articular injection as a type of joint fluid replacement in the treatment of osteoarthritis. It is a sterile, viscoelastic solution prepared from chicken combs (the fleshy growths on top of chicken heads).
GOUT
Gout is an arthritic syndrome caused by an inflammatory response to crystals of monosodium urate monohydrate in joints, renal tubules, and other tissues. The deposition of these crystals occurs as a consequence of hyperuricemia, which can result from overproduction or underexcretion of uric acid. Other risk factors for gout include obesity, alcohol consumption, and hypertension.
Acute gout is treated with an NSAID or colchicine to relieve joint inflammation. Subsequent attacks of gout can be prevented by long-term therapy with a drug that either increases uric acid excretion or inhibits uric acid formation and thereby reduces the serum level of uric acid.
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
MECHANISM OF ACTION
The nonsteroidal anti-inflammatory drugs comprise a large family of weak acidic drugs whose pharmacological effects result primarily from the inhibition of cyclooxygenase (COX), an enzyme that catalyzes the first step in the synthesis of prostaglandins from arachidonic acid and other precursor fatty acids (see Chapter 26). COX is a microsomal enzyme, existing as a dimer (two molecules linked to form a functional unit) in the lumen and membrane of the endoplasmic reticulum. NSAIDs decrease COX activity primarily by competitive inhibition; however, aspirin forms a covalent, irreversible inhibition of COX (Fig. 30–2). The net effect of NSAID administration is a decrease in the production of prostaglandins and other autacoids.
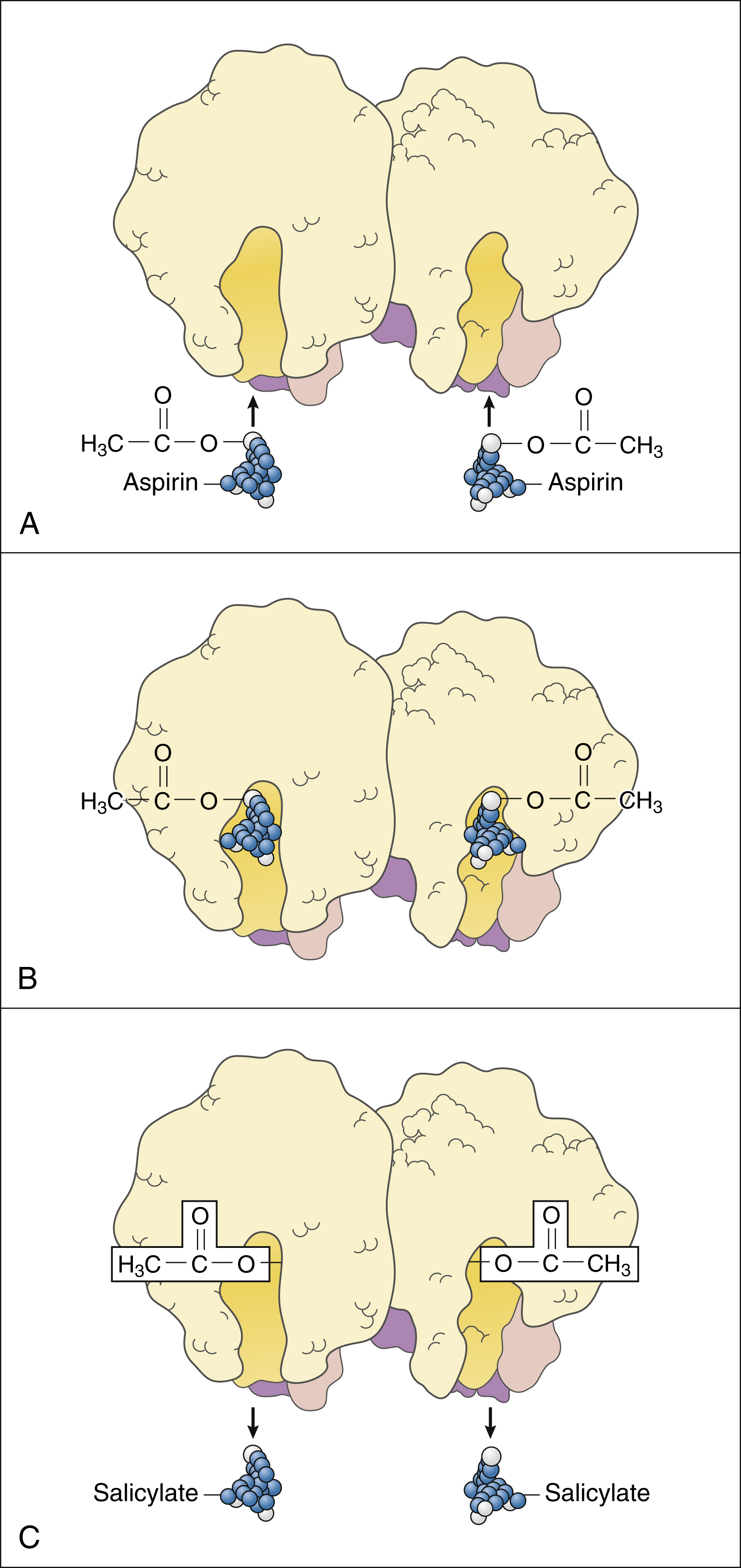
Figure 30–2 Mechanism of action for aspirin. (A) The COX enzyme exists as a homodimer with two interlocking identical molecules each providing an active channel for its substrates. Aspirin is chemically known as acetylsalicylic acid. (B) Aspirin molecules enter the active channel of COX and add an acetyl group to an amino acid R-group in the active site of COX. The acetyl group is attached to serine at the 530 position in COX-1 and to serine at the 516 position in COX-2. This produces irreversible or noncompetitive inhibition. (C) The aspirin molecule without the acetyl group is called salicylic acid or salicylate, which is a competitive inhibitor of COX.
Prostaglandin Effects
Prostaglandins play an important role in the development of pain, inflammation, and fever. Prostaglandins are released from cells in response to chemical stimuli or physical trauma. They sensitize sensory nerve endings to nociceptive stimuli and thereby amplify the generation of pain impulses. They also promote tissue inflammation by stimulating inflammatory cell chemotaxis, causing vasodilation and increasing capillary permeability and edema.
Fever, defined as the elevation of body temperature to a level above 37° C (98.6° F), often results from an alteration of hypothalamic thermoregulatory mechanisms. Bacterial toxins and other pyrogens stimulate the production of cytokines by leukocytes, and these cytokines increase prostaglandin synthesis in the preoptic area of the hypothalamus. The prostaglandins then act to reset the body’s thermostat to a new point above 37° C. This, in turn, activates temperature-raising mechanisms, such as a reduction in heat loss via cutaneous vasodilation, and causes the temperature to rise. All NSAIDs relieve fever by inhibiting prostaglandin synthesis in the hypothalamus, but these drugs are not capable of reducing body temperature below normal.
Cyclooxygenase Isozymes
Cyclooxygenase is now known to occur in two major isoforms: cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). COX-1 is a constitutive or “housekeeping” enzyme that is found in relatively constant levels in various tissues. COX-1 participates in the synthesis of prostaglandins that have a cytoprotective effect on the gastrointestinal (GI) tract. It also catalyzes the formation of thromboxane A2 in platelets, leading to platelet aggregation and hemostasis. In contrast, COX-2 is an inducible enzyme. Its levels are normally very low in most tissues but are rapidly up-regulated during the inflammatory process by proinflammatory substances (e.g., cytokines, endotoxins, and tumor promoters). Both COX-1 and COX-2 appear to participate in renal homeostasis.
Most of the NSAIDs available today are nonselective inhibitors of COX-1 and COX-2. The discovery of COX isozymes led to the development of selective COX-2 inhibitors, the first one being celecoxib. These selective inhibitors are effective anti-inflammatory drugs, and they produce less GI bleeding and ulcers than do the nonselective COX inhibitors.
A third COX isozyme (COX-3), which was recently discovered, appears to be an alternative splice variant of COX-1. Acetaminophen potently inhibits COX-3, and this finding likely explains the reason that acetaminophen has little anti-inflammatory action.
SPECIFIC AGENTS
Nonselective Cyclooxygenase Inhibitors
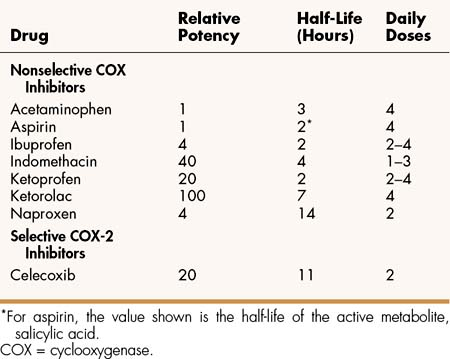
Among the nonselective COX inhibitors are many well-known NSAIDs that are available without a prescription, including aspirin, ibuprofen, ketoprofen, and naproxen. Acetaminophen is a weak anti-inflammatory agent, but it is also included in this class of drugs because it exerts analgesic and antipyretic effects via inhibition of COX. As shown in Table 30–1, NSAIDs vary greatly in potency and half-life, but most of them are administered from two to four times a day with food.
Lower doses of NSAIDs are usually sufficient to treat mild to moderate pain and counteract fever, whereas higher doses are generally needed to relieve inflammation associated with arthritic disorders and injuries. NSAIDs are particularly effective in relieving pain caused by tissue inflammation or bone or joint trauma, and they can be combined with opioid analgesics to obtain a greater analgesic effect and reduce the need for higher doses of opioids. For example, NSAIDs are widely used in the treatment of postoperative pain, either alone or in combination with an opioid.
Although NSAIDs are effective in relieving the pain of chronic disorders, their long-term use is associated with a number of adverse effects, including GI bleeding, peptic ulcers, and renal and hepatic dysfunction. Acetaminophen produces fewer GI problems than do other nonselective COX inhibitors, but it also lacks significant antiplatelet and anti-inflammatory activity. Although acetaminophen can be used concurrently with another NSAID for supplemental analgesia, alternative combinations of two NSAIDs should generally be avoided, not only because they increase the risk of GI and other side effects but also because they sometimes have adverse interactions. For example, aspirin and other salicylates displace some NSAIDs (e.g., ketorolac) from plasma proteins and thereby increase their serum levels significantly.
The NSAIDs can interact with a large number of other drugs through pharmacokinetic and pharmacodynamic mechanisms. Most NSAIDs inhibit the renal excretion of lithium and can increase lithium serum levels and toxicity. NSAIDs can reduce the clearance of methotrexate and aminoglycoside drugs. NSAIDs can also interfere to varying degrees with the antihypertensive effect of diuretics, β-adrenoceptor antagonists, angiotensin inhibitors, and other antihypertensive drugs. When given with potassium-sparing diuretics, NSAIDs can cause potassium retention and lead to hyperkalemia. Some drug interactions are only associated with a particular NSAID. For example, high doses of salicylates exert a hypoglycemic effect that can alter the effects of antidiabetic drugs. Indomethacin reduces the natriuretic effect of diuretics and can cause nephrotoxicity when given with triamterene.
Low doses of acetaminophen can be safely used for analgesia and antipyresis during pregnancy. The use of other NSAIDs during the second half of pregnancy is generally not recommended, however, because of potential adverse effects to the fetus. These effects result from prostaglandin inhibition and include GI bleeding, platelet inhibition, renal dysfunction, and premature closure of the ductus arteriosus.
Aspirin and Other Salicylates
The therapeutic value of salicylates was originally recognized when they were identified as the active ingredients of willow bark and other plant materials used in folk medicine to relieve pain and fever. Aspirin was synthesized in 1899 during a search for a salicylate derivative that would be less irritating to the stomach than salicylic acid. Aspirin soon became widely used around the world as an analgesic, antipyretic, and anti-inflammatory drug.
Salicylic acid derivatives include aspirin (acetyl-salicylic acid) and several nonacetylated drugs, such as salsalate, choline magnesium salicylate, and methyl salicylate (oil of wintergreen).
PHARMACOLOGICAL EFFECTS AND INDICATIONS
In adults, the salicylates can be used in the management of pain, fever, and inflammation, as well as in the prophylaxis of myocardial infarction, stroke, and other thromboembolic disorder. In children, the use of salicylates should be avoided, because the risk of Reye’s syndrome appears to be increased in virus-infected children who are treated with these drugs.
The analgesic, antipyretic, and anti-inflammatory effect of aspirin and other salicylates result from nonspecific inhibition of COX in peripheral tissues and the CNS. Aspirin irreversibly acetylates platelet COX and has a longer-lasting effect on thromboxane synthesis than do other salicylates. The antiplatelet effect of aspirin persists for about 14 days, whereas that of most other NSAIDs is much shorter. The effect is long-lived, because platelets lack a nucleus and do not make new COX enzyme.
The salicylates are usually administered orally, but formulations are also available for topical and rectal administration. The oral dosage of aspirin that is needed to inhibit platelet aggregation is somewhat lower than the oral dosage needed to obtain analgesic and antipyretic effects, and it is much lower than the oral dosage needed to relieve inflammation caused by arthritic and other inflammatory disorders. Figure 30–3 shows the relationship between the dosage of aspirin and the pharmacologic and toxic effects of the drug.

PHARMACOKINETICS
Aspirin is well absorbed from the gut. Although its concurrent administration with antacids may slow its absorption rate, it does not significantly reduce its bioavailability. Aspirin is rapidly hydrolyzed to salicylic acid (salicylate) by plasma esterase, and this accounts for its short plasma half-life (about 15 minutes). Most of the pharmacologic effects of aspirin are attributed to its salicylate metabolite, which has a half-life of about 2 hours. Aspirin itself, however, is responsible for irreversible inhibition of platelet COX and platelet aggregation.
Most of the salicylic acid formed from aspirin and other salicylate drugs is conjugated with glycine to form salicyluric acid. This substance is then excreted in the urine, along with about 10% of free salicylate and a similar amount of glucuronide conjugates. The rate of excretion of salicylate is affected by urine pH. For this reason, alkalinization of the urine by administration of sodium bicarbonate has been used to increase the ionization and elimination of salicylic acid in cases of drug overdose (see Chapter 2).
When a therapeutic dose of aspirin or other salicylate drug is ingested, the rate of metabolism and the rate of excretion of salicylate are proportional to the drug’s plasma concentration (first-order elimination). When an excessive dose is taken, the elimination pathways become saturated, giving rise to zero-order elimination. For this reason, larger doses can rapidly elevate plasma salicylate concentrations to toxic levels, especially in the elderly, who are at greatest risk of aspirin toxicity.
ADVERSE EFFECTS
The use of aspirin in children with chickenpox and other viral infections has been associated with Reye’s syndrome. As mentioned, treatment with salicylates, therefore, should be avoided in children.
Therapeutic doses of aspirin can cause gastric irritation and contribute to GI bleeding and peptic ulcers. Moderately high therapeutic doses can cause tinnitus, which is described as an abnormal auditory sensation or buzzing noise, and considered an early sign of salicylate toxicity.
Excessive doses of aspirin produce the toxic effects shown in Figure 30–3. Hyperventilation is caused by direct and indirect stimulation of the respiratory center in the medulla, and it often leads to increased exhalation of carbon dioxide and respiratory alkalosis. Higher plasma salicylate concentrations can cause fever, dehydration, and severe metabolic acidosis. If not treated promptly, these events can culminate in shock, coma, organ system failure, and death. Excessive doses of aspirin also cause hypoprothrombinemia, which is an impairment of hemostasis and causes bleeding.
Aspirin hypersensitivity is an uncommon but serious condition that can result in severe and potentially fatal anaphylactic reactions. Symptoms of aspirin intolerance include vasomotor rhinitis, angioedema, and urticaria (hives). Aspirin sensitivity occurs most frequently in persons with asthma, nasal polyps, or chronic urticaria. Persons who have had a severe hypersensitivity reaction to aspirin or another salicylate should not be treated with another type of NSAID, because a 5% risk of cross-sensitivity exists between salicylates and other NSAIDs.
TREATMENT OF SALICYLATE OVERDOSE
The treatment of salicylate poisoning may include the following: (1) induction of vomiting and gastric lavage to remove unabsorbed drug; (2) intravenous administration of sodium bicarbonate to counteract metabolic acidosis, increase the ionization of salicylate in the kidneys, and thereby enhance the rate of excretion of salicylate; and (3) administration of fluids, electrolytes, and other supportive care, as needed.
Acetaminophen
PHARMACOLOGICAL EFFECTS AND INDICATIONS
For more than 100 years, acetaminophen has been available for the treatment of mild pain and fever. The drug is a p-aminophenol derivative that exerts analgesic and antipyretic effects at doses that are well tolerated and produce remarkably few adverse effects during short-term administration. Unlike aspirin use, acetaminophen use has not been associated with Reye’s syndrome, so acetaminophen can be safely given to children with fever caused by viral illnesses.
Acetaminophen has only weak anti-inflammatory activity, partly because it is inactivated by peroxides produced in the cells of inflamed tissue. Recent evidence suggests the existence of a third COX isoform, designated COX-3, with roles in mediating pain and fever, and subject to inhibition by acetaminophen. Acetaminophen has little effect on COX-1 or COX-2 and, thus, lacks anti-inflammatory activity. Although acetaminophen is not considered a first-line drug for patients with arthritic disorders, it is sometimes used as an analgesic in those with mild arthritis. Because acetaminophen lacks the ability to inhibit thromboxane synthesis and platelet aggregation, it is not used for the prophylaxis of myocardial infarction, stroke, or other thromboembolic disorders.
PHARMACOKINETICS
Acetaminophen is rapidly absorbed from the gut, exhibits minimal binding to plasma proteins, and is widely distributed to peripheral tissues and the CNS.
As shown in Figure 30–4, acetaminophen is extensively metabolized by several pathways in the liver. Most of the drug is conjugated with sulfate and glucuronide, and these metabolites are excreted in the urine. A small amount of acetaminophen is converted by cytochrome P450 to a potentially hepatotoxic quinone intermediate. When a therapeutic dose of acetaminophen is taken, the quinone intermediate is rapidly inactivated by conjugation with glutathione. Toxic doses of acetaminophen, however, deplete hepatic glutathione, cause accumulation of the quinone intermediate, and lead to hepatic necrosis. To prevent liver damage, patients who ingest an overdose and are determined to be at risk for hepatotoxicity can be given acetylcysteine, a sulfhydryl compound that conjugates the quinone intermediate and renders it harmless.
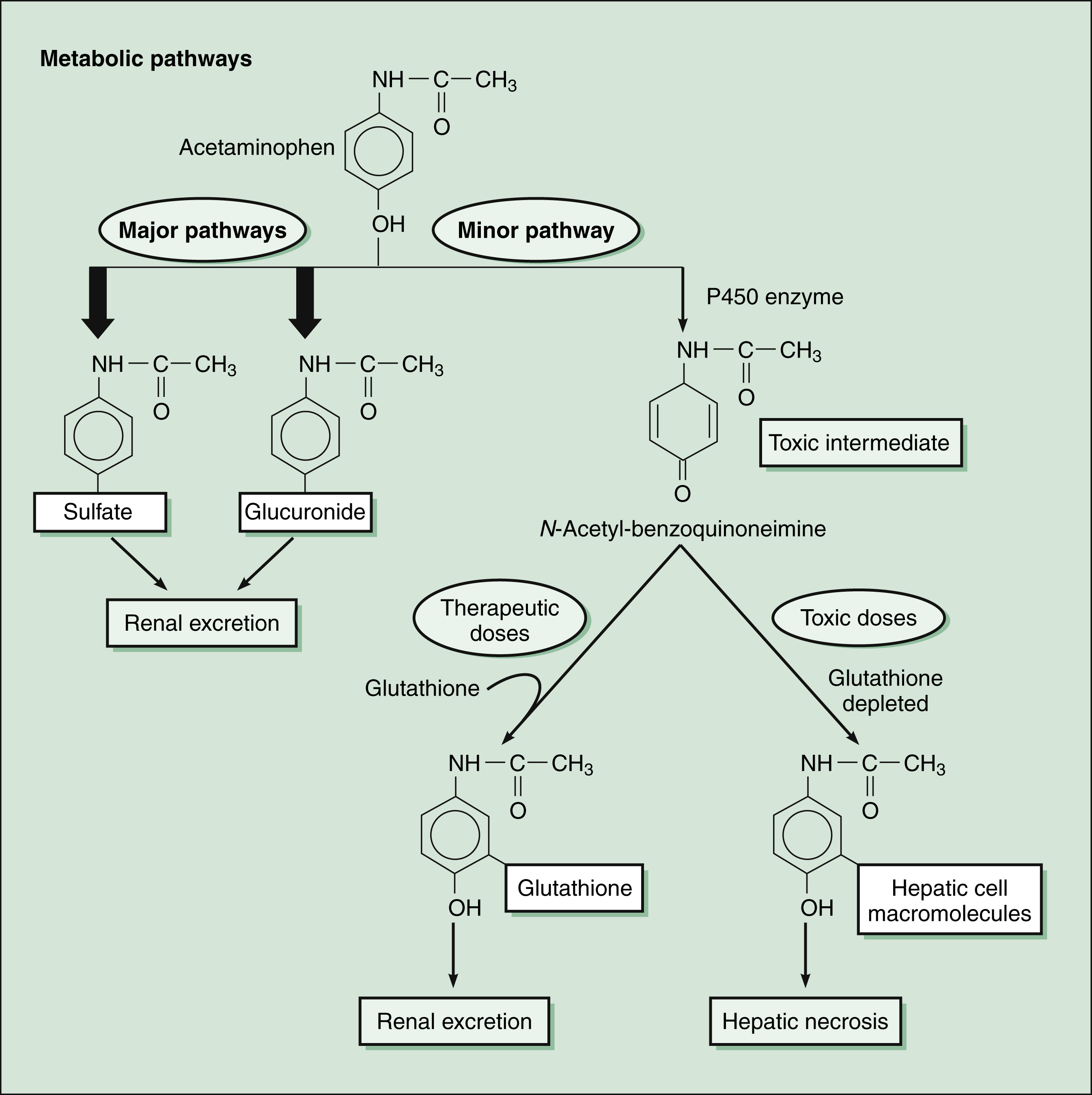
Figure 30–4 Acetaminophen metabolism and hepatotoxicity. Acetaminophen is primarily metabolized by conjugation with sulfate and glucuronide. A minor pathway involves oxidation of acetaminophen by cytochrome P450 enzymes (CYP1A2, CYP2E1, and CYP3A isozymes) to form a potentially toxic intermediate, N-acetyl-benzoquinoneimine. When a therapeutic dose of acetaminophen is taken, this quinone intermediate is conjugated with glutathione and excreted in the urine. When a toxic dose of acetaminophen is taken, glutathione stores are depleted, and the quinone intermediate attacks hepatic cell macromolecules. This process results in hepatic necrosis. Acetylcysteine (Mucomyst) given to treat acetaminophen overdose protects the liver by maintaining or restoring the glutathione levels, or by acting as an alternate substrate for conjugation with, and thus, detoxification of the reactive metabolite.
ADVERSE EFFECTS
Some epidemiologic evidence indicates that long-term use of acetaminophen is associated with an increased risk of renal dysfunction.M
Although therapeutic doses of acetaminophen are remarkably nontoxic, the ingestion of 20 to 30 tablets is sufficient to cause life-threatening hepatotoxicity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


