Figure 20–1 Patterns on electroencephalogram (EEG) in the normal state and during seizures. The locations of seizure foci are shown as shaded areas on the cerebral hemispheres. (A) In the normal state, the EEG shows asynchronous alpha (8–12 Hz) and beta (12–30 Hz) rhythms originating in the cortex of the frontal (F), temporal (T), and occipital (O) lobes. (B) During a partial seizure, synchronous discharges are observed in various areas of the brain. In this example, they are seen in the left frontal and left temporal lobes, but they are not seen in other lobes. (C) During a generalized tonic-clonic seizure, the tonic phase is characterized by low-frequency and high-amplitude waves, whereas the clonic phase shows synchronous oscillations. (D) During a generalized absence seizure, a synchronous 3-Hz spike-and-wave pattern is seen throughout the cortex.
About 60% of epileptic seizures are partial seizures. Electroencephalographic abnormalities are seen in one or more lobes of a cerebral hemisphere, and the patient may exhibit motor, sensory, and autonomic symptoms. In simple partial seizures, consciousness is not altered. In complex partial seizures, however, patients have an altered consciousness and exhibit repetitive behaviors (automatisms). Complex partial seizures often originate in the temporal lobe, in which case the disorder is called either temporal lobe epilepsy or psychomotor epilepsy. Some partial seizures progress along anatomical lines as the electrical discharges spread across the cortex. For example, a seizure may first involve the fingers, then the hand, and finally the entire arm. This is characteristic of jacksonian epilepsy, or “jacksonian march.” Partial seizures can also evolve into generalized seizures.
The two main types of generalized seizures are tonic-clonic seizures and absence seizures. Generalized tonic-clonic seizures, which were formerly called grand mal seizures, begin with a brief tonic phase that is followed by a clonic phase with muscle spasms lasting 3 to 5 minutes, and they conclude with a postictal period of drowsiness, confusion, and a glazed look in the eyes. Status epilepticus is a condition in which patients experience recurrent episodes of tonic-clonic seizures without regaining consciousness or normal muscle movement between episodes. Generalized absence seizures, or petit mal seizures, are characterized by abrupt loss of consciousness and decreased muscle tone, and they can include a mild clonic component, automatisms, and autonomic effects. On the EEG, generalized absence seizures exhibit a synchronous 3-Hz (three cycles per second) spike-and-dome pattern that usually lasts 10 to 15 seconds.
Less common types of generalized seizures include myoclonic seizures and atonic seizures (see Table 20–1).
Neurobiology of Seizures
Epileptic seizures are caused by synchronous neuronal discharges within a particular group of neurons, or seizure focus, which is often located in the cerebral cortex but can be found in other areas of the brain. Once initiated, the abnormal discharges spread to other parts of the brain and produce abnormal movements, sensations, or thoughts. The neuronal mechanisms that initiate a seizure are not fully understood, but growing evidence indicates the involvement of excessive excitatory neurotransmission mediated by glutamate (Fig. 20–2). Investigators believe that excessive activation by glutamate of n-methyl-d-aspartate (NMDA) receptors displaces Mg2+ ions from the NMDA receptor–calcium ion channel and thereby facilitates calcium entry into neurons. Calcium contributes to the long-term potentiation of excitatory glutamate neurotransmission by activating the synthesis of nitric oxide.
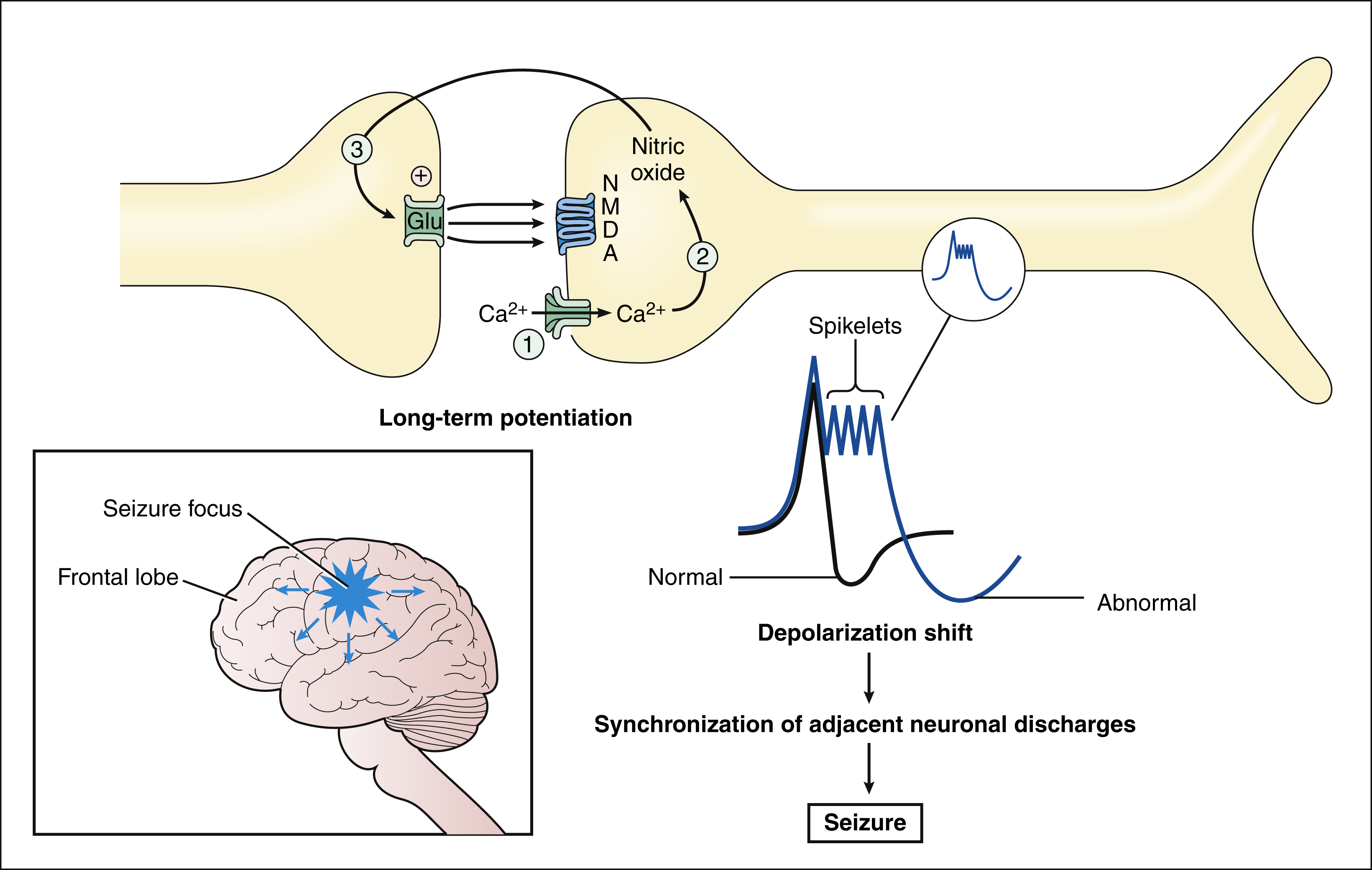
Figure 20–2 Neuronal mechanisms underlying seizures. In this example, a seizure is caused by the synchronous discharge of a group of neurons (focus) in the cortex. Activation of NMDA receptors increases calcium influx and nitric oxide synthesis. Nitric oxide then diffuses to the presynaptic neuron and increases the release of glutamate via formation of cyclic guanosine monophosphate. Increased excitatory glutamate neurotransmission leads to long-term potentiation. Long-term potentiation is believed to facilitate a depolarization shift, characterized by prolonged depolarizations with spikelets. The depolarization shift can cause adjacent neurons to discharge synchronously and thereby precipitate a seizure.
Nitric oxide is a gas that can diffuse backward to the presynaptic neuron, where it facilitates glutamate release via stimulation of a G protein that activates the synthesis of cyclic guanosine monophosphate. These actions further increase NMDA receptor activation and calcium influx, which are believed to contribute to the depolarization shift that is observed in seizure foci. The depolarization shift consists of abnormally prolonged action potentials (depolarizations) that have spikelets. The shift recruits and synchronizes depolarizations by surrounding neurons and thereby initiates a seizure.
Several other mechanisms can be involved in seizures. One is the suppression of inhibitory neurotransmission of γ-aminobutyric acid (GABA), and another is an increase in calcium influx via T-type calcium channels in thalamic neurons.
Mechanisms of Antiepileptic Drugs
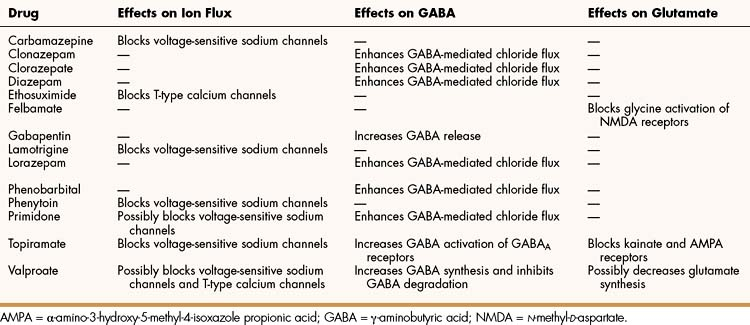
Antiepileptic drugs are believed to suppress the formation or spread of abnormal electrical discharges in the brain. As shown in Table 20–2, the currently available drugs accomplish these actions via three mechanisms: (1) inhibition of the sodium or calcium influx responsible for neuronal depolarization, (2) augmentation of inhibitory GABA neurotransmission, and (3) inhibition of excitatory glutamate neurotransmission.
Effects on Ion Channels
Under normal circumstances, voltage-sensitive (voltage-gated) sodium channels are rapidly opened when the neuronal membrane potential (voltage) reaches its threshold. This causes rapid depolarization of the membrane and the conduction of an action potential along the neuronal axon. When the action potential reaches the nerve terminal, it evokes the release of a neurotransmitter. After the neuronal membrane is depolarized, the sodium channel is inactivated by closure of the channel’s inactivation gate. The inactivation gate must be opened before the next action potential can occur.
Many antiepileptic drugs, including carbamazepine, lamotrigine, phenytoin, and topiramate, prolong the time that the sodium channel’s inactivation gate remains closed, and this delays the formation of the next action potential. These drugs bind to the channel when it is opened. Because rapidly firing neurons are opened a greater percentage of the time than are slowly firing neurons, the drugs exhibit use-dependent blockade. For this reason, the drugs suppress abnormal repetitive depolarizations in a seizure focus more than they suppress normal neuronal activity. By these actions, carbamazepine and other drugs prevent the spread of abnormal discharges in a seizure focus to other neurons.
A few drugs, such as ethosuximide and valproate, block T-type (low-threshold) calcium channels that are located in thalamic neurons and participate in the initiation of generalized absence seizures.
Effects on GABAergic Systems
Antiepileptic drugs facilitate GABA neurotransmission by various means. Benzodiazepines, like clonazepam and barbiturates, such as phenobarbital, enhance GABA activation of the GABAA receptor-chloride ion channel (see Chapter 19). Topiramate also activates the GABAA receptor. Gabapentin increases GABA release, whereas valproate inhibits GABA degradation. Drugs that augment GABA may serve to counteract the excessive excitatory neurotransmission responsible for initiating and spreading abnormal electrical discharges.
Effects on Glutaminergic Systems
A few antiepileptic drugs, including felbamate, topiramate, and valproate, inhibit glutamate neurotransmission, and other drugs that work via this mechanism are under development. This is an attractive mechanism of action because it may affect the formation of a seizure focus and thereby terminate a seizure at an early stage of its development.
TREATMENT OF SEIZURE DISORDERS
As indicated in the drugs listed in the box at the beginning of this chapter, some are active against only one or two types of seizures. In contrast, valproate has a broad spectrum of activity and is active against most types of seizures. The newer agents, such as lamotrigine, topiramate, tiagabine, levetiracetam, zonisamide, and pregabalin, are considered adjunct agents and primarily used in combination with older drugs for the treatment of partial seizures. These newer agents are particularly useful because complex partial seizures are more resistant to treatment than are other types of seizures.
The pharmacologic properties of antiepileptic drugs are described below, whereas the mechanisms of action, adverse effects, contraindications, and drug interactions are given in Tables 20–2, 20–3, and 20–4.

Drugs for Partial and Generalized Tonic-Clonic Seizures
The first-line drugs for partial seizures and generalized tonic-clonic seizures are carbamazepine, phenytoin, and valproate. Carbamazepine and phenytoin have a similar mechanism of action and clinical effectiveness, and both drugs induce cytochrome P450 enzymes and increase drug metabolism. Valproate acts by a different mechanism and inhibits cytochrome P450 enzymes. Two other drugs that are effective against partial seizures as well as generalized tonic-clonic seizures are phenobarbital and primidone.
Carbamazepine
PHARMACOKINETICS
Carbamazepine is adequately absorbed after oral administration, and it is biotransformed to an active metabolite, carbamazepine epoxide. Almost all the drug is excreted as metabolites in the urine and feces.
MECHANISMS AND EFFECTS
Carbamazepine blocks voltage-sensitive sodium channels in neuronal cell membranes. As described, blockade of these channels inhibits the spread of abnormal electrical discharges from the seizure focus to other neurons by preventing the release of excitatory neurotransmitters from nerve terminals. Carbamazepine has additional mechanisms of action, but their contributions to its antiepileptic effects are unknown. For example, carbamazepine blocks adenosine receptors in a way that leads to up-regulation of these receptors, and it blocks norepinephrine reuptake in the same way that tricyclic antidepressants block it. The latter action is probably responsible for the mood-elevating effect of carbamazepine.
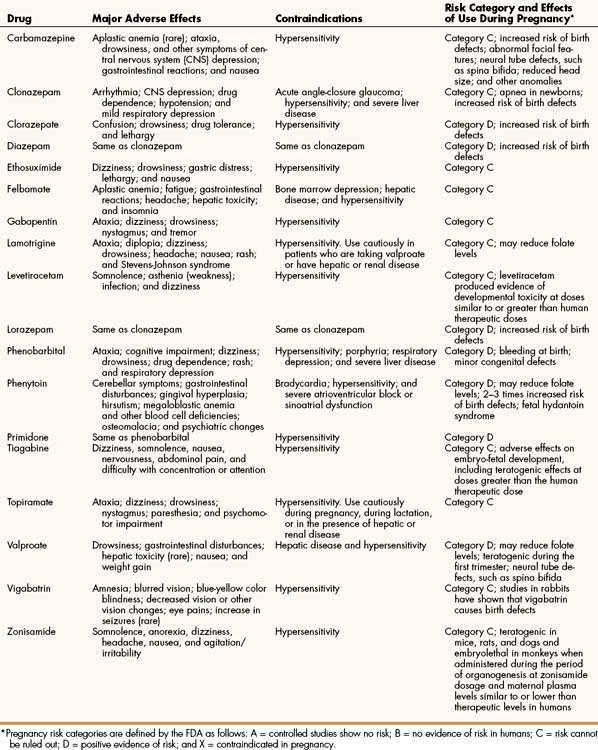
Carbamazepine can cause drowsiness, ataxia, and other symptoms of central nervous system (CNS) depression, as well as gastrointestinal reactions. Rarely, its use has been associated with aplastic anemia. In general, carbamazepine usually produces fewer adverse effects than does phenytoin.
INTERACTIONS
Carbamazepine is a potent inducer of cytochrome P450 enzymes that metabolize a wide range of drugs. Carbamazepine accelerates its own metabolism as well as that of many other drugs, including lamotrigine, phenytoin, topiramate, and valproate. For this reason, it decreases the serum level and effects of these drugs. Carbamazepine can also increase lithium toxicity.
INDICATIONS
In addition to its use in treating partial seizures and generalized tonic-clonic seizures, carbamazepine is the drug of choice for trigeminal neuralgia (tic douloureux), a condition that can cause chronic and intense pain on one or both sides of the face. Carbamazepine is also effective as an alternative to lithium in the treatment of bipolar disease, a mood disorder discussed in Chapter 22.
Phenytoin and Fosphenytoin
PHARMACOKINETICS
Phenytoin is a hydantoin derivative formerly called diphenylhydantoin. It is poorly soluble in water, and different pharmaceutical formulations of it may have different bioavailability. Hence, it is prudent to avoid switching from one formulation to another. If a switch must be made, serum drug levels should be monitored. A new formulation, called fosphenytoin, has become available for parenteral administration. Fosphenytoin is more soluble in water, and this prevents precipitation of the drug after intramuscular or intravenous administration.
Phenytoin is converted to an inactive hydroxylated metabolite by cytochrome P450 enzymes. The drug exhibits dose-dependent kinetics, whereby lower concentrations are eliminated by a first-order process, but higher concentrations saturate biotransformation enzymes and exhibit zero-order kinetics. Phenytoin hydroxylation also exhibits genetic polymorphism. These factors are responsible for the considerable patient variation in the plasma drug concentrations produced by a given dose. Because of this variation, serum drug levels should be monitored at the start of therapy and whenever toxicity or therapeutic failure occurs.
MECHANISMS AND EFFECTS
As with carbamazepine, phenytoin blocks voltage-sensitive sodium channels by prolonging the inactivation state of these channels. This enables phenytoin to inhibit the repetitive firing of neurons in a seizure focus.
Phenytoin can cause a number of adverse effects. The drug interferes with folate metabolism, and this can lead to megaloblastic anemia. Folate antagonism can also contribute to birth defects such as those seen in fetal hydantoin syndrome. This syndrome is characterized by cardiac defects; malformation of ears, lips, palate, mouth, and nasal bridge; mental retardation; and microcephaly. By impairing cerebellar function, phenytoin can cause ataxia, diplopia, nystagmus, and slurred speech. By interfering with vitamin D metabolism and decreasing calcium absorption from the gut, phenytoin sometimes causes osteomalacia. Phenytoin adversely affects collagen metabolism and thereby contributes to gingival hyperplasia, a condition in which the gums can extend down over the teeth if good dental hygiene is not practiced. It can also cause excessive hair growth, known as hirsutism. Because of these adverse effects, phenytoin use in children should generally be avoided.
INTERACTIONS
Phenytoin induces the CYP3A4 isozyme and accelerates the metabolism of other antiepileptic agents, including felbamate, lamotrigine, topiramate, and valproate. It can also reduce levels of digoxin, steroids, vitamin K, and other drugs. In patients who are being treated with phenytoin, vitamin K supplements are given to prevent hypoprothrombinemia and bleeding. Carbamazepine induces the metabolism of phenytoin and decreases its serum levels, whereas cimetidine and other drugs inhibit the metabolism of phenytoin and increase its serum levels.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


