Figure 14–1 Relationships between ion currents, cardiac action potentials, and the findings on surface ECG. The normal heartbeat originates in the SA node. The impulse is conducted through internodal fibers to the AV node and then through the bundle of His, bundle branches, and Purkinje fibers to the ventricular muscle. On the ECG, the P wave represents atrial depolarization, the QRS complex represents ventricular depolarization, and the T wave represents ventricular repolarization. The PR interval is primarily related to the conduction time through the AV node, and the QT interval represents the time between ventricular depolarization and repolarization. In phase 0, ventricular depolarization is caused by sodium influx through the fast sodium channel. In phase 1, the membrane is transiently repolarized as a result of potassium efflux. In phase 2, the membrane potential is relatively stable because of the concurrent influx of calcium and efflux of potassium. In phase 3, repolarization is caused by continued potassium efflux as calcium influx declines. In phase 4, the ion balance is returned to normal by the action of the sodium pump (Na+,K+-adenosine triphosphatase [ATPase]). Calcium is removed from the cell by the sodium-calcium exchanger and the calcium ATPase (not shown).
The SA node, located in the right atrium, is the site of origin of the normal heartbeat. The SA node spontaneously depolarizes to form an impulse that is conducted through the atrium to the AV node and then through the bundle of His, bundle branches, and Purkinje fibers to the ventricular muscle.
The spontaneous depolarization of the SA node is caused by the influx of sodium and calcium as the efflux of potassium subsides. When the threshold potential (TP) is reached at about −40 mV, the SA node is more rapidly depolarized by sodium and calcium influx to generate an impulse that can be conducted to the rest of the heart. As the impulse is conducted to atrial and ventricular muscle cells, these cells are rapidly depolarized by the influx of sodium through the fast sodium channel. As the cells depolarize to about −40 mV, the slow calcium channels open. The influx of calcium through these channels during phase 2 serves to activate muscle contraction. Cardiac tissues become repolarized during phase 3 as a result of the efflux of potassium through several types of rectifier potassium channels, an efflux that occurs when the influx of calcium declines.
As shown in Figure 14–1, the surface ECG is a summation of action potentials generated by the heart during the cardiac cycle. The P wave represents atrial depolarization, whereas the PR interval corresponds to the time required to conduct the action potential through both the atria and the AV node. The QRS complex and the T wave represent ventricular depolarization and ventricular repolarization, respectively. Hence, the QT interval represents the duration of the ventricular action potential.
Pathophysiology of Arrhythmias
Arrhythmias can be caused by coronary ischemia and tissue hypoxia, electrolyte disturbances, overstimulation of the sympathetic nervous system, general anesthetics, and other conditions or drugs that perturb cardiac transmembrane potentials and lead to abnormal impulse formation or abnormal impulse conduction.
Abnormal Impulse Formation
Abnormal impulse formation can generate extrasystoles and result in tachycardia. The two mechanisms that are primarily responsible for abnormal impulse formation are increased automaticity and the occurrence of afterdepolarizations. These are depicted in Box 14–1.
BOX 14–1 THE ELECTROPHYSIOLOGIC BASIS OF ARRHYTHMIAS
Abnormal Impulse Formation
The two mechanisms primarily responsible for abnormal impulse formation are increased automaticity and afterdepolarizations.
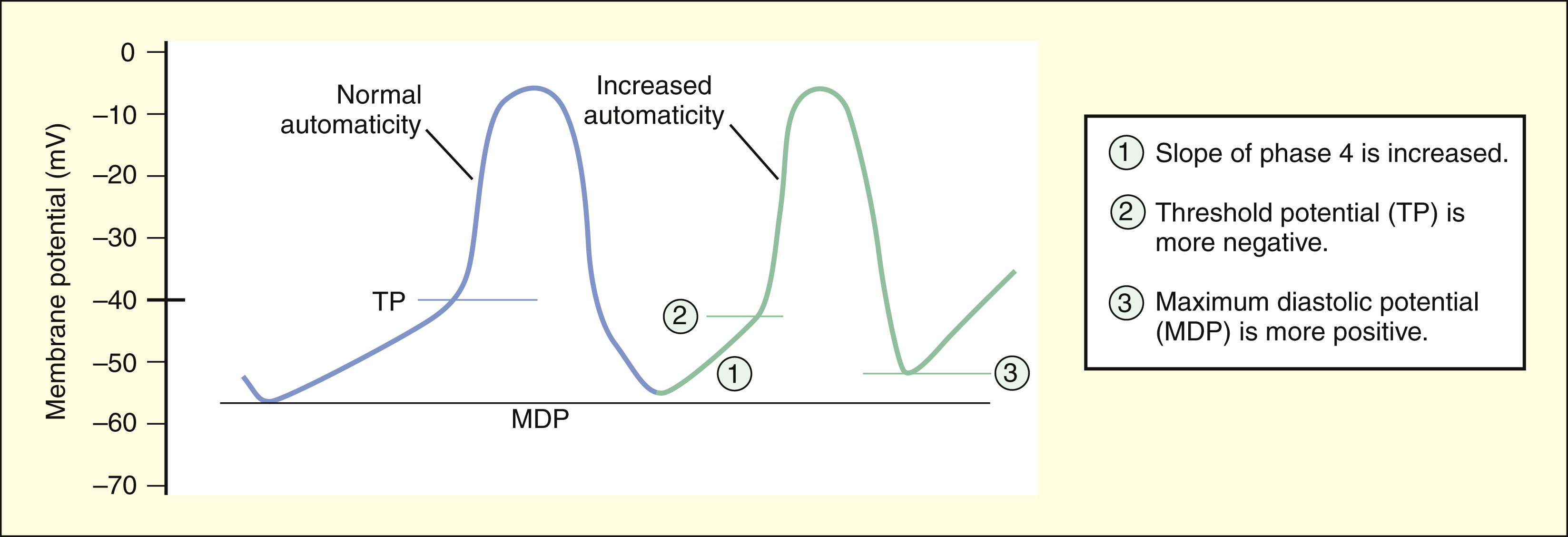
Increased automaticity can be caused by any change that decreases the time required for depolarization from the maximal diastolic potential (MDP) to the TP. Increased automaticity occurs if the rate of diastolic depolarization (the slope of phase 4) in the SA node or in latent pacemakers is increased. It also occurs if a shift of the TP occurs to a more negative value or if a shift occurs of the MDP to a more positive value.
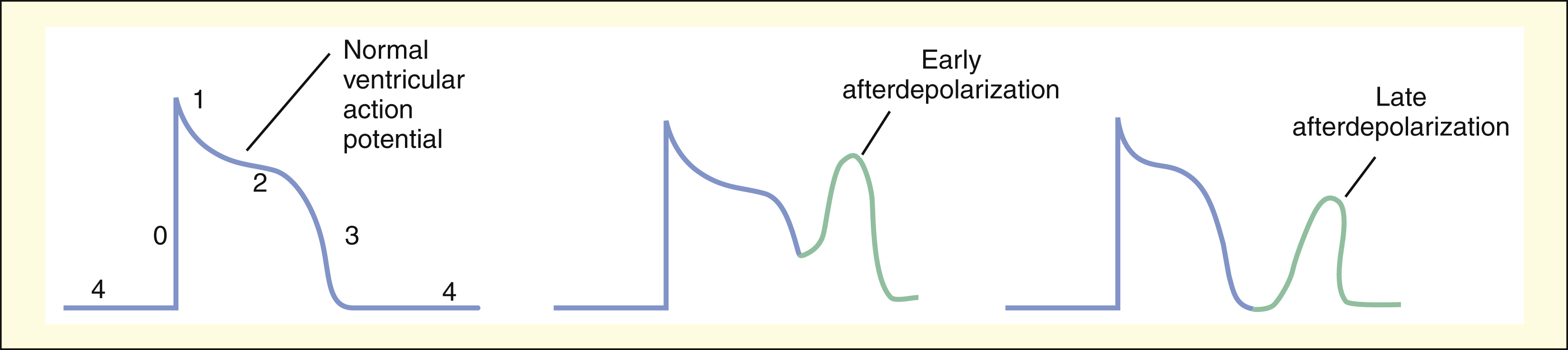
Afterdepolarizations are believed to result from abnormal calcium influx into cardiac cells during or immediately after phase 3 of the ventricular action potential. Afterdepolarizations can lead to extrasystoles and tachycardia.
Abnormal Impulse Conduction
Reentry is characterized by the retrograde conduction of an impulse into previously depolarized tissue. It is usually caused by the presence of a unidirectional conduction block in a bifurcating conduction pathway. For reentry to occur, the conduction time through the retrograde pathway must exceed the refractory period of the reentered tissue.
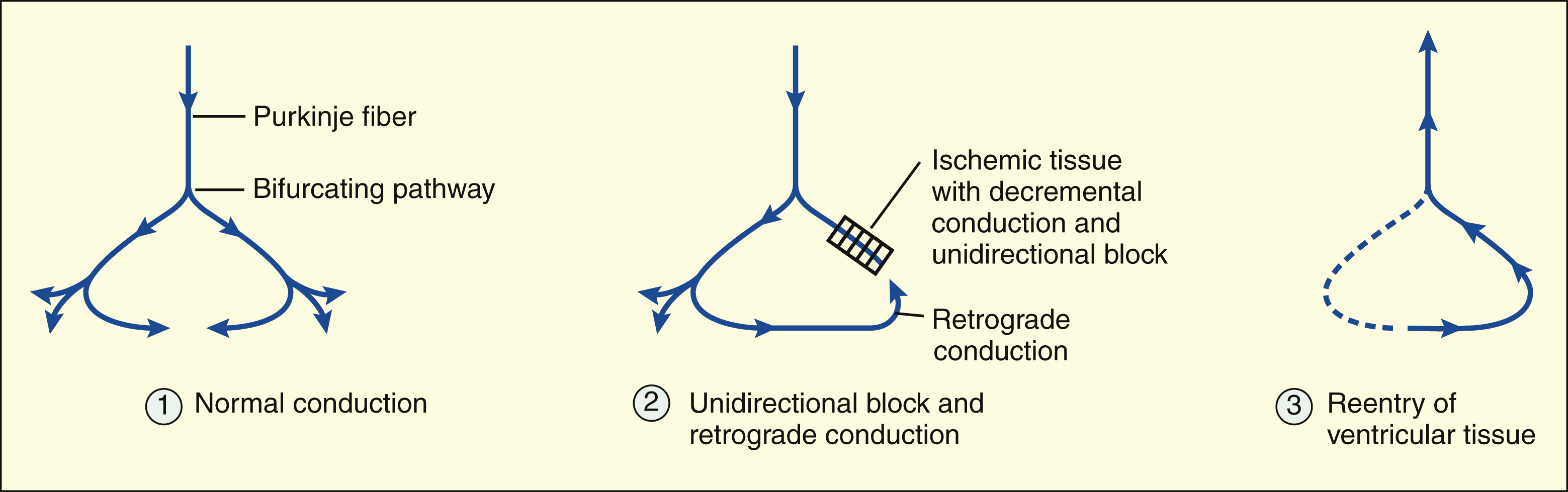
In ventricular tissue, the unidirectional block is often caused by decremental conduction of the impulse in the anterograde direction, with normal conduction of the impulse in the retrograde direction.
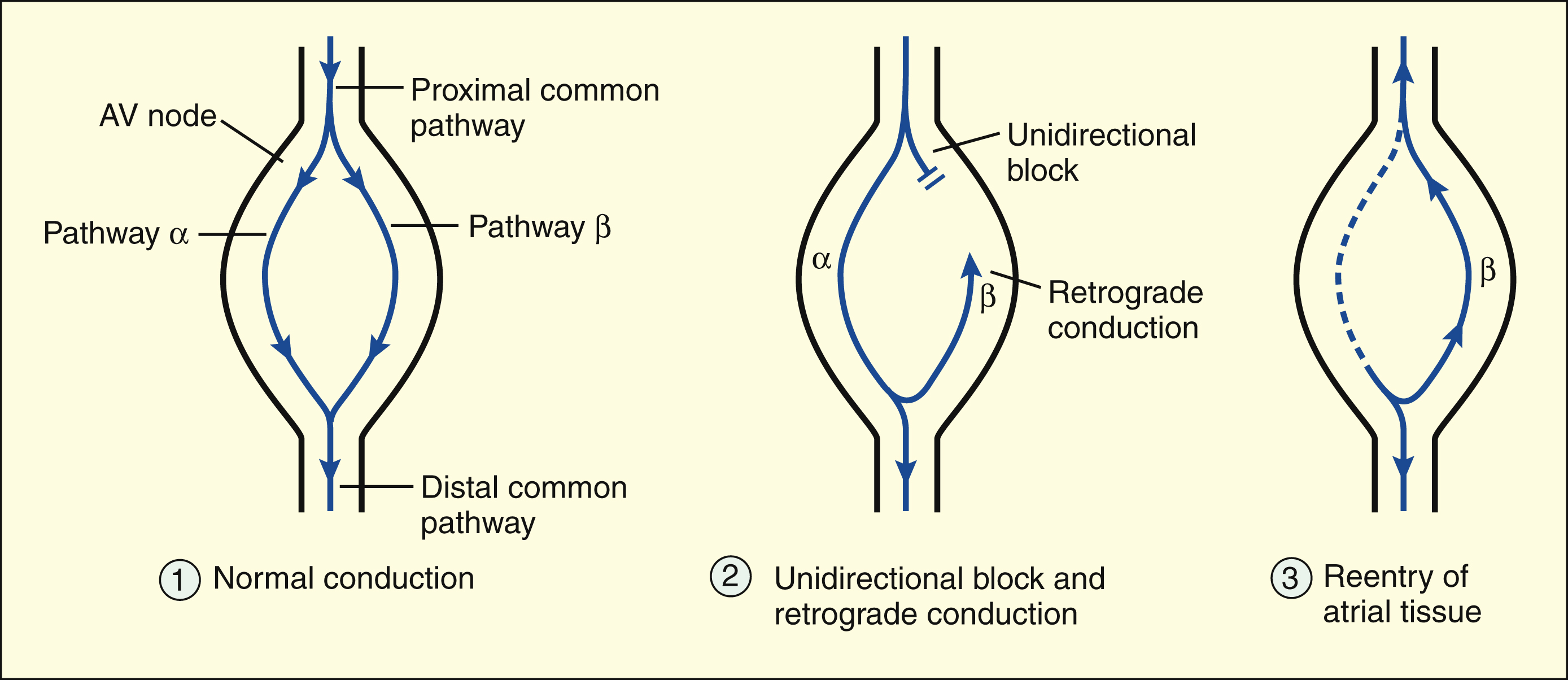
Reentry in the AV node is the most common electrophysiologic mechanism responsible for PSVT. Reentry occurs when a premature atrial depolarization arrives at the AV node and finds that one pathway (β) is still refractory from the previous depolarization. The other pathway (α), however, is able to conduct the impulse to the ventricle. Retrograde conduction of the impulse through pathway β leads to reentry of the atrium and results in tachycardia. In the AV node, the unidirectional block results from the β pathway’s longer refractory period, which blocks anterograde conduction but permits retrograde conduction after it has recovered its excitability.
INCREASED AUTOMATICITY
Spontaneous phase 4 depolarization generates an action potential that can be propagated to other parts of the heart. The SA node is the usual site of spontaneous impulse initiation (automaticity), but other cardiac tissues, including the AV node and the His-Purkinje system tissues, are also capable of spontaneous depolarization. Pathologic conditions or drugs can cause these tissues to depolarize more rapidly and thereby generate abnormal impulses. For example, overstimulation of the sympathetic nervous system or use of sympathomimetic drugs increases automaticity and can cause tachyarrhythmia.
The rate at which action potentials are generated in the SA node and elsewhere in the heart depends on the time required to depolarize the tissue from the maximal diastolic potential (MDP) to the TP. Automaticity is increased if the MDP becomes more positive or if the TP becomes more negative. Serum electrolyte abnormalities, hypoxia, and other pathologic changes can affect the MDP or TP in this manner and lead to arrhythmias.
AFTERDEPOLARIZATIONS
Afterdepolarizations are abnormal impulses resulting from the spontaneous generation of action potentials during or immediately after phase 3 repolarization. Depending on the time they occur, the abnormal impulses are designated as early afterdepolarizations or late afterdepolarizations. Afterdepolarizations are believed to be triggered by abnormal calcium influx and can be provoked by digitalis glycosides and by other drugs or pathologic events that prolong cardiac repolarization and the QT interval.
Abnormal Impulse Conduction
Abnormal impulse conduction is the basis for the formation of arrhythmias by the process of reentry, a process that involves reexcitation of a particular zone of cardiac tissue by the same impulse. Reentry is believed to be the most common mechanism responsible for the genesis of arrhythmias. It is usually caused by the presence of a unidirectional conduction block in a bifurcating conduction pathway (see Box 14–1).
REENTRY IN VENTRICULAR TISSUE
In ventricular tissue, ischemia and tissue hypoxia can cause a reduction in the resting membrane potential and a decrease in membrane responsiveness. Under these conditions, the cells do not depolarize as rapidly or completely during phase 0, and this reduces the rate at which the impulse is conducted to surrounding ventricular tissue. When a cardiac impulse is conducted through ischemic or infarcted tissue, the conduction velocity gradually slows until conduction ceases. This phenomenon, called decremental conduction, is the most common cause of a unidirectional block in ventricular tissue. If the unidirectional block occurs in one arm of a bifurcating pathway, the impulse can continue through the other arm in the normal (anterograde) direction and then reenter the ventricular tissue by retrograde conduction.
The reason that the impulse is blocked in the anterograde direction but is conducted in the retrograde direction is related to the characteristics of decremental conduction. In this type of conduction, the impulse encounters increasing resistance as it moves in the anterograde direction, and its velocity slows until the impulse is finally extinguished. The retrograde impulse has full velocity as it encounters the area of greatest resistance, and this enables it to jump across the ischemic area without being extinguished.
REENTRY IN THE ATRIOVENTRICULAR NODE
Reentry in the AV node is the most common electrophysiologic mechanism responsible for paroxysmal supraventricular tachycardia (PSVT). Of the several forms of PSVT, the form that occurs in patients with Wolff-Parkinson-White syndrome involves an accessory AV node conduction pathway through the bundle of Kent. In patients with the most common form of PSVT, however, the reentrant circuit is located entirely within the AV node. In the common form of PSVT, a premature atrial impulse is blocked in one pathway, is conducted through the AV node via the other pathway, and reenters the atrium by retrograde conduction. In AV node reentry, the unidirectional block does not result from decremental conduction but, instead, is caused by the difference in the refractory periods of the two pathways.
Drug-Induced Arrhythmias
Drugs can induce arrhythmias by several mechanisms.
Sympathomimetic drugs can increase the automaticity of the SA node, AV node, or His-Purkinje fibers and thereby produce tachyarrhythmias.
Digitalis glycosides sometimes evoke afterdepolarizations by increasing calcium influx into cardiac cells, and they can also impair AV node conduction and cause AV block.
Other drugs cause arrhythmias via their effects on ventricular conduction and repolarization. Drugs that slow ventricular repolarization and cause QT prolongation can evoke a form of polymorphic ventricular tachycardia called torsades de pointes (based on a French term meaning “fringe of pointed tips”). In this disorder, each QRS complex has a configuration that differs from the preceding one, and QT prolongation probably predisposes the ventricular tissue to afterdepolarizations that produce extrasystoles and tachycardia. Types of drugs that have been reported to induce torsades de pointes include antiarrhythmic drugs (e.g., quinidine and sotalol), histamine antagonists (e.g., astemizole and terfenadine), psychotropic drugs (e.g., phenothiazines), and other agents (e.g., cisapride).
Mechanisms and Classification of Antiarrhythmic Drugs
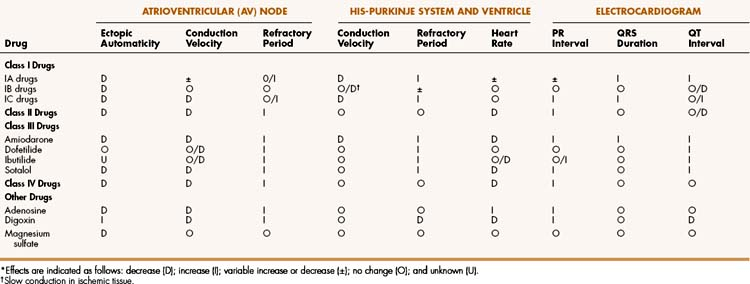
Antiarrhythmic drugs act primarily by suppressing or preventing abnormal impulse formation or conduction. Drugs that block sodium or calcium channels can reduce abnormal automaticity and slow conduction of the cardiac impulse. Drugs that block potassium channels can prolong repolarization and the action potential duration and thereby increase the refractory period of cardiac tissue. Drugs that block β-adrenoceptors reduce the sympathetic stimulation of cardiac automaticity and conduction velocity and thereby prevent the overstimulation that contributes to some arrhythmias.
On the basis of these mechanisms, Vaughan-Williams has divided the antiarrhythmic drugs into four main classes, with Class I consisting of sodium channel blockers, Class II consisting of β-adrenoceptor antagonists (β-blockers), Class III consisting of potassium channel blockers and other drugs that prolong the action potential duration, and Class IV consisting of calcium channel blockers. Although this classification system is helpful, a few drugs (e.g, adenosine) do not fit into any of these categories, and some drugs (e.g., amiodarone) could be included in more than one category.
SODIUM CHANNEL BLOCKERS
Class I, the largest group of antiarrhythmic drugs, consists of sodium channel blockers. These drugs bind to sodium channels when the channels are in the open and inactivated states, and they dissociate from the channels during the resting state (Box 14–2). The sodium channel blockers have the most pronounced effect on cardiac tissue that is firing rapidly, because sodium channels in this tissue spend more time in the open and inactivated states than in the resting state. This is called use-dependent blockade. Because of use-dependent blockade, sodium channel blockers suppress cardiac conduction more in a person with tachycardia than in a person with a normal heart rate.
BOX 14–2 ELECTROPHYSIOLOGIC PROPERTIES OF SODIUM CHANNEL BLOCKERS
During phase 0 of the ventricular action potential, the sodium channels open to depolarize the cell. The channels are then inactivated and no longer permit sodium entry during phases 1, 2, and 3. The channels must return to the resting state (phase 4) before they can open again during the next action potential.
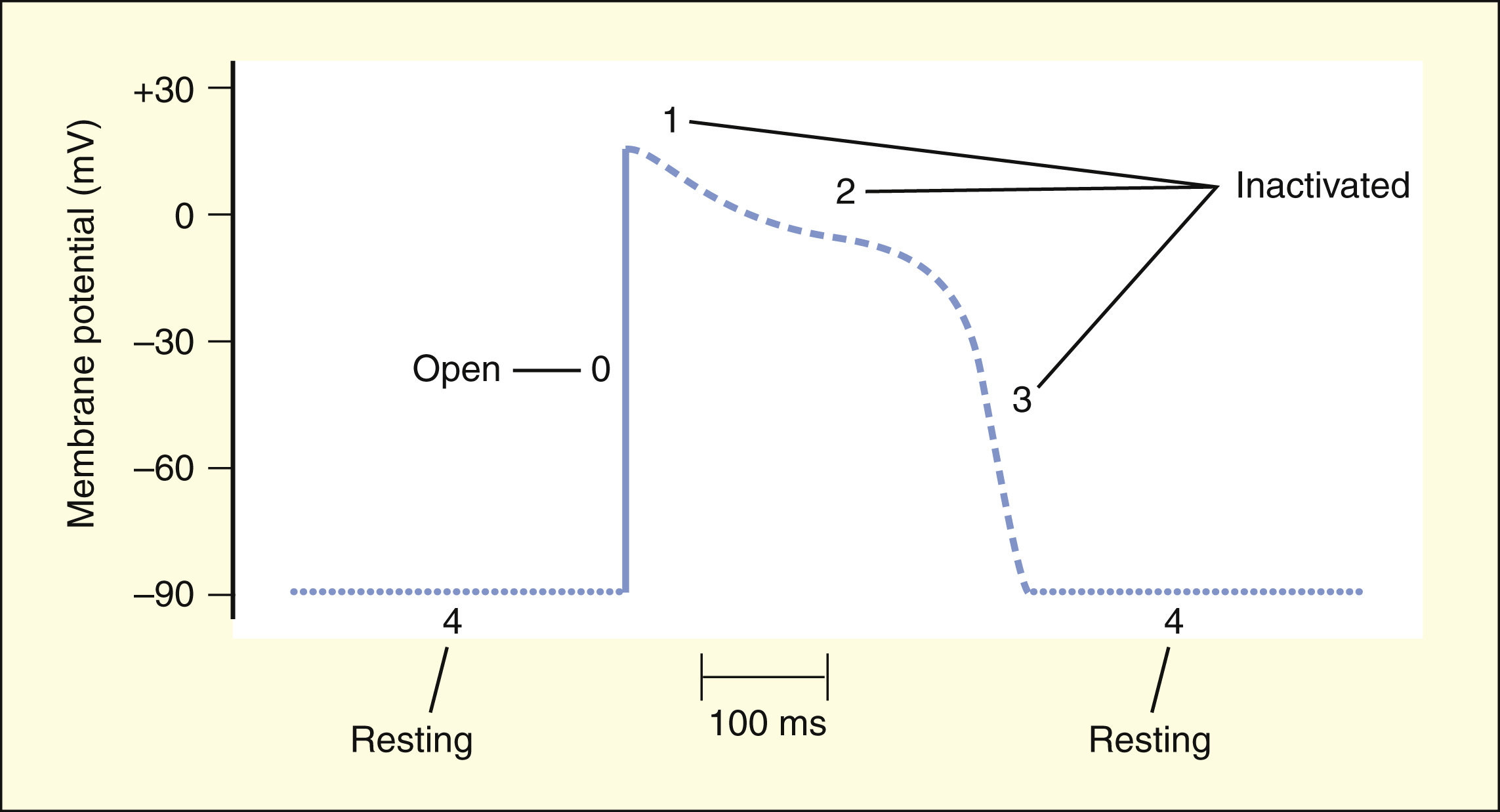
Drugs dissociate from the sodium channels at different rates (recovery). Drugs with a slow recovery have a greater effect on cardiac conduction velocity.

The drugs in Class I have been subdivided into three groups (IA, IB, and IC), based on whether they have greater affinity for the open state or the inactivated state and based on their rate of dissociation from sodium channels (rate of recovery). As shown in Box 14–2, Class IA drugs have greater affinity for the open state and have a slow recovery; Class IB drugs have greater affinity for the inactivated state and have a rapid recovery; and Class IC drugs have greater affinity for the open state and a very slow recovery.
Class IA Drugs
Disopyramide, procainamide, and quinidine are Class IA drugs. These drugs have similar electrophysiologic effects and clinical indications, but they differ in their pharmacokinetic properties and adverse effects.
Drug Properties
The Class IA drugs block the fast sodium channel and also block potassium channels. Therefore, they slow phase 0 depolarization and phase 3 repolarization in ventricular tissue (Fig. 14–2). These actions decrease the ventricular conduction velocity and prolong the ventricular action potential duration and refractory period (Table 14–1). On the ECG, this increases the QRS duration and prolongs the QT interval. Class IA drugs suppress abnormal (ectopic) automaticity, but they usually do not significantly affect SA node automaticity and the heart rate.
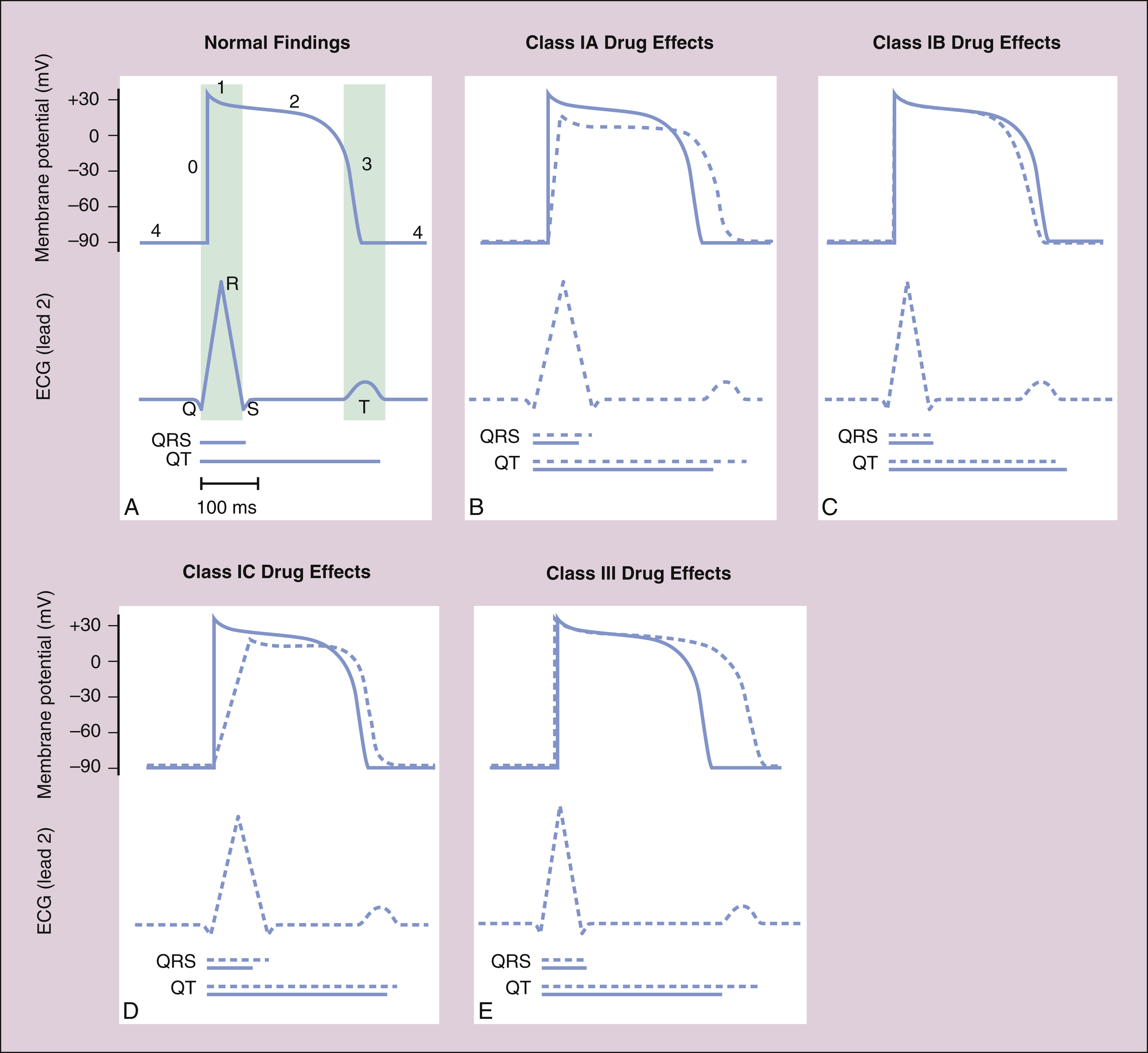
Figure 14–2 Effects of Class I and Class III antiarrhythmic drugs on the ventricular action potential duration and on the ECG. In each panel, the membrane potential scale is in millivolts (mV), and the time scale is in milliseconds (ms). Blue tracings and blue lines for the time scale depict normal findings; dashed lines and tracings depict the effects of drug administration. (A) Lightly shaded vertical bars show the relationship between the ventricular action potential duration and the findings on ECG in the normally functioning heart. (B) Class IA drugs slow phase 0 depolarization and phase 3 repolarization, thereby increasing the QRS duration and the QT interval. (C) Class IB drugs have little effect on normal cardiac tissue, but they can accelerate phase 3 repolarization and decrease the QT interval slightly. (D) Class IC drugs have the greatest effect on phase 0 depolarization and increase the QRS duration markedly, but they have little effect on phase 3 and the QT interval. (E) Class III drugs have no effect on phase 0, but they markedly prolong phase 3 and increase the QT interval.
All of the Class IA drugs have some degree of antimuscarinic (atropine-like) activity and may inhibit parasympathetic (vagal) effects on the SA and AV nodes. Disopyramide has the greatest antimuscarinic effect, procainamide has the least, and quinidine has an intermediate effect.
Quinidine
Quinidine is an isomer of quinine, an alkaloid obtained from the bark of the cinchona tree that is used to treat fever and malaria. The pharmacokinetic properties of quinidine and other antiarrhythmic drugs are summarized in Table 14–2. Quinidine is usually administered orally. It undergoes hepatic biotransformation and is excreted in the urine as the parent compound and metabolites. Because it has a moderately short half-life, quinidine is often given as a sustained-release preparation. The most common adverse effect of quinidine is diarrhea, which occurs in up to 30% of patients taking the drug and is often responsible for their discontinuation of its use. Less commonly, quinidine causes excessive prolongation of the QT interval and torsade de pointes, which can cause syncope secondary to a reduction in cardiac output. Thrombocytopenia has also been reported with quinidine use. Higher doses of quinidine can cause cinchonism, characterized by a constellation of neurologic symptoms that include tinnitus, dizziness, and blurred vision. Although its use has declined, quinidine is occasionally used to suppress supraventricular and ventricular arrhythmias.
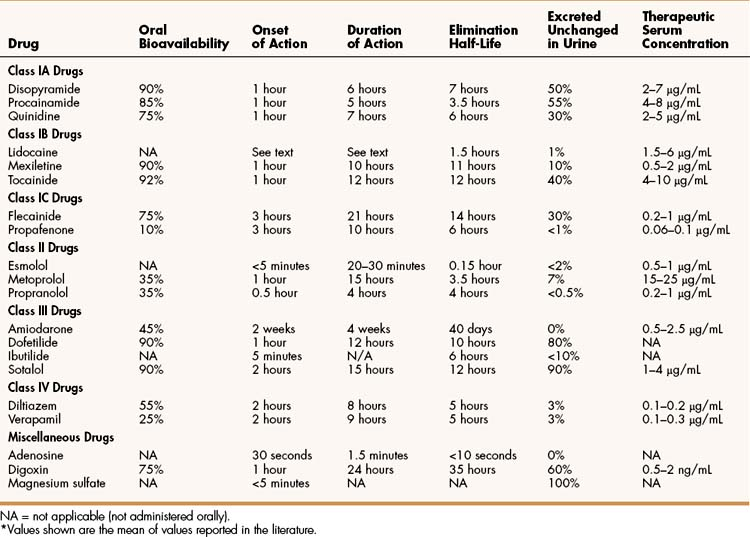
Procainamide
Procainamide is the amide derivative of the local anesthetic procaine. It is well absorbed from the gut and is converted to an active metabolite, N-acetylprocainamide. Long-term use of procainamide often causes a syndrome that resembles lupus erythematous, presents with arthralgia and a butterfly rash on the face, and is reversible. This syndrome can be distinguished from idiopathic lupus on the basis of serologic tests for anti-DNA antibodies. It is often responsible for discontinuation of the drug. The clinical use of procainamide has declined in recent years. Although not a first-line drug for any arrhythmia, procainamide is occasionally used to terminate acute ventricular arrhythmias, and it may be given orally for long-term suppression.
Class IB Drugs
Lidocaine, mexiletine, and tocainide are Class IB drugs. Class IB drugs have a greater affinity for inactivated sodium channels than for open channels.
Drug Properties
CHEMISTRY AND PHARMACOKINETICS
Lidocaine is a local anesthetic that also has antiarrhythmic activity. It undergoes extensive first-pass hepatic inactivation after oral administration and is not suitable for administration by this route. Mexiletine and tocainide are lidocaine congeners that are not susceptible to first-pass inactivation and are intended for oral administration. Lidocaine is rapidly inactivated by hepatic enzymes, whereas mexiletine and tocainide are more slowly metabolized and have much longer half-lives (see Table 14–2).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


