Allele type
Repeat range
Methylation status
Normal
5–44
Not methylated
Intermediate/gray zone
45–54
Not methylated
Premutation
55–200
Not methylated
Full mutation
>200
Methylated
Methylation mosaic
>200
Variable methylation
Premutation/full-mutation mosaic (repeat size mosaic)
Mixed premutation and full mutation
Full mutation may be methylated
Clinical Utility of Testing
Due to the presence of unrecognized FMR1 alterations in unaffected carrier mothers, the first indication of FXS within a family is usually the diagnosis of an affected child. Unfortunately, many families do not learn the FXS diagnosis for long periods after first concerns about their child’s development or behavior, and many have subsequent pregnancies before diagnosis for their first child. Such situations highlight the importance of diagnosing FXS so that children and families can receive the benefits of genetic counseling and early intervention services. Other than the infrequent deletion or point mutation, which often are spontaneous and not inherited from a parent, mothers of all FXS patients are either premutation or full mutation carriers. In turn, at least one of the mother’s parents has an FMR1 alteration. Consequently, FMR1 mutations may be present in siblings of an affected individual as well as other extended family members. It is important to remember that each daughter of unaffected males with a premutation (transmitting males) is an obligate carrier of a premutation, and that their offspring are at risk for FXS. Many extended families have been documented in which an FMR1 mutation has been transmitted through numerous generations and into family branches unknown to one another.
For DD children, FMR1 molecular testing is diagnostic, as FXS affects development from infancy; however, the nonspecific nature of FXS during early development makes the testing approach one of ruling out FXS in most situations. The hallmark finding in almost all patients with FXS is ID, but the physical and behavioral features of males with FXS are variable prior to puberty. Physical features not readily recognizable in preschool-age boys and become more obvious with age: long face, prominent forehead, large ears, prominent jaw, and enlarged testicles (macroorchidism). Motor milestones and speech are frequently delayed, and temperament often is affected (e.g., hyperactivity, hand flapping, hand biting, temper tantrums, and occasionally autism). Females with FXS usually have milder manifestations and as a result are more difficult to diagnose clinically. FXS always should be suspected in males with mild to moderate ID and females with mild ID until shown otherwise by negative FMR1 analysis.
Women who are full-mutation or premutation carriers have a 50 % risk of transmitting their abnormal allele in each pregnancy. While transmission of a full mutation always leads to a child with a full mutation, the risk of a premutation transmission resulting in an affected offspring with a full mutation is proportional to the maternal allele size. Empirically, the 50 % risk of a female carrier producing an affected male child is reduced to 7 % if the premutation contains 56–59 repeats, 10 % for 60–69 repeats, 29 % for 70–79 repeats, 36 % for 80–89 repeats, and 47 % for 90–99 repeats; it reaches the maximum, 50 %, when a premutation has >100 and up to 200 repeats. Because females have approximately 50 % penetrance, the risk of having an affected female child is half that of having an affected male child in any premutation repeat interval category.
Prenatal testing for FMR1 mutations is available in some clinical molecular laboratories. Genomic DNA isolated from amniocytes obtained during amniocentesis at 16–18-week gestation or from chorionic villus sampling (CVS) at 10–12-week gestation can be used for testing. Prenatal molecular analysis proceeds in much the same fashion as that performed on DNA obtained from adult peripheral blood. However, the DNA analysis of CVS may be more complex, as chorionic villi are extraembryonic. Hypermethylation in CVS may be incomplete and not representative of the true FMR1 methylation status in fetal tissues; therefore, a follow-up amniocentesis occasionally may be required to resolve ambiguous CVS test results.
General population screening for FMR1 mutations has been proposed but remains controversial. In comparison to most disorders already screened for in the newborn period, FXS is more prevalent and testing is highly reliable; however, before population screening is practical for newborns and women of reproductive age, the relatively high costs and the technical complexities of testing must be resolved. Several recent studies demonstrate that higher throughput testing for population-screening purposes is becoming increasingly possible, both from a cost and a time perspective [6, 7]. Protein testing of FMRP may also be useful for screening populations with ID.
Available Assays
Routine clinical testing for FMR1 mutations includes molecular assessment of both the trinucleotide repeat number and the FMR1 methylation status. Traditional approaches to this testing include two concurrent analyses: (1) double-digest Southern blot analysis using a methylation-sensitive restriction enzyme such as EagI, BssHII, or NruI along with a methylation-insensitive restriction enzyme such as EcoRI or HindIII [8]; and (2) polymerase chain reaction (PCR) assays specific for the CGG repeat segment of FMR1 (Fig. 7.1) [9]. When used in conjunction with PCR, Southern blot analysis provides a more complete inspection of the trinucleotide repeat region by detecting multiple possible molecular events, including repeat expansion, DNA methylation, and the relatively rare FMR1 deletions. Specialized fragile X chromosome cytogenetic analysis, using special culture techniques to induce fragile sites, is no longer used for diagnosis of FXS due to low sensitivity. While only a very few FXS patients with point mutations in FMR1 have been identified, clinical molecular testing does not routinely investigate this gene for point mutations, deletions, insertions, or inversions downstream of the repeat segment.
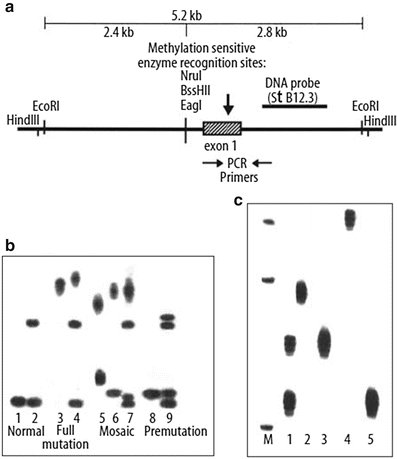
Figure 7.1
Repeat expansion and methylation in FMR1. (a) Restriction enzyme map of FMR1, with locations of restriction enzyme sites, DNA probe, and PCR primers used in molecular testing. Top line indicates DNA fragments generated using EcoRI and EagI as depicted in the Southern blot analysis in b (2.8 kb and 5.2 kb fragments are detected by the DNA probe StB12.3). The vertical arrow indicates the location of the CGG repeat in exon 1. (b) Southern blot analysis of FMR1. Only the 2.8 kb fragment is detected in normal males (lane 1), while both the 2.8 kb and 5.2 kb fragments are detected in normal females (lane 2) due to methylation associated with normal X-inactivation that prevents cutting of the inactivated methylated allele with the methylation-sensitive restriction enzyme EagI, BssHII, or NruI. Completely methylated full mutations are depicted in lane 3 (affected male with full mutation) and lane 4 (affected female with full mutation contained on one of her X chromosomes; normal allele on her other X chromosome). Smeary signals occur due to variable repeat expansion within peripheral lymphocytes used for DNA isolation. Mosaic patterns are illustrated in lane 5 (male with partial methylation of full mutation), lane 6 (male with premutation/full-mutation mosaicism), and lane 7 (female with premutation/full-mutation mosaicism). Lane 8 illustrates a transmitting male with a premutation and lane 9 illustrates a female with a premutation. Both premutations contain approximately 75 repeats. (c) PCR analysis of FMR1 repeats from five individuals separated on a 6 % polyacrylamide gel. Lane 1 contains PCR products from a female with 20 and 30 repeats, respectively, contained within her two normal alleles. Lanes 2, 3, and 5 are males with normal repeat alleles (40, 30, and 20 repeats, respectively), while lane 4 illustrates a male with a 65-repeat premutation allele. Smeary signals result from DNA polymerase stuttering during the PCR amplification.
In most clinical laboratories, PCR is used to size normal and smaller premutation-sized alleles with a typical sensitivity of up to approximately 120 repeats. PCR product yield is inversely proportional to the number of trinucleotide repeats such that with traditional PCR methods, little or no product can be obtained when larger repeats are present. Since the inception of diagnostic testing for FXS, different testing strategies to facilitate the transition away from Southern blots have been met with limited success. Although some PCR-based testing protocols have higher sensitivity regarding detection of larger repeats, most laboratories have been reluctant to adopt these practices due to technical difficulties. The ability to rapidly identify trinucleotide expansions in an efficient and cost-effective manner has been revisited in recent years and several very similar protocols to address this challenge have been developed [6, 10–13]. Each of these new methods relies on a triplet repeat-primed PCR reaction to separate alleles with apparent CGG expansions from those without expansions. Typically, this is based on a threshold of 55 trinucleotide repeats; however, the threshold can be adjusted according to the needs of the user (Fig. 7.2). Patients with expanded alleles detected by this PCR screening test then can be reflex tested by Southern blot analysis for further evaluation of expansion size and methylation status. This considerable advancement in testing allows the opportunity to significantly reduce the number of Southern blots needed, thereby significantly decreasing the turnaround time for most samples tested. Although only routinely performed in a few clinical laboratories, protein-based testing for FMRP can be utilized. Since severity of the FXS phenotype appears to inversely correlate with FMRP expression, assessment of FMRP production in patients with methylation mosaicism may be a useful prognostic indicator of disease severity [14].
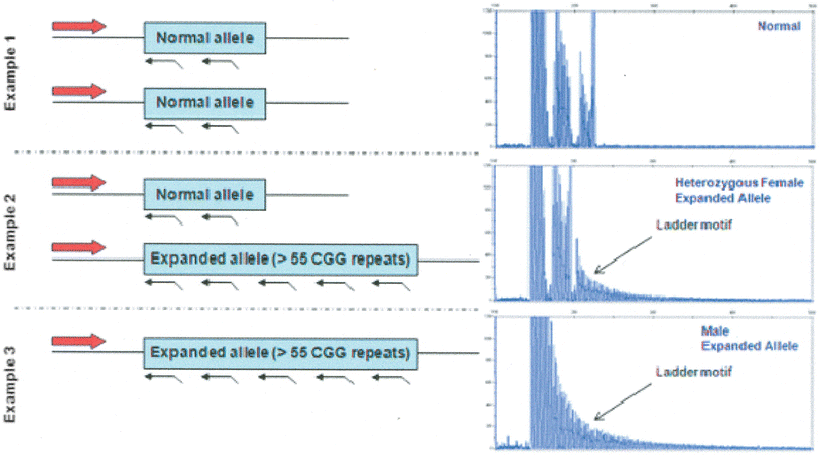
Figure 7.2
Triplet repeat-primed FMR1 PCR screening test for fragile X alleles. The forward PCR primer is located upstream of the FMR1 CGG region while the fluorescently-labeled reverse primer randomly binds inside the FMR1 CGG repeat region. These PCR primers generate different sized amplicons depending on the size of the CGG repeat region present. The presence or absence of a trinucleotide “ladder” can be easily identified and a threshold of 55 CGG repeats typically is used to define expanded FMR1 alleles. Example 1 shows the trace for a female with two normal alleles, neither of which results in the characteristic ladder motif present for expanded alleles. Example 2 illustrates the typical pattern for a female with one normal allele and one expanded allele while Example 3 illustrates the typical pattern for a male with an expansion at the FMR1 locus. From Basehore MJ, Marlowe NM, Jones JR, Behlendorf DE, Laver TA, Friez MJ. Validation of a screening tool for the rapid and reliable detection of CGG trinucleotide repeat expansions in FMR1. Genet Test Mol Biomarkers. 2012 Jun;16(6):465–70. doi: 10.1089/gtmb.2011.0134. Epub 2012 Jan 6. Reprinted with permission from Mary Ann Liebert, Inc.
Interpretation of Test Results
Because FMR1 appears to be the only disease-causing gene for FXS, test specificity is 100 %. Using both Southern blot analysis and PCR specific for the FMR1 locus, test sensitivity for repeat expansion can be estimated to be nearly 99 %, as only rare point mutations, small deletions/insertions remote from the repeat segment, or gene inversions would be missed. Non-repeat expansion molecular alterations may be under-ascertained in FMR1 since gene regions downstream of the repeat segment in exon 1 are rarely investigated. When identified, the presence of cellular mosaicism, in either repeat size or methylation status, presents potential problems for the prediction of FXS severity. Essentially all patients with mutations resulting in reduction of FMRP are impaired, but expression of variable amounts of FMRP may allow some individuals to function at a higher level than expected; therefore, these individuals may occasionally have higher intelligence quotients (IQ > 70). Prognostication of severity based on testing of a young child should be predicated with great caution during genetic counseling, because no long-term study exists following the development of individuals with methylation mosaicism.
Laboratory Issues
If the etiology of ID in an individual is unknown, DNA analysis for FXS should be performed as part of a comprehensive genetic evaluation that includes routine cytogenetic analysis (see Chap. 6), since cytogenetic abnormalities have been identified at least as frequently as FMR1 mutations in individuals with ID. In addition, the use of Southern blotting on DNA isolated from amniocytes for prenatal FMR1 analysis, with typical 2- to 3-week turnaround times, may lead to stressful situations on occasion regarding the timing of possible pregnancy termination. Utilization of CVS specimens provides additional time for possible pregnancy termination, but equivocal results sometimes occur due to incomplete methylation when a full mutation is present, requiring follow-up amniocentesis.
PCR-based kits used to estimate repeat copy number are available through some commercial vendors but not widely utilized. Many laboratories use laboratory-developed methods for both FMR1 Southern blot analysis and PCR. Patient control cell lines may be purchased from the Coriell Institute (http://coriell.umdnj.edu/). Testing for FXS is routinely included in proficiency tests administered by the College of American Pathologists.
Angelman Syndrome and Prader-Willi Syndrome
Molecular Basis
Angelman syndrome (AS) is a neurodevelopmental disorder characterized by severe DD/ID, gait ataxia, microcephaly, seizures, and a happy demeanor that includes frequent laughing, smiling, and excitability [15]. AS is caused by deficient expression of the maternally inherited copy of the UBE3A (ubiquitin-protein ligase E3A) gene which can result from one of several different genetic abnormalities involving the proximal part of the long arm of chromosome 15 (15q11–q13). This two megabase (Mb) region of chromosome 15 contains multiple imprinted genes, meaning that their expression is dependent on the parent of origin. Within this region of chromosome 15, the MKRN3, MAGEL2, NECDIN, and SNURF–SNRPN genes, as well as a cluster of C/D box small nucleolar RNAs (snoRNAs) are paternally expressed, while the UBE3A and ATP10C genes are maternally expressed. Imprinting of genes in this domain is coordinately controlled by a bipartite imprinting center (IC) which overlaps the SNRPN promoter and extends approximately 35 kb upstream. Flanking this imprinted region are several low-copy repeats that predispose the region to chromosomal rearrangement by unequal crossing over.
Imprinted UBE3A expression is tissue specific, displaying predominantly maternal expression in the human fetal brain and adult frontal cortex but biparental expression in other tissues. UBE3A encodes a protein that is involved in the ubiquitination pathway which targets certain proteins for degradation [16]. Four known genetic mechanisms cause loss of UBE3A expression, and explain 85–90 % of AS cases [17]. A 4 Mb deletion of the maternal chromosome 15q11–q13 region occurs sporadically in 65–70 % of cases and is due to unequal crossing over. Paternal uniparental disomy (UPD) of chromosome 15 is detected in about 7 % of cases and is likely to be postzygotic in origin. Approximately 3 % of AS cases involve an imprinting defect caused by microdeletions of the IC and result in lack of expression of the maternally inherited UBE3A gene in the brain. Point mutations within UBE3A (mostly truncating mutations) are found in approximately 11 % of cases [17]. Currently, approximately 10 % of patients with a clinical diagnosis of AS have no identifiable chromosomal or molecular abnormality, most likely due to an undetected abnormality affecting the UBE3A gene or a mutation in another gene within the ubiquitination pathway.
While the AS phenotype results from loss of expression of maternally expressed UBE3A, another developmental disorder, Prader-Willi syndrome (PWS), is due to loss of expression of paternally expressed genes within this 15q11–q13 region. PWS is characterized by infantile hypotonia, hypogonadism, dysmorphic appearance, small hands and feet, hyperphagia and obesity, DD, and ID. Approximately 70 % of PWS cases involve a 4 Mb deletion of the paternal chromosome 15q11–13, while 20 % of PWS cases are due to maternal UPD of chromosome 15. Another 1–5 % of cases are thought to be due to an imprinting defect. Recent evidence suggests that loss of expression of one or more of the C/D box snoRNAs, including SNORD116, encoded within the SNRPN locus may cause the PWS phenotype [18, 19]. The following section reviews the available diagnostic testing for AS, as well as PWS, since the primary testing method used to diagnose AS also will diagnose PWS.
Clinical Utility of Testing
Infants with AS commonly present with non-specific findings such as DD and/or seizures. These findings may result in a broad differential diagnosis, which can include inborn errors of metabolism, mitochondrial encephalopathy, cerebral palsy, and other neurodevelopmental syndromes including Rett syndrome, Christianson syndrome, and Pitt-Hopkins syndrome, which will be reviewed later in this chapter. Analysis of parent-specific DNA methylation imprints in the 15q11–q13 chromosome region is typically the first test that is ordered as it will detect approximately 80 % of individuals with AS, including those with a deletion, paternal UPD, or an IC defect. This analysis also is used to diagnose PWS and will detect nearly 100 % of those cases. Methylation analysis for AS/PWS typically investigates the methylation status of CpG dinucleotide sites within the promoter region of the SNRPN gene. Once the diagnosis of AS (or PWS) is established through an abnormal DNA methylation pattern, the specific genetic mechanism involved can be investigated for the purposes of genetic counseling. Fluorescent in situ hybridization (FISH) or array-based comparative genomic hybridization (aCGH) analysis can be used to detect a deletion, while DNA microsatellite analysis can be performed to investigate the presence of UPD. For patients with an AS phenotype but normal DNA methylation, sequence analysis of the UBE3A gene can be used for further evaluation of the diagnosis. Collectively, molecular testing for AS through DNA methylation studies and sequencing of UBE3A will identify an alteration in approximately 90 % of individuals with clinical AS/PWS.
The recurrence risk for AS in a family and the type of prenatal testing that is available will vary according to the chromosome or molecular defect that is ultimately detected in the proband. For this reason, prenatal diagnosis should be undertaken only after the specific genetic mechanism in the proband has been determined and the parents have received genetic counseling. In cases where de novo deletions or UPD have been demonstrated, recurrence risk will be low, although prenatal testing may be offered for reassurance purposes. For cases of IC and UBE3A mutations, recurrence risk can be as high as 50 %. Since germline mosaicism has been reported in mothers of AS patients with UBE3A mutations, prenatal testing should be offered even if a mutation is not identified in the mother. Because methylation status within the promoter region of the SNRPN gene is established in early embryonic development, DNA methylation analysis on cells obtained from CVS is theoretically possible; however, most clinical molecular laboratories currently performing prenatal methylation testing prefer to use amniocytes for testing due to the relative hypomethylation of cells derived from the placenta [15]. Prenatal testing is appropriate for families without a previous child with AS if a deletion of chromosome 15q11–q13 is suspected on chromosome analysis of CVS or amniotic fluid, or if trisomy 15 is noted on CVS but a normal karyotype is found on amniotic fluid, which would suggest that a trisomy rescue event may have occurred (possibly resulting in UPD). Prenatal testing also would be indicated if either a de novo chromosome 15 translocation or a supernumerary marker chromosome is found by karyotype analysis.
Parents of patients with deletions, specifically the mothers of AS patients, should be tested by chromosome and FISH analysis to determine if they carry balanced chromosome rearrangements or deletions which are not expressed as an abnormal phenotype in that parent. Chromosome analysis is appropriate for parents of patients with UPD combined with a Robertsonian translocation to determine whether the translocation is inherited or de novo. Parents also should be tested for mutations that are identified in the proband, such as IC deletions or UBE3A mutations. If a parent of an AS patient is found to be a carrier of a mutation or a chromosomal translocation, then the siblings of that parent should be offered testing.
Available Assays
DNA Methylation Analysis
Standard molecular techniques for the methylation analysis of CpG sites within the SNRPN gene promoter region include (1) double-digest Southern blot analysis using a methylation-sensitive enzyme such as NotI along with a methylation-insensitive enzyme such as XbaI [20]; (2) PCR amplification of the SNRPN promoter region following either NotI or mcrBC digestion; and (3) methylation-specific PCR (MS-PCR) which is based on modifying DNA with bisulfite, which converts unmethylated cytosine (C) to uracil (U), followed by amplification using primers specific for the unmethylated and methylated alleles [21].
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) provides a method to simultaneously semiquantitatively analyze copy number changes and DNA methylation status at numerous sites across the 15q11–q13 region. Oligonucleotide probes specific to the SNRPN gene are utilized, of which a subset contain a HhaI restriction site. After hybridization and ligation, the methylation-sensitive restriction enzyme HhaI is used to digest the unmethylated DNA. PCR amplification using a universal primer is performed, followed by separation of the amplification products by capillary electrophoresis. If the site is not methylated, the HhaI digestion will prevent amplification of the MS-MLPA probe. If the CpG sites are methylated, the HhaI enzyme is unable to digest the DNA, and PCR products are generated and detected. Individuals with AS will have two unmethylated (maternal) alleles and accordingly no MS-MLPA signal will be present. Individuals with no methylation abnormality will show a 50 % reduction in the MS-MLPA signal due to the presence of both a methylated and an unmethylated allele. Individuals with PWS will have two methylated (paternal) alleles and will show a 50 % increase in the MS-MLPA signal compared to normal controls.
Pyrosequencing is a quantitative method available to assess the CpG methylation status of the SNRPN gene [22]. As in MS-PCR, the genomic DNA is treated with bisulfite to convert unmethylated C to U, whereas methylated cytosine (mC) will remain unchanged. During PCR amplification of the bisulfite-treated DNA, the U will be amplified as thymine (T) and the mC will be amplified as C. Pyrosequencing, which is a “sequencing-by-synthesis” method based on the luminometric detection of a pyrophosphate release that occurs upon the incorporation of nucleotides into the sequence, is then utilized to discriminate C and mC by the presence of a T or C at each CpG site analyzed, respectively. This information is used to determine the percent methylation at each CpG site, which will be approximately 50 % for normal individuals who have both a methylated (paternal) and unmethylated (maternal) allele. Individuals with AS will have 0 % methylation at each site, and individuals with PWS will have 100 % methylation at each site.
Uniparental Disomy Analysis
For individuals who exhibit methylation abnormalities but are negative for a deletion by FISH or aCGH, DNA microsatellite analysis to test for whole or segmental UPD of chromosome 15 is available for further evaluation of the diagnosis. UPD analysis requires DNA from the proband as well as both parents. Polymorphic repetitive regions (microsatellites) along chromosome 15 are analyzed to determine if the proband’s genetic constitution reflects contribution from both parents at all loci tested (biparental) or from only one parent (UPD). Paternal UPD of chromosome 15 is consistent with AS while maternal UPD of chromosome 15 is consistent with PWS. Alternatively, SNP-based arrays can be used to detect segmental and whole chromosome UPD, but may not identify all cases of UPD, because the segment of UPD may be too small to be detected by SNPs designed to evaluate UPD of larger chromosomal regions. Abnormal methylation studies and biparental inheritance of chromosome 15 is suggestive of an imprinting center defect.
Targeted UBE3A Analysis
When DNA methylation testing is normal, UBE3A sequence analysis should be considered. The majority of mutations identified in the UBE3A gene are protein truncating mutations [23, 24]. These mutations must be present on the maternal allele in order to be pathogenic since UBE3A expression occurs only from the maternal allele. In some cases, small intragenic deletions of UBE3A have been detected, so gene-specific deletion and duplication testing should be considered for individuals who test negative for UBE3A gene sequencing [25, 26].
Interpretation of Test Results
If the methylation pattern or methylation-specific amplification is characteristic of only paternal inheritance in the individual tested, then a diagnosis of AS is confirmed (Fig. 7.3, lane 8). Conversely, a methylation pattern characteristic of only maternal inheritance is diagnostic for PWS (Fig. 7.3, lane 7). Methylation assays detect virtually all cases of PWS and AS that are caused by large deletions, UPD, and IC defects; however, they will not detect rare small deletions that do not involve the SNRPN locus or sequence alterations within the UBE3A gene. Therefore, methylation analysis will detect approximately 80 % of AS cases. Sequencing of the UBE3A gene will detect another 11 % of cases. Approximately 10 % of AS cases will not be detected with the currently available tests.
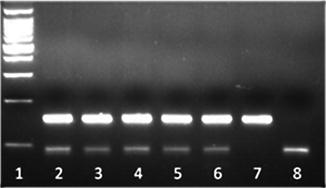
Figure 7.3
Methylation-sensitive PCR (MS-PCR) analysis of SNRPN DNA of patient samples referred to a clinical molecular laboratory. PCR products are amplified from the methylated and unmethylated alleles of the SNRPN locus. Normal individuals exhibit a biparental inheritance pattern (both upper methylated and lower unmethylated allele PCR products present; lanes 2–6), whereas patients with PWS show a pattern of only maternal inheritance (only upper methylated allele PCR product present; lane 7) and patients with AS show a pattern of only paternal inheritance (only lower unmethylated allele PCR product present; lane 8). Molecular weight ladder present in lane 1.
Laboratory Issues
If the etiology of DD in a patient is unknown, DNA analysis should be performed as part of a comprehensive genetic evaluation that includes assessment of single gene disorders by molecular and cytogenetic approaches. Although every laboratory does not need to provide all testing methods, a smooth progression through the various tests may be necessary to determine the genetic mechanism causing AS. Testing is facilitated by a clinical molecular laboratory that is able to offer DNA methylation, FISH, UPD analysis and sequencing of the UBE3A gene, as well as gene-specific deletion/duplication testing. Mutation analysis of the IC is available only on a research basis.
The SNRPN probe for Southern blot analysis is available from American Type Culture Collection (http://www.atcc.org). Caution should be exercised when interpreting MS-PCR results, as allele dropout has been reported [27]. Because of the risk of allele dropout, multiple MS-PCR primer sets should be considered to minimize the possibility of a false-positive result. An MS-MLPA kit is available from MRC-Holland (http://www.mrc-holland.com). A pyrosequencing kit to evaluate methylation status of the AS/PWS critical region is available from Qiagen (http://www.qiagen.com). There are many microsatellite markers available for testing of the 15q11–q13 region, but caution should be exercised because some previously used markers are now considered problematic, such as DS15S113 and D15S817 [28, 29].
Rett Syndrome and Other MECP2-Related Disorders
Molecular Basis
Rett syndrome is an X-linked neurodevelopmental disorder that almost exclusively affects females. In the classic form of the disease, affected girls appear to develop normally until the age of 6–18 months followed by a characteristic pattern of regression, which includes deceleration of head growth leading to acquired microcephaly, autistic features, loss of speech and purposeful hand use, irregular breathing patterns, stereotypical hand wringing, and seizures [30]. There are well-documented criteria for the clinical diagnosis of Rett syndrome which include the clinical features that are considered as either necessary or supportive for the diagnosis [31]. The frequency of classic Rett syndrome is approximately 1 in 10,000 females [32]. In 1999, mutations in the methyl-CpG-binding protein 2 (MECP2) gene located at Xq28 were reported to be the underlying cause for Rett syndrome [33]. Since that time, a broader range of clinical phenotypes have also been associated with mutations in MECP2 [34]. This section focuses on MECP2 variants causing Rett and atypical-Rett syndromes which are X-linked dominant conditions. The other MECP2-related phenotypes primarily affect males and are considered to be X-linked recessive disorders.
Since being associated with MECP2, the field of Rett syndrome research has focused attention on determining the function of the MECP2 protein to better understand the underlying pathophysiology of the condition. The current perspective of MECP2 function is complex and is based on its apparent involvement as both a transcriptional repressor and activator [35]. MECP2 binds preferentially to methylated DNA via its methyl-CpG-binding domain, and silences transcription by recruiting corepressor complexes through its transcriptional repression domain. More recent studies have demonstrated that loss of MECP2 leads to reduced expression of numerous genes, implying a role in upregulating gene expression, although the mechanisms involved are not well understood [36]. This is an area of great interest given that Rett syndrome is not associated with any sort of gross anatomic abnormalities of the brain. MECP2 clearly affects transcriptional regulation more dramatically in certain regions of the brain than other regions. The physiologic significance of these differences needs to be understood in order for individuals affected by Rett syndrome to benefit from promising new therapeutic options in the future.
Clinical Utility of Testing
Independent studies have confirmed MECP2 as the major causative gene for Rett syndrome by the identification of pathogenic mutations in approximately 95 % of classic cases. Approximately 85 % of classic Rett patients have either point mutations or small insertions/deletions within the MECP2 coding region, while larger deletions have been identified in approximately 10 % of classic cases [37]. Nearly 50 % of Rett patients have one of eight recurrent point mutations, while most of the remainder has one of the many other described pathogenic alterations. It should be noted that greater than 99 % of mutations in individuals with classic Rett syndrome are de novo, with the large majority occurring on the paternally inherited allele [38]. Mutations also have been found in atypical mild variant cases and in severe early-onset cases of Rett syndrome. In addition, MECP2 mutations have been documented in patients with an Angelman-like presentation and in individuals with autistic phenotypes. Affected males with variable phenotypes ranging from lethal neonatal encephalopathy to uncharacterized ID have been shown to be caused by MECP2 mutations. The presentation of Rett syndrome also has been documented in males with Klinefelter (47, XXY) syndrome, as well as those with mosaic MECP2 mutations [39]. Definitive genotype-phenotype correlations have not been consistent, although several findings related to specific mutations appear to be reproducible [40]. Some studies have investigated multiple individuals with the same mutation but variable degrees of clinical severity, demonstrating that additional factors beyond the specific MECP2 alteration play significant roles in the presentation of Rett syndrome [41]. In males, larger duplications of Xq28 that include MECP2 have a consistent set of clinical features that include hypotonia, severe ID, absence of speech, seizures, and recurrent respiratory infections that often lead to death at an early age [42]. Given the spectrum of neurodevelopmental phenotypes associated with MECP2 mutations, the clinical utility of molecular testing is significant.
Available Assays
The MECP2 gene is composed of four exons, which give rise to two distinct MECP2 isoforms [43]. Diagnostic testing for Rett syndrome should begin with DNA sequence analysis of the entire MECP2 coding region given the profile of known mutations. In the past, mutation scanning using denaturing high performance liquid chromatography (DHPLC) was used, but very few laboratories still utilize this method given the relative ease of sequencing MECP2. Sanger sequencing with capillary electrophoresis continues to be considered the gold standard for point mutation detection. Approximately 85 % of classic Rett syndrome patients have mutations that are detectable by sequencing of MECP2, which is performed by numerous clinical molecular laboratories (http://www.genetests.org/).
To increase the overall mutation detection for the MECP2 gene, additional testing is available for larger MECP2 dosage alterations, present in approximately 10 % of classic Rett patients. Deletions and duplications involving all or part of the gene have been identified by dosage-sensitive testing methods such as multiplex ligation-dependent probe amplification (MLPA), quantitative real-time PCR analysis, and targeted aCGH.
Interpretation of Test Results
Rett syndrome is commonly considered in females with DD, making the volume of MECP2 testing performed worldwide significant. Molecular confirmation of Rett syndrome is straightforward when one of the eight common MECP2 mutations is present. Interpreting other less common alterations can be assisted by use of available mutation databases. RettBASE, the database dedicated to MECP2 (http://mecp2.chw.edu.au/), and the Human Gene Mutation Database (HGMD) are currently the most comprehensive resources. Some of the changes reported in the databases are not clinically correlated, leaving their pathogenicity uncertain. When other novel variants are identified, evaluation of additional family members to determine the segregation pattern may allow the clinical significance of an alteration to be interpreted with greater certainty. For females with variants of unknown clinical significance, the most common strategy is to test both parents. Alterations that are shown to be de novo are normally accepted as pathogenic mutations. In some rare cases, two de novo alterations are present with uncertainty which change is pathogenic, or whether their collective impact on protein function is causative of the symptoms. A variant of unknown significance inherited from the father is interpreted as not being clinically significant given the expectation that the variant would have an even greater phenotypic consequence in a male with a single X chromosome. When variants are inherited from the mother, the interpretation becomes more challenging and typically requires additional testing which often includes both X-inactivation testing of the mother and variant analysis of additional family members. If the proband and her mother carry the same alteration and both have normal/random X-inactivation patterns, the variant typically is interpreted as a benign variant with no clinical significance. Testing of extended family members has the potential to identify the change in a phenotypically normal male which also confirms that the change is not clinically relevant. When testing males, the interpretation issues change to some degree and the strategy for these will be discussed later in the X-linked intellectual disability section of this chapter.
Laboratory Issues
One of the issues encountered in sequence-based clinical testing is the interpretation of novel sequence variations, particularly alleles of uncertain pathogenic significance. A valuable resource for laboratories and referring clinicians is provided by the American College of Medical Genetics, which has issued updated recommended standards and guidelines for interpretation of sequence variations [44]; the recommendations are available at http://www.acmg.net. Another current issue pertaining to molecular testing for Rett syndrome is the value of two-tier testing (sequencing followed by dosage analysis) to provide comprehensive mutation analysis of MECP2. Identification of the mutation in the proband facilitates prenatal testing in subsequent pregnancies, although the majority of MECP2 point mutations are de novo and of paternal origin, with extremely low rates of recurrence. Prenatal testing should be offered to females who are known to carry a point mutation or large rearrangement in MECP2. In cases where the mother is not a carrier, prenatal testing may be sought for parental reassurance due to rare reports of germline mosaicism [45]. The current focus on point mutation analysis within the coding region by sequence analysis does not rule out potential mutations in regulatory elements or other important noncoding regions of MECP2. Equally important to consider are the other conditions that phenotypically overlap with Rett syndrome and should be given further consideration when clinical testing for MECP2 is negative. Several of these syndromes and their respective gene associations are discussed in the following section.
Angelman/Rett Syndrome Second-Tier Testing
Molecular Basis
AS and Rett syndromes are clinically similar conditions that often present with numerous overlapping phenotypic features. Clinicians with significant clinical experience typically can distinguish the two conditions, but in many cases clinical diagnosis can be challenging. As discussed earlier, AS and Rett syndromes are caused by disruption or loss of normal gene function of the UBE3A and MECP2 genes, respectively. Testing for these conditions is common given their prevalence and the number of individuals with DD suggestive of one or both of these diagnoses. Accordingly, many patients have clinical presentations that mimic AS and Rett syndrome and have normal clinical test results for both disorders. In these instances, additional testing of genes causing diseases with overlapping phenotypes may provide the molecular diagnosis needed. Some of the most common conditions that should be considered in the differential diagnosis for AS and Rett syndrome for which diagnostic testing is available are discussed in this section. The syndromes detailed in this section are not meant to be an all-inclusive list, but rather as a starting point for second tier testing for patients with features in this clinical spectrum of ID/DD.
Clinical Utility of Testing
The molecular confirmation of a clinical diagnosis allows the physician to provide recurrence risks and genetic counseling, as well as prenatal diagnosis, to the proband and additional family members. Once the presence of a causative mutation has been established in an individual, targeted analysis of fetal samples is available for prenatal testing, as well as for other family members.
Molecular Genetic Testing
CDKL5 Gene
Mutations in the CDKL5 (cyclin-dependent kinase-like 5) gene have been identified in individuals with early-onset infantile spasms and clinical features that overlap other neurodevelopmental disorders, such as AS and Rett syndromes [46]. Subsequently, mutations in CDKL5 (also known as Serine Threonine Kinase 9 or STK9) have been associated with an atypical variant of Rett syndrome, which includes ID and other severe neurological symptoms including severe early-onset seizures, regression of communication and skills, and global developmental delay. Rett-like stereotypies such as hand-wringing have been identified in individuals with CDKL5 mutations, further indicating a shared clinical differential.
Mutations identified in CDKL5 show haploinsufficiency, including missense and splicing mutations, small deletions and insertions, as well as nonsense mutations. De novo mutations and evidence of germline mosaicism have been reported, suggesting that the presence of an affected proband in a family is sufficient to consider prenatal diagnosis. Large deletions have also been detected, thus deletion and duplication testing should be considered if an individual tests negative for sequence mutations.
FOXG1 Gene
Point mutations and deletions in the FOXG1 (forkhead box G1) gene are associated with a developmental disorder known as the congenital variant of Rett syndrome. This disorder exhibits features of classic Rett syndrome, but with an earlier onset within the first months of life. Reports of individuals with mutations in the FOXG1 gene show a clinical profile consisting of postnatal microcephaly, apraxia, absent language, stereotypical hand and mouth movements, seizures, poor sleep, and hypoplasia of the corpus callosum with decreased white matter volume [47–49]. Unlike Rett syndrome, individuals with a FOXG1 mutation do not have periods of normal development; thus regression is not usually a feature.
In addition to sequence mutations, deletions and duplications have been reported in the FOXG1 gene. Deletions have been identified in a cohort of patients with severe mental retardation, microcephaly, absent language, and/or brain anomalies [50]. Duplications have been associated with epilepsy, mental retardation, and speech impairment [51]. For patients suspected to have the congenital variant of Rett syndrome, sequence analysis is recommended as the first step in mutation identification. For patients in whom mutations are not identified by gene sequencing, deletion/duplication testing is appropriate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


