Figure 43–1 Mechanisms of action of nucleoside analogues used in the treatment of viral infections. Acyclovir and other nucleoside analogues are converted to active nucleoside triphosphates by viral and host cell kinases. These active nucleoside triphosphates compete with the corresponding endogenous nucleoside triphosphates and competitively inhibit viral DNA polymerase. Acyclovir and the nucleoside reverse transcriptase inhibitors (NRTIs) are incorporated into viral DNA and cause chain termination because they lack the 3′-hydroxyl group required to attach the next nucleoside. Ganciclovir and penciclovir do not cause chain termination.
PHARMACOKINETICS AND INDICATIONS
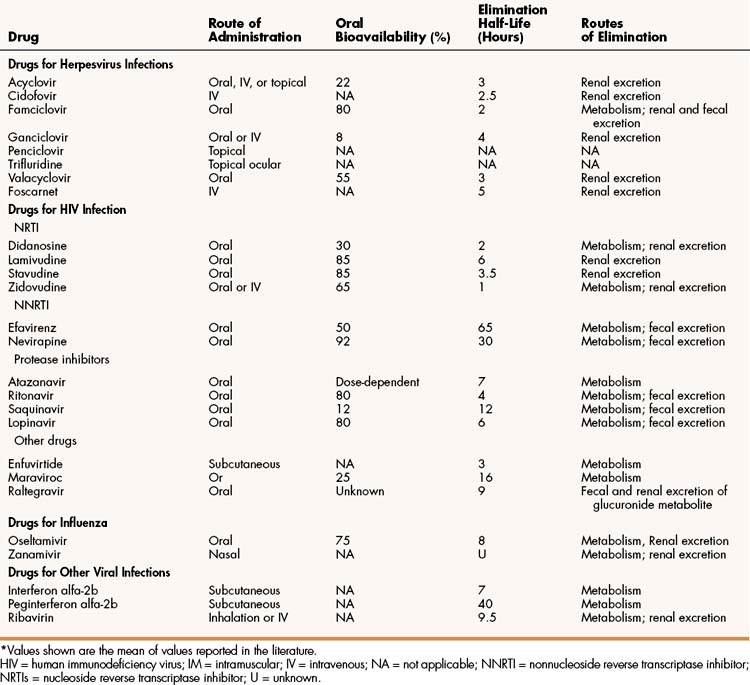
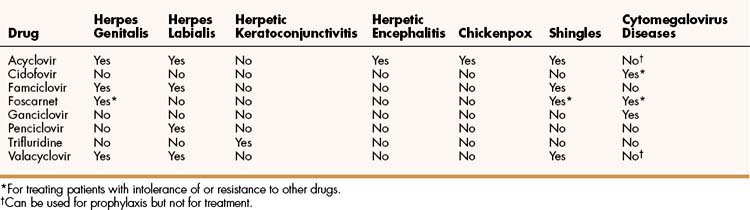
The properties and clinical uses of individual drugs for herpesvirus infections are compared in Tables 43–1 and 43–2.
VIRAL RESISTANCE
The incidence of resistance to the nucleoside analogues varies with the drug and viral pathogen.
Resistance of HSV and VZV to acyclovir is not common, and resistant strains are usually less infective than are sensitive strains. Furthermore, most acyclovir-resistant HSV and VZV strains are not resistant to other nucleoside analogues or to foscarnet. Most acyclovir-resistant strains have been recovered from immunocompromised patients. Loss of thymidine kinase activity is the major cause of innate and acquired resistance to acyclovir.
Resistance of CMV to ganciclovir is a more serious clinical problem than is HSV resistance, but most ganciclovir-resistant CMV strains are sensitive to cidofovir and foscarnet. Loss of a virus-specific protein kinase is the major cause of resistance to ganciclovir.
Acyclovir, Famciclovir, and Valacyclovir
Acyclovir, famciclovir, and valacyclovir are nucleoside analogues that are effective in the treatment of various HSV and VZV infections (see Table 43–2). These drugs are not sufficiently active against CMV to be effective in treating CMV infections, but acyclovir and valacyclovir can be used for prophylaxis of CMV infections, such as in bone marrow and organ transplant recipients and in persons with HIV infection. All three drugs are available for oral use. In addition, acyclovir is available for intravenous and topical use.
The intravenous form of acyclovir is the most effective treatment for serious herpesvirus infections, including herpetic encephalitis and severe HSV and VZV infections in immunocompromised patients.
The topical form of acyclovir can be used to treat herpes genitalis and mild mucocutaneous infections in immunocompromised patients. In cases of herpes genitalis, however, the topical form is less effective than the oral form of acyclovir.
In its oral form, acyclovir has a relatively low bioavailability (22%). Valacyclovir, which was developed later than acyclovir, is a prodrug that is rapidly converted to acyclovir by intestinal and hepatic enzymes and is more completely absorbed than acyclovir. Because of its greater bioavailability (55%), valacyclovir requires less frequent administration than acyclovir. Famciclovir has the greatest bioavailability (80%) and is rapidly hydrolyzed to penciclovir following its absorption.
When acyclovir, famciclovir, or valacyclovir is given orally for the treatment of herpes genitalis, it prevents the replication of HSV and thereby reduces pain and other symptoms of acute infection. It also shortens the time to healing of lesions and reduces the amount of viral shedding. It does not eliminate the virus, and recurrent episodes of infection are common. Shorter courses of therapy are usually sufficient for these episodes, because recurrent infections are usually milder than the initial infection. Severe herpes genitalis may require intravenous acyclovir therapy.
When acyclovir, famciclovir, or valacyclovir is given orally for the treatment of shingles, it shortens the duration of acute illness, acute pain, and postherpetic pain (neuralgia). In patients with shingles, famciclovir and valacyclovir appear to be more effective than acyclovir. The newer drugs allow for less-frequent administration and provide higher serum drug levels because of their greater oral bioavailability. A new vaccine has been shown to reduce the incidence and severity of herpes zoster infections in older adults.
Acyclovir is available in an oral suspension for the treatment of children with chickenpox. The drug has a good safety record in this setting.
Acyclovir, famciclovir, and valacyclovir are well tolerated when given orally, and they do not have significant interactions with other drugs. Gastrointestinal disturbances, headache, and rash are the most common side effects. Intravenous administration of acyclovir can produce phlebitis and reversible renal dysfunction.
Penciclovir
Penciclovir, the active metabolite of famciclovir, is now available in a topical cream formulation for the treatment of herpes labialis. In a study of patients with a history of frequent herpes labialis episodes, use of penciclovir was found to shorten the time to healing and the duration of pain by about a day. It also was found to decrease the duration of viral shedding.
Ganciclovir and Cidofovir
Ganciclovir and cidofovir are nucleoside analogues used to prevent and treat CMV diseases, including retinitis, esophagitis, and colitis. Both drugs are available for intravenous use, and ganciclovir is also available for oral use. Ganciclovir is usually given initially, whereas cidofovir is generally reserved for diseases that are resistant to ganciclovir or other drugs. Ganciclovir has a relatively low oral bioavailability, so oral administration is used only for long-term suppression of CMV retinitis. Cidofovir can be given intravenously for this purpose.
Ganciclovir is about 100 times more active against CMV than is acyclovir. Ganciclovir, however, produces a much higher incidence of adverse effects than do acyclovir and famciclovir. The most common serious adverse effects of ganciclovir are leukopenia and thrombocytopenia. Severe myelosuppression is more likely if the drug is given concurrently with zidovudine. Other adverse effects of ganciclovir include retinal detachment, liver and renal dysfunction, rash, fever, and gastrointestinal disturbances.
Cidofovir sometimes causes nephrotoxicity, neutropenia, metabolic acidosis, and other serious adverse effects. About 25% of patients have discontinued cidofovir because of serious adverse effects. The drug is contraindicated in patients who are taking other nephrotoxic drugs, such as aminoglycosides or amphotericin B.
Trifluridine
Trifluridine is administered topically to treat ocular herpesvirus infections. It is the most widely used nucleoside analogue in patients with herpetic keratoconjunctivitis and epithelial keratitis (inflammation caused by infection of the cornea), and it is usually effective in treating infections that are not responsive to idoxuridine or vidarabine. The drug is generally well tolerated but can cause superficial ocular irritation and hyperemia.
Other Drugs for Herpesvirus Infections
Foscarnet is a pyrophosphate derivative that blocks the pyrophosphate-binding sites on viral DNA polymerase and prevents attachment of nucleotide precursors to DNA. Unlike the nucleoside analogues used to treat herpesvirus infections, foscarnet does not require activation by viral or host cell kinases.
Foscarnet is active against CMV, VZV, and HSV. It must be administered intravenously and is used to treat CMV retinitis in patients with AIDS and to treat acyclovir-resistant HSV infections and shingles. Foscarnet can be combined with ganciclovir to treat infections that are resistant to either drug alone.
Adverse reactions to foscarnet include renal impairment and acute renal failure, hematologic deficiencies, cardiac arrhythmias and heart failure, seizures, and pancreatitis. Renal toxicity can be minimized by administering intravenous fluids to induce diuresis before and during foscarnet treatment.
DRUGS FOR HUMAN IMMUNODEFICIENCY VIRUS INFECTION
Remarkable advances have been made in the treatment of HIV infection and AIDS. Many new drugs have been introduced, and the combined use of two or more drugs from different classes has been shown to markedly reduce viral loads and improve survival in many HIV-positive individuals. This type of multidrug treatment for HIV infection has been called highly active antiretroviral therapy (HAART). Initially, HAART regimens were quite complicated and required multiple doses of several drugs throughout the day. In recent years, emphasis has been placed on developing and using drug regimens that require only a few doses a day. This has been accomplished by developing longer-acting drugs and combination drug products.
Sites of Drug Action
HIV is an RNA retrovirus. Its replication and sites of drug action are depicted in Figure 43–2. Viral replication begins when glycoprotein 120 on the surface of HIV-1 binds to the CD4 (cell differentiation-4) antigen on the surface of HIV-specific helper lymphocytes (CD4 cells). Binding of glycoprotein 120 to CD4 causes a conformational change in glycoprotein 120, enabling it to interact with the chemokine co-receptor (CCR5 or CXCR4) on the lymphocyte surface. These events expose a virus fusion protein, glycoprotein 41, which undergoes a conformational change so it can insert a hydrophobic tail into the host cell membrane and bind host cell integrins, leading to fusion of the viral and host cell membranes and transfer of the viral genome into the cytoplasm.
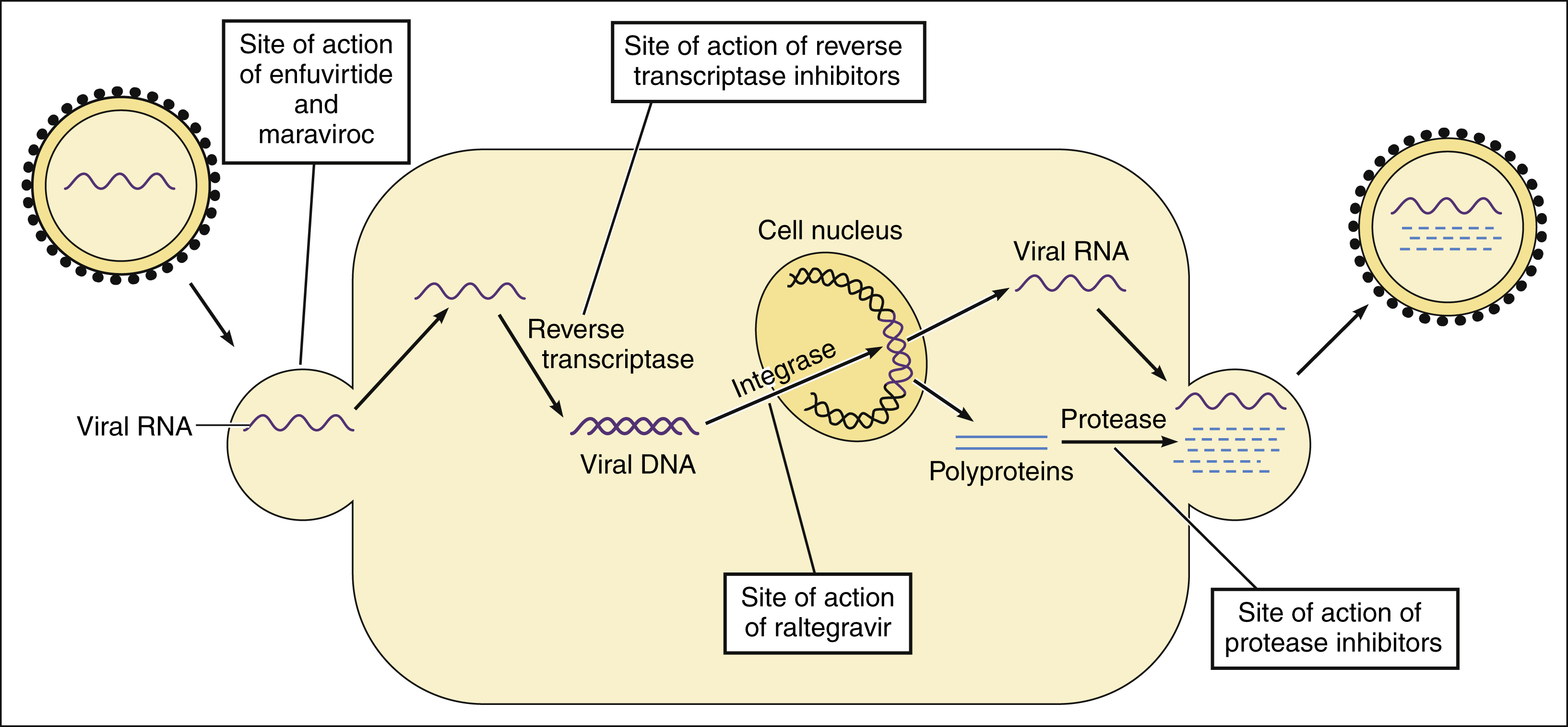
Figure 43–2 Sites of action of drugs for human immunodeficiency virus (HIV) infection. Enfuvirtide inhibits the fusion of HIV with host CD4 cell membranes. After the virus penetrates the host cell and becomes uncoated, the viral RNA is transcribed by reverse transcriptase to form viral DNA. Viral DNA is incorporated into the host genome in the cell nucleus by HIV integrase. The viral DNA is then transcribed to RNA. Viral RNA is incorporated into new virions and is translated to synthesize polyproteins. The polyproteins are cleaved into viral proteins by HIV protease as the new virions are released from the cell.
Once HIV enters the CD4 cell, viral RNA serves as a template to produce a complementary doubled-stranded DNA in a reaction catalyzed by viral reverse transcriptase (RNA-dependent DNA polymerase). The viral DNA then enters the host cell nucleus and is incorporated into the host genome in a reaction catalyzed by HIV integrase. Eventually, the viral DNA is transcribed and translated to produce large, nonfunctional polypeptides called polyproteins. These polyproteins are packaged into immature virions at the cell surface. An enzyme called HIV protease cleaves the polyproteins into smaller, functional proteins in a process called viral maturation as the virions are released into the plasma.
The drugs now available for treatment of HIV infection include those that inhibit fusion and entry, reverse transcriptase, integrase strand transfer, and protease.
Reverse Transcriptase Inhibitors
The two most important types of reverse transcriptase inhibitors are the nucleoside reverse transcriptase inhibitors (NRTIs) and the nonnucleoside reverse transcriptase inhibitors (NNRTIs). Small amounts of the NRTI are converted to their active triphosphate metabolites by host cell kinases. The triphosphate metabolites (nucleotides) compete with the corresponding endogenous nucleoside triphosphates for incorporation into viral DNA in the reaction catalyzed by reverse transcriptase. Once incorporated into DNA, the NRTIs cause DNA chain termination in the same manner as described earlier for acyclovir (see Fig. 43–1). The NRTIs also inhibit host cell DNA polymerase to varying degrees, and this may account for some of their toxic effects (e.g., anemia).
Unlike the NRTIs, the nonnucleoside drugs bind directly to reverse transcriptase and disrupt the catalytic site. Hence, the NNRTIs do not require phosphorylation for activity. Because they act by different mechanisms, the NRTIs and NNRTIs exhibit synergistic inhibition of HIV replication when they are given concurrently.
Tenofovir disoproxil fumarate, a diphosphonate diester of a nucleoside drug, is classified as a nucleotide reverse transcriptase inhibitor (a nucleotide being a phosphate ester of a nucleoside). As a nucleotide prodrug, tenofovir disoproxil fumarate is hydrolyzed in the body to form tenofovir, and then tenofovir is converted to tenofovir diphosphate by CD4 cell kinases. Tenofovir diphosphate competes with deoxyadenosine 5′-triphosphate and is incorporated into viral DNA by reverse transcriptase, causing DNA chain termination. Hence, tenofovir is similar to the NRTI in its manner of activation and mechanism of action.
Nucleoside Reverse Transcriptase Inhibitors
The NRTIs were the first class of drugs developed for the treatment of HIV-positive individuals, and they are included in almost all HIV treatment regimens. Although all NRTIs have the same basic mechanism of action, different drugs in the class serve as antimetabolites of different purine and pyrimidine bases of DNA. For this reason, an NRTI is often more effective when it is given in combination with another NRTI than when it is given alone. As shown in Table 43–3, two NRTIs are usually combined with either an NNRTI or a protease inhibitor.
TABLE 43–3 Regimens for Initial Treatment of HIV Infection in Adults and Adolescents∗
To construct an antiretroviral regimen, select one component from column A and one component from column B.
| COLUMN A NNRTI OR PI OPTIONS | COLUMN BDUAL NRTI OPTIONS | |
|---|---|---|
| Preferred Components | NNRTI: efavirenz PI: atazanavir, fosamprenavir, or lopinavir; each in combination with ritonavir | abacavir + lamivudine or tenofovir + emtricitabine |
| Alternatives to Preferred Components | NNRTI: nevirapine PI: atazanavir, fosamprenavir, lopinavir, or saquinavir; each in combination with ritonavir | zidovudine + lamivudine or didanosine + (emtricitabine or lamivudine) |
NRTI = nucleoside reverse transcriptase inhibitor; NNRTI = nonnucleoside reverse transcriptase inhibitor; PI = protease inhibitor.
∗ Issued January 29, 2008. For details and updates, see http://AIDSinfo.nih.gov.
Drug Properties
CHEMISTRY AND PHARMACOKINETICS
The NRTIs are synthetic derivatives of naturally occurring nucleosides. Didanosine is a purine base congener, whereas lamivudine, stavudine, emtricitabine, and zidovudine are pyrimidine base congeners. All of the NRTIs can be given orally, and zidovudine can also be given intravenously. The NRTIs cross the blood-brain barrier and are distributed to the cerebrospinal fluid. The drugs are eliminated primarily by renal excretion, and renal impairment will prolong their plasma elimination half-life and may necessitate a reduction in dosage.
SPECTRUM AND INDICATIONS
The NRTIs are the foundation of chemotherapy for HIV infection. In addition to inhibiting the replication of human and animal retroviruses, some of the NRTIs have demonstrated activity against hepatitis B virus and Epstein-Barr virus.
VIRAL RESISTANCE
Resistance to NRTIs can develop during therapy and is more likely to occur in persons receiving single-drug therapy for 6 months or longer. Studies of zidovudine indicate that HIV type 1 (HIV-1) acquires resistance to the drug in a stepwise manner involving four or five specific mutations in the gene that encodes the reverse transcriptase enzyme. Because the virus undergoes frequent mutations, the only way to prevent resistance is to prevent HIV replication by using combination drug therapy. Resistance to lamivudine is associated with a mutation at codon 184 in the HIV reverse transcriptase gene. Reduced sensitivity of hepatitis B virus to lamivudine is related to mutations in the catalytic domain of hepatitis B virus DNA polymerase.
ADVERSE EFFECTS AND INTERACTIONS
As shown in Table 43–4, the NRTIs differ in their major toxicities and in their interactions with other drugs. Zidovudine produces bone marrow suppression and can cause anemia and neutropenia. Didanosine and stavudine can cause pancreatitis, and didanosine can also cause peripheral neuropathy. Abacavir is more likely to cause a hypersensitivity reaction, whereas tenofovir produces renal impairment in some patients.
TABLE 43–4 Most Important Adverse Effects and Interactions of Drugs for Human Immunodeficiency Virus (HIV) Infection
| Drug | Adverse Effects and Interactions |
|---|---|
| NRTI | |
| All NRTI | Lactic acidosis, hepatic steatosis, and lipodystrophy (all higher with stavudine) |
| Abacavir | Hypersensitivity reactions |
| Didanosine | Pancreatitis, peripheral neuropathy, gastrointestinal intolerance |
| Stavudine | Pancreatitis, peripheral neuropathy |
| Tenofovir | Headache, gastrointestinal intolerance, renal impairment |
| Zidovudine | Headache, gastrointestinal intolerance, bone marrow suppression |
| NNRTI | |
| All NNRTI | Rash, drug interactions |
| Efavirenz | Neuropsychiatric reactions, teratogenic |
| Nevirapine | Hepatotoxicity, rash including Stevens-Johnson syndrome, induces metabolism of protease inhibitors and contraceptive steroids |
| Protease Inhibitors | |
| All protease inhibitors | Lipodystrophy (fat accumulation), hyperlipidemia, insulin resistance and diabetes, liver dysfunction and hepatitis; inhibit metabolism of other drugs including protease inhibitors, antiarrhythmic agents, opioids, and tricyclic antidepressants |
| Atazanavir | PR interval prolongation |
| Fosamprenavir | Gastrointestinal intolerance, rash |
| Lopinavir, ritonavir | Gastrointestinal intolerance |
| Other Drugs | |
| Enfuvirtide | Injection site reactions, hypersensitivity reactions |
| Maraviroc | Upper respiratory symptoms, possible hepatotoxicity |
| Raltegravir | Headache, diarrhea, nausea, vomiting |
NRTI = nucleoside reverse transcriptase inhibitor; NNRTI = nonnucleoside reverse transcriptase inhibitor.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


