Figure 45–1 Cell cycle activity of antineoplastic drugs. The cycle of cell replication includes the sequential phases of G1 (gap 1), S (DNA synthesis), G2 (gap 2), and M (mitosis). Differentiated cells can enter a resting state called G0. Growth factors may stimulate resting cells to reenter the cell cycle. Progress through the cycle is promoted by proteins called cyclins (A, B, D, E), which are controlled by cyclin-dependent kinases. Two check points, which often malfunction in cancer cells, control entry into the critical phases of DNA synthesis and mitosis. Cell cycle–specific drugs primarily act during the designated phase of the cycle. Cell cycle–nonspecific drugs act throughout the cell cycle.
Increased understanding of the regulation of cancer cell proliferation has led to the development of drugs that target specific growth factors involved in this process, including agents that inhibit protein kinases and growth factor receptors involved in cell cycle kinetics and metastasis. Drugs that increase cancer cell apoptosis (programmed cell death) are being developed, and it may be eventually possible to develop agents that reverse the malignant transformation of cells.
Antineoplastic drugs can be divided into two broad categories: (1) cytotoxic agents that nonspecifically inhibit DNA replication or mitosis, and (2) targeted anticancer drugs that inhibit specific proteins involved in tumor cell growth. The introduction of new anticancer drugs has accelerated with the release of a growing number of monoclonal antibodies and a plethora of “small molecule” inhibitors of protein kinases linked to growth factor receptors. New cytotoxic agents have also contributed to the explosion of antineoplastic drugs. Significant advances in cancer biology will further accelerate the introduction of new drugs. Because of the growing number of similar antineoplastic agents, this chapter focuses on important examples of each class of anticancer drugs.
PRINCIPLES OF CANCER CHEMOTHERAPY
Uses and Goals of Treatment
Antineoplastic drugs are used to treat hematologic cancers that cannot be surgically excised, such as leukemia and lymphoma. They are also used in combination with surgery or radiation therapy to treat solid tumors. In patients with solid tumors that can be surgically removed, chemotherapy may eliminate micrometastases and prevent recurrence of the malignant growth. In patients with inoperable tumors, chemotherapy is often palliative rather than curative. Palliative therapy is intended to prolong life and reduce incapacitating symptoms.
Treatment Regimens and Schedules
Drug regimens for cancer chemotherapy are designed to optimize the synergistic effects of drug combinations while minimizing toxicity. The regimens often use drugs that have different toxicities and different mechanisms of action to maximize cytotoxic effects on tumor cells while sparing host tissue.
In the treatment of some types of cancer, specific drug regimens are used for induction of remission, consolidation therapy, and maintenance therapy. Induction therapy, such as that used in acute lymphocytic leukemia, seeks to produce a rapid reduction in the tumor cell burden and thereby produce a symptomatic response in the patient. Consolidation therapy seeks to complete or extend the initial remission, and it often uses a different combination of drugs than that used for induction. Maintenance therapy aims to sustain the remission as long as possible, and it can use less-frequent courses of chemotherapy and different classes of drugs than were used for induction and consolidation.
Cancer chemotherapy regimens are probably the most complicated form of drug therapy in use today. These regimens often employ multiple drugs administered as intermittent courses of therapy rather than as continuous therapy. Intermittent therapy allows the bone marrow and other normal host cells to recover between treatment courses and reduces the level of toxicity. The selection of drugs is largely based on clinical trials comparing the effectiveness of various drug combinations in patients with particular stages of a specific type of cancer, and the treatments for each stage of a particular cancer sometimes vary considerably. First-line drugs are used for the initial treatment of tumors, whereas other drugs are indicated for patients who have relapsed after first-line therapy. These regimens are continuously evolving with the introduction of new drugs and completion of new trials. The dosage and frequency of treatment are typically based on such factors as drug kinetics, tumor cell cycle kinetics, and drug toxicity.
Cell Cycle Specificity
The cytotoxic antineoplastic drugs can be classified as cell cycle–specific and cell cycle–nonspecific agents. As shown in Figure 45–1, the cycle of cell replication includes the G1, S, G2, and M phases. DNA is replicated during the S phase, and mitosis occurs during the M phase. Because early cytologists observed no activity between the S and M phases, they referred to the period before S as G1 (gap 1) and to the period before M as G2 (gap 2). It is now known that cells are actively preparing for DNA synthesis and mitosis during the G1 and G2 phases, respectively. Cyclins are growth factors that regulate the progression of cells through the cell cycle and are targets of new drug development. Examples of cyclins are shown in Figure 45–1.
Drugs that act during a specific phase of the cell cycle are called cell cycle–specific drugs, whereas drugs that are active throughout the cell cycle are called cell cycle-nonspecific drugs. Cell cycle–specific drugs include all DNA synthesis inhibitors and mitotic inhibitors. Cell cycle-nonspecific drugs include all DNA alkylating agents and most DNA intercalating agents.
Limitations of Cancer Chemotherapy
Most antineoplastic drugs have three major limitations: susceptibility to tumor cell resistance, production of host toxicity, and an inability to suppress metastasis. Newer drugs have had some success in overcoming these obstacles, though a cure for most cancers is yet to be developed.
Drug Resistance
Drug resistance is a major cause of cancer treatment failure. As with microbial drug resistance, tumor cell resistance can be innate or acquired. Acquired drug resistance can result from genomic mutations or abnormal gene expression as cancer cells continuously evolve. The mechanisms of tumor cell resistance include induction of drug efflux pumps, decreased affinity or overexpression of target enzymes, decreased drug activation or increased drug inactivation, altered expression of proapoptotic and antiapoptotic molecules, and increased tumor cell repair.
Drug resistance can occur through failure of the drug to reach its target because of drug efflux from tumor cells. Two important efflux pumps are the P-glycoprotein (Pgp) and the multidrug-resistance protein (MRP). Both pumps are members of the adenosine triphosphate–binding cassette family. Pgp is the product of the multidrug resistance-1 gene and acts to transport many naturally occurring drugs out of neoplastic cells, including anthracyclines, taxanes, and vinca alkaloids (Fig. 45–2). Induction of Pgp by antineoplastic drugs can lead to multidrug resistance.
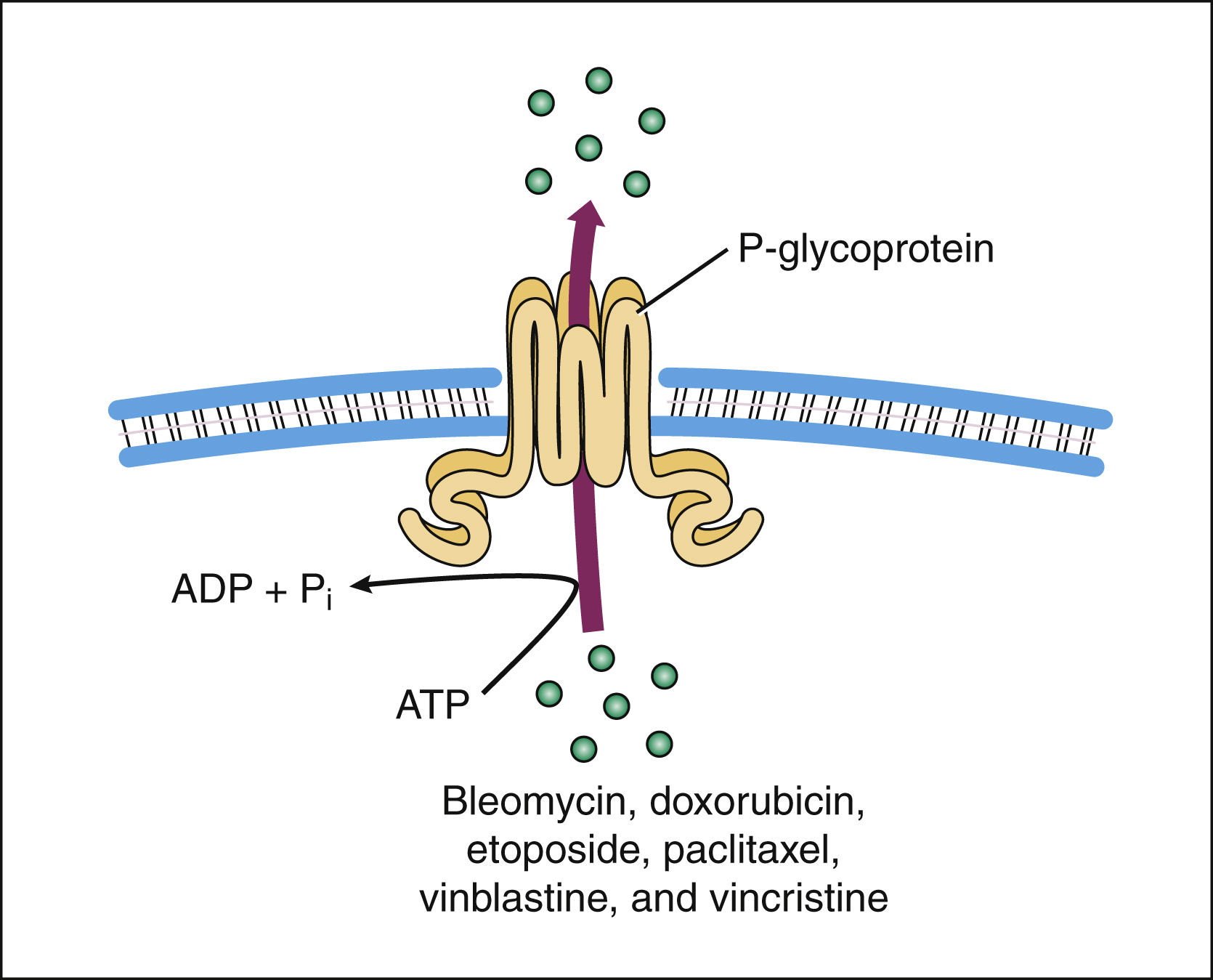
Figure 45–2 The P-glycoprotein mechanism of drug efflux. P-glycoprotein utilizes adenosine triphosphate to actively export bleomycin and many other naturally occurring antineoplastic drugs from the cell. Because some agents (e.g., verapamil) inhibit the P-glycoprotein pump and thereby allow antineoplastic drugs to stay in the cell, these agents are being studied as potential adjuncts to cancer chemotherapy. ADP = adenosine diphosphate; ATP = adenosine triphosphate.
MRP removes drugs from tumor cells after conjugation of drugs with glutathione. The drugs transported by MRP are similar to those transported by Pgp except that the taxane drugs are poorly transported by MRP. Several drugs that block both Pgp and MRP are undergoing clinical trials as antineoplastic drug enhancers.
Other examples of acquired drug resistance include topoisomerase mutations that convey resistance to topoisomerase inhibitors (e.g., etoposide). Resistance to methotrexate can occur through mutations in its target enzyme, dihydrofolate reductase, or through overexpression of the enzyme. Mutations in genes for tubulin or microtubule-associated proteins can cause resistance to the vinca alkaloids and taxane drugs. Finally, expression of antiapoptotic proteins (e.g., Bcl-2) can produce resistance to many drugs by interfering with the cell death signal induced by an antineoplastic agent.
Drug Toxicity
The most common toxicities of traditional antineoplastic drugs (Table 45–1) result from nonspecific inhibition of cell replication in the bone marrow, gastrointestinal epithelium, and hair follicles. Many antineoplastic drugs also stimulate the chemoreceptor trigger zone in the medulla and thereby elicit nausea and vomiting.
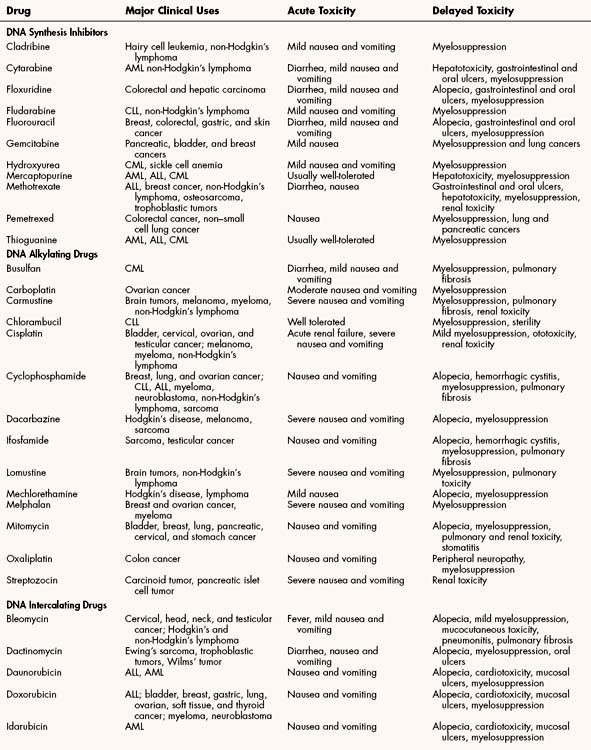
The myelosuppression (bone marrow suppression) produced by many antineoplastic drugs often results in leukopenia and thrombocytopenia, although anemia can also occur. Leukopenia predisposes patients to serious infections, whereas thrombocytopenia can lead to bleeding. The onset of leukopenia is delayed because of the time required to clear circulating cells before the effect that drugs have on precursor cell maturation in the bone marrow becomes evident. With many drugs, including methotrexate, fluorouracil, and cyclophosphamide, the leukocyte count reaches its nadir in about 7 days and recovery occurs in 2 to 4 weeks. Nitrosourea drugs (e.g., carmustine) produce a more delayed and long-lasting suppression of leukocyte production. Bleomycin, cisplatin, and vincristine produce less myelosuppression than do other antineoplastic drugs, so they are often used in combination with myelosuppressive drugs.
The nausea and vomiting caused by antineoplastic drugs range from mild to severe. Among the antineoplastic drugs, the most emetic are cisplatin and carmustine. Their adverse effects can be prevented or substantially reduced, however, by pretreatment with a combination of antiemetic drugs, including serotonin antagonists (e.g., ondansetron) and corticosteroids (e.g., dexamethasone). The antiemetics are discussed in greater detail in Chapter 28.
Alopecia is a cosmetically distressing but less-serious adverse effect of chemotherapy. It is generally reversible after treatment ends, although the hair might differ in texture and appearance from its previous condition.
Several antineoplastic drugs have characteristic organ system toxicities that appear unrelated to inhibition of cell division. For example, use of doxorubicin and other anthracyclines can cause cardiotoxicity; use of cyclophosphamide and ifosfamide can cause hemorrhagic cystitis; use of cisplatin can cause renal toxicity; use of bleomycin or busulfan can cause pulmonary toxicity; and use of vincristine, paclitaxel, and other vinca alkaloids and taxanes can cause neurotoxicity.
Agents have been developed to prevent some of these organ system toxicities. For example, dexrazoxane was developed to prevent anthracycline-induced cardiotoxicity. Another cytoprotective drug, mesna, was developed to prevent cyclophosphamide-induced hemorrhagic cystitis. Cisplatin-induced renal toxicity can be partly prevented by administering fluids, along with mannitol and sodium thiosulfate. Mannitol maintains renal blood flow and tubular function, whereas sodium thiosulfate inactivates the drug in the kidneys. No specific agents currently exist to prevent pulmonary toxicity and neurotoxicity; therefore, patients at risk should be closely monitored so that treatment can be discontinued if these toxicities develop.
DNA SYNTHESIS INHIBITORS
Most of the DNA synthesis inhibitors are analogues of purine or pyrimidine bases found in DNA or of folic acid. They act as antimetabolites to inhibit enzymes catalyzing various steps in DNA synthesis. The uses and adverse effects of selected inhibitors are listed in Table 45–1.
Folate Antagonists
Methotrexate
Nearly 50 years ago, methotrexate (MTX) was used successfully to induce remission in patients with acute childhood leukemia. Today, it is the most widely used antimetabolite in cancer chemotherapy, and it is also used as an immunosuppressive drug in the treatment of rheumatoid arthritis, lupus erythematosus, and other conditions (see Chapter 30).
CHEMISTRY AND MECHANISMS
The structures of MTX and folic acid are similar. MTX, however, has an amino group substituted for a hydroxyl group on the pteridine ring, and it also has an additional methyl group.
Methotrexate is actively transported into mammalian cells and inhibits dihydrofolate reductase, the enzyme that normally converts dietary folate to the tetrahydrofolate form required for thymidine and purine synthesis (Fig. 45–3).
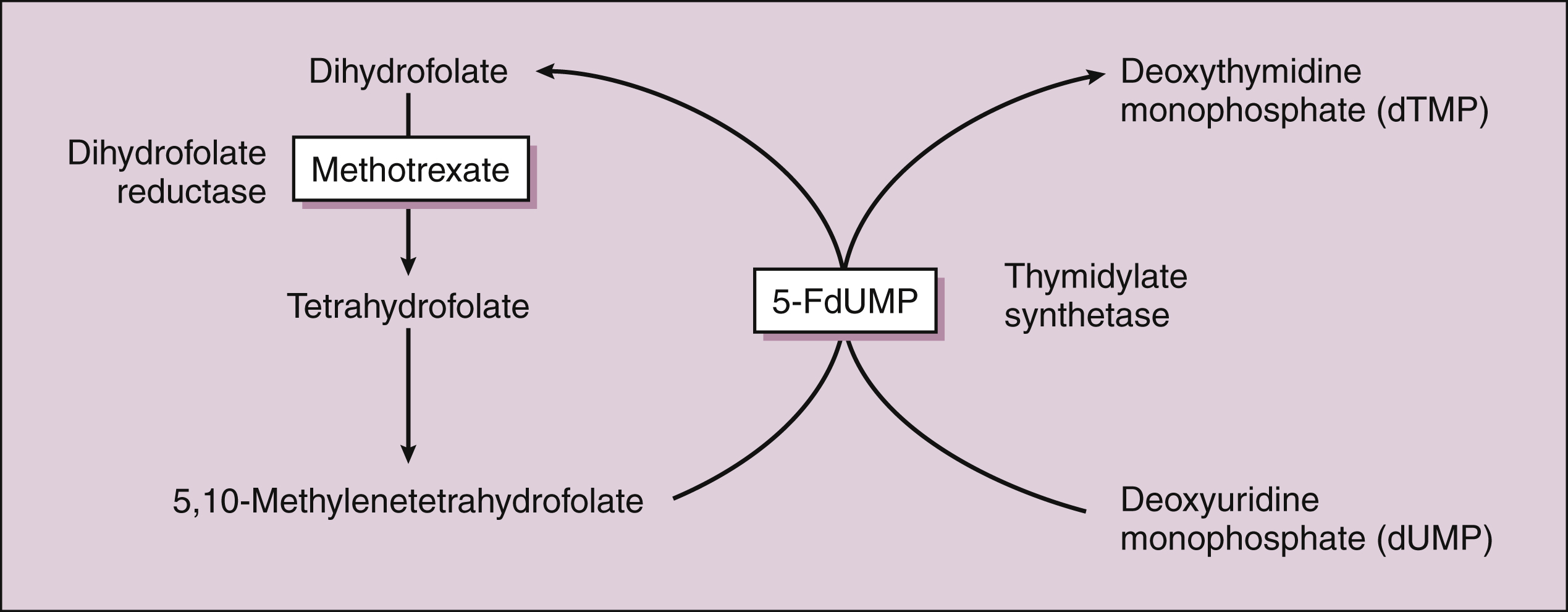
Figure 45–3 Inhibition of DNA synthesis by methotrexate and fluorouracil. Methotrexate inhibits dihydrofolate reductase and the conversion of dihydrofolate to tetrahydrofolate. This reduces the supply of 5,10-methylenetetrahydrafolate, a substance required for the synthesis of deoxythymidine monophosphate (dTMP). The conversion of deoxyuridine monophosphate (dUMP) to dTMP, a critical step in DNA synthesis, is catalyzed by thymidylate synthetase. This enzyme is inhibited by 5-fluorodeoxyuridine monophosphate (5-FdUMP), which is the active form of fluorouracil.
PHARMACOKINETICS
MTX can be administered orally or parenterally. The oral bioavailability of the drug is dose-dependent. Low doses are completely absorbed, whereas higher doses undergo significantly less absorption. Because MTX does not penetrate the central nervous system (CNS), it must be administered intrathecally for the prevention or treatment of CNS disease.
Methotrexate is rapidly distributed throughout the total body water, and it is partly degraded in the intestine (by intestinal flora) and in the liver. The drug has a biphasic elimination pattern. It is primarily eliminated as the parent compound and metabolites in the urine. The terminal half-life reflects redistribution from tissues and “third space” fluids, such as ascites and pleural effusions.
INDICATIONS
The use of MTX in the treatment of choriocarcinoma, a trophoblastic tumor, was the first demonstration of curative chemotherapy. Today, MTX has a variety of uses, including the treatment of trophoblastic tumors, breast cancer, non-Hodgkin’s lymphomas, and osteosarcoma. It is also effective for treating acute lymphocytic leukemia and the meningeal metastases of a wide range of tumors.
ADVERSE EFFECTS
The bone marrow and gastrointestinal mucosa are the normal tissues most sensitive to MTX toxicity. Although myelosuppression is the primary dose-limiting toxicity of the drug, severe oral ulceration (stomatitis) can also occur and necessitate dosage reduction. Administration of a fully activated form of folic acid called leucovorin (folinic acid) can prevent these adverse reactions, but it cannot reverse them after they have occurred. Leucovorin rescue is normally used with high doses of MTX (doses exceeding 100 mg/m2 body surface area). Assays for MTX in plasma are readily available, and drug level monitoring provides a useful guide to the duration of leucovorin administration.
Methotrexate can cause hepatotoxicity, especially with long-term low-dose therapy for psoriasis and rheumatoid conditions. At high doses, MTX can crystallize in the urine and cause renal damage. To prevent renal toxicity, dosage reduction is necessary in persons with renal insufficiency.
Pemetrexed
Pemetrexed is a novel antifolate drug. It is rapidly metabolized to active polyglutamate forms that inhibit several tetrahydrofolate-dependent enzymes involved in the synthesis of purine and pyrimidine bases, including thymidylate synthase. Although the drug can cause considerable and even life-threatening toxicities when given alone, investigators found that these toxicities can be minimized by giving folic acid and vitamin B12 without impairing drug efficacy. Pemetrexed has broad antitumor activity, and initial studies found that it prolongs survival in patients with advanced colorectal cancer. Early clinical trials show that it is also active against non–small-cell lung cancer and pancreatic cancer. Neutropenia and other forms of bone marrow suppression are the primary adverse effects of pemetrexed.
Purine Analogues
The first purine analogue to be used in cancer chemotherapy was mercaptopurine, a drug introduced almost 60 years ago for the treatment of acute lymphocytic leukemia. Mercaptopurine and thioguanine are the thio analogues of the purine bases hypoxanthine and guanine, respectively. Both drugs are converted to nucleotides by the addition of ribose phosphate, a reaction catalyzed by hypoxanthine guanine phosphoribosyltransferase. Tumor cells may acquire resistance to these drugs by deleting this enzyme, and cross-resistance is usually observed between the two drugs. The active metabolites of mercaptopurine and thioguanine inhibit several steps in the biosynthesis of purine bases (adenine and guanine) and in purine recycling pathways that supply purine precursors, thereby impairing DNA synthesis.
Mercaptopurine and thioguanine are given orally, and their bioavailability is variable, incomplete, and reduced by food. Mercaptopurine is given with methotrexate to maintain remission in patients with acute lymphocytic leukemia. Thioguanine is used to maintain remission in patients with acute lymphocytic and acute myeloid leukemia. Mercaptopurine is metabolized by xanthine oxidase, whereas thioguanine is degraded by other enzymes.
Mercaptopurine and thioguanine are usually well tolerated. Myelosuppression is generally mild with mercaptopurine but can be dose-limiting with thioguanine. Long-term mercaptopurine use can cause hepatotoxicity, and both drugs contribute to the development of other cancers (therapy-related cancer). Doses of mercaptopurine must be reduced by at least 50% in patients who are taking allopurinol, which inhibits xanthine oxidase and thereby elevates plasma levels of mercaptopurine. Allopurinol is often given to patients undergoing cancer chemotherapy because it inhibits the synthesis of uric acid and thereby prevents hyperuricemia and gout. Cancer chemotherapy places patients at risk for these problems because the destruction of cancer cells increases purine catabolism and uric acid formation.
Purine Nucleoside Analogues
Fludarabine and cladribine are halogenated purine nucleoside analogues whose triphosphate metabolites are incorporated into nascent DNA, causing DNA chain termination. Fludarabine is a highly active agent in the treatment of chronic lymphocytic leukemia and low-grade non-Hodgkin’s lymphoma. Cladribine is primarily used for hairy cell leukemia. Clofarabine and nelarabine are new purine nucleoside antagonists for treating refractory or relapsed acute lymphocytic leukemia.
Pyrimidine Antagonists
Cytarabine, Fluorouracil, and Related Drugs
Cytarabine (cytosine arabinoside) and fluorouracil are commonly used pyrimidine antimetabolites. Cytarabine is composed of cytosine and the sugar arabinose. Fluorouracil is an analogue of thymine in which the methyl group is replaced by a fluorine atom. Floxuridine is the deoxyribonucleoside derivative of fluorouracil, whereas capecitabine is converted in the body to fluorouracil and its active metabolites.
The active triphosphate metabolite of cytarabine blocks DNA synthesis by several actions, including the inhibition of DNA polymerase and incorporation of the drug into nascent DNA, causing DNA chain termination. Cytarabine crosses the blood-brain barrier and reaches cerebrospinal fluid levels that are 40% to 50% of plasma levels. Fluorouracil has two active metabolites: 5-fluorodeoxyuridine monophosphate (5-FdUMP) and 5-fluorodeoxyuridine triphosphate (5-FdUTP). 5-FdUMP inhibits thymidylate synthetase and prevents the synthesis of thymidine, a major building block of DNA (see Fig. 45–3). 5-FdUTP is also incorporated into RNA by RNA polymerase and interferes with RNA function. When the drugs are given parenterally, they are extensively metabolized before undergoing renal excretion.
Cytarabine can be administered intravenously or subcutaneously and is used in combination with daunorubicin to treat acute myeloid leukemia. Fluorouracil is administered intravenously to treat solid tumors, especially breast, colorectal, and gastric tumors and squamous cell tumors of the head and neck. Regional delivery of the drug via the hepatic artery can produce a sustained response in patients whose colorectal cancer has metastasized to the liver. Topical application of fluorouracil is used to treat actinic keratoses and noninvasive skin cancers. Capecitabine is indicated for colorectal and breast cancer, whereas floxuridine is used in the treatment of colorectal and hepatic carcinoma.
The pyrimidine antimetabolites can cause nausea and vomiting, myelosuppression, and oral and gastrointestinal ulceration. Nausea and vomiting are usually mild. With fluorouracil, myelosuppression is more problematic after bolus injections, whereas mucosal damage is dose-limiting with continuous infusions. High doses of these drugs can damage the liver, heart, and other organs.
Gemcitabine
Gemcitabine, an S-phase specific inhibitor of DNA synthesis, is a fluorinated cytidine analogue. After conversion to gemcitabine diphosphate and triphosphate, these metabolites inhibit the synthesis of deoxynucleoside triphosphates and the incorporation of deoxynucleoside triphosphates into DNA. In addition, gemcitabine triphosphate itself is incorporated into DNA, causing DNA chain termination. The drug is indicated as a first-line treatment for pancreatic carcinoma and for use with cisplatin as first-line therapy for inoperable, non–small-cell lung cancer. Gemcitabine has also been used for biliary tract, gallbladder, breast, and ovarian cancer. As with other DNA synthesis inhibitors, gemcitabine can cause myelosuppression leading to anemia, leukopenia, and thrombocytopenia. It also causes alopecia and various gastrointestinal disturbances.
Ribonucleotide Reductase Inhibitor
Hydroxyurea inhibits ribonucleotide reductase, the enzyme that converts ribonucleotides to deoxyribonucleotides. It thereby stops DNA synthesis and causes cells to accumulate in the S phase of the cell cycle.
Hydroxyurea is given orally to treat chronic myelogenous leukemia, ovarian cancer, and melanoma. Hydroxyurea is also used in the management of sickle cell anemia. In this condition, hydroxyurea elevates the concentration of fetal hemoglobin and decreases the frequency of sickle cell crises. The dose-limiting toxicity of hydroxyurea is rapid-onset myelosuppression.
DRUGS THAT CROSS-LINK DNA
A DNA alkylating drug is an agent that cross-links DNA strands by forming covalent bonds between alkyl groups of the drug and guanine bases of DNA. The DNA alkylating drugs include nitrogen mustards, nitrosourea drugs, and several other agents. Platinum compounds (e.g., cisplatin) do not contain alkyl groups but cross-link DNA by forming intrastrand and interstrand covalent bonds with DNA bases.
Nitrogen Mustards
The nitrogen mustards are nitrogen analogues of the sulfur mustards that were used as chemical warfare agents in World War I. The cytotoxic effects of nitrogen mustards were discovered in the early 1940s, and the drugs were soon introduced into clinical use. They are considered the first effective antineoplastic drugs.
The nitrogen mustards are bifunctional alkylating agents that undergo spontaneous conversion to active metabolites in body fluids or are enzymatically converted to active metabolites in the liver. The strong electrophilic intermediates formed by these reactions attack the N7 nitrogen of guanine and thereby form covalent bonds with this base. Sequential attachment to two guanine residues results in cross-linking of DNA, and this prevents DNA replication and transcription (Fig. 45–4A). The alkylating drugs act throughout the cell replication cycle.
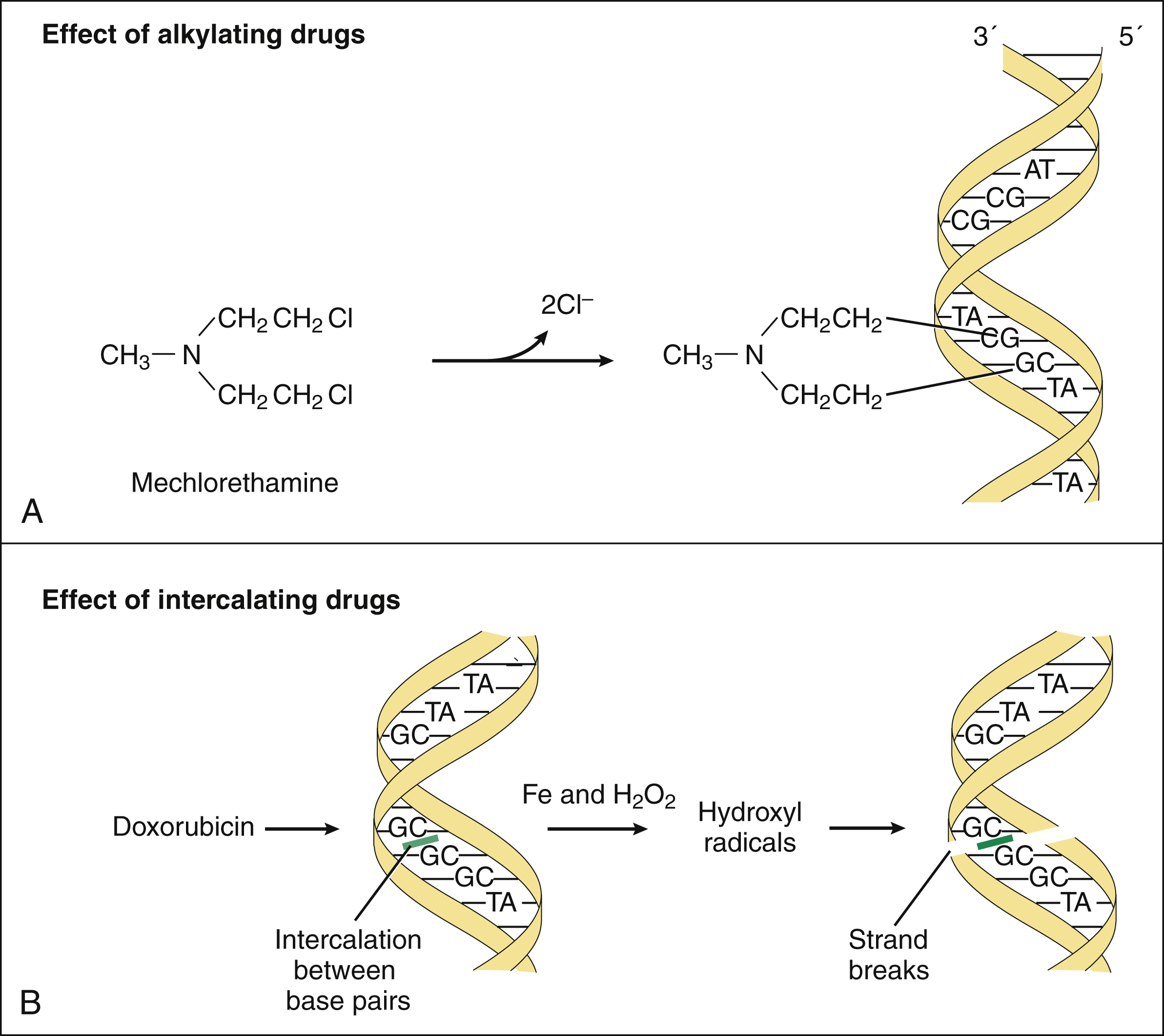
Figure 45–4 Mechanisms of action of DNA alkylating drugs and DNA intercalating drugs. (A) Mechlorethamine and other bifunctional alkylating drugs form electrophilic intermediates that attack the N7 nitrogen of guanine residues in DNA, thereby cross-linking DNA strands and preventing replication and transcription. (B) Doxorubicin and other anthracycline drugs intercalate between DNA base pairs. Anthracyclines are reduced to intermediates that donate electrons to oxygen to form superoxide. Superoxide then reacts with itself to make hydrogen peroxide, which is cleaved in the presence of iron to form the destructive hydroxyl radical that cleaves DNA.
Cyclophosphamide and Ifosfamide
CHEMISTRY AND PHARMACOKINETICS
Cyclophosphamide and ifosfamide are prodrugs that must be converted to active metabolites by hepatic mixed-function oxidase (cytochrome P450) enzymes. The active alkylating metabolite of cyclophosphamide is thought to be phosphoramide mustard. Cyclophosphamide and ifosfamide are partly converted to acrolein, which is probably responsible for hemorrhagic cystitis, an adverse effect that sometimes occurs with use of these drugs. Both drugs can be administered intravenously; cyclophosphamide can also be given orally. Cyclophosphamide is completely absorbed after oral administration.
INDICATIONS
Cyclophosphamide is the most widely used nitrogen mustard because of its broad spectrum of activity. It is used in the treatment of acute and chronic lymphocytic leukemia; non-Hodgkin’s lymphoma; breast, lung, and ovarian cancers; and a variety of other cancers (see Table 45–1). Because cyclophosphamide is a potent immunosuppressant, it is used in the management of rheumatoid disorders and autoimmune nephritis. It is also used in preoperative regimens for bone marrow transplantation.
Ifosfamide is primarily used to treat patients with sarcoma and patients with testicular cancer that is refractory to first-line treatments.
ADVERSE EFFECTS
Adverse effects of cyclophosphamide and ifosfamide include alopecia, nausea, vomiting, myelosuppression, and hemorrhagic cystitis. Nausea and vomiting are usually mild when cyclophosphamide is given orally, but they can be severe when given intravenously. The dose-limiting toxicity of cyclophosphamide is myelosuppression, whereas that of ifosfamide is usually hemorrhagic cystitis. This type of cystitis is characterized by symptoms of urinary frequency and irritation and by blood loss from the bladder. Ingestion of large amounts of fluid and the administration of mesna, a sulfhydryl reagent, can significantly reduce the incidence of cystitis. Mesna binds to acrolein, the toxic metabolite that causes cystitis, and converts it to an inactive substance.
Chlorambucil, Mechlorethamine, and Melphalan
Chlorambucil, mechlorethamine, and melphalan are nitrogen mustards that act via the mechanisms described earlier.
Chlorambucil is orally administered, has selective cytotoxicity for lymphocyte cell lines, and is primarily used to manage chronic lymphocytic leukemia. It is well tolerated but can cause dose-limiting myelosuppression and sterility. Long-term therapy is associated with a high incidence of secondary acute leukemia.
Mechlorethamine is a highly reactive and vesicant drug that is rapidly and spontaneously converted to its alkylating intermediate in body fluids after it is given intravenously. Mechlorethamine is used for treating Hodgkin’s and non-Hodgkin’s lymphomas. Melphalan is a nitrogen mustard that is primarily used to treat multiple myeloma (plasma cell myeloma) and breast cancer.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


