Viruses, Cell Transformation, and Cancer
Daniel DiMaio
Hung Fan
General Principles of Viral Transformation
In 1908, Ellerman and Bang143 reported that cell-free filtrates from chickens with leukemia could transmit the disease to healthy birds. The ability of the causative agent to pass through a fine filter identified it as a virus, providing the first evidence that viruses could cause cancer as well as acute contagious diseases. Three years later, Peyton Rous382 showed that a solid tumor in chickens, sarcoma, could also be transmitted by cell-free filtrates. In the 100 years since these discoveries, tumor viruses have been studied intensely in the belief that thorough understanding of these relatively simple agents would provide mechanistic insight into carcinogenesis, identify the causes of some human malignancies, and suggest novel strategies to prevent and treat cancer. All of these expectations have been met. Tumor viruses dysregulate or exploit crucial regulatory nodes in cells in order to replicate and persist in their host. Because these nodes regulate cell proliferation and survival, these viruses can cause cancer. Furthermore, because of their intimate association with this central cellular machinery, tumor viruses have proven to be prime tools for unraveling the complexities of all human cancers, not just those induced by viruses themselves. For example, studies of tumor viruses led to the discovery of cellular oncogenes and tumor suppressor proteins and to the elucidation of many important aspects of signal transduction, cell cycle control, cellular biochemistry, and carcinogenesis. Indeed, the concept that cancer is a genetic disease emerged largely from studies of tumor viruses. We have also learned that tumor viruses are associated with specific types of human cancer and that tumor virus infection is responsible for approximately 15% of human cancer deaths.310 Vaccines have been developed and deployed that prevent infection by certain tumor viruses and inhibit the formation of precancerous lesions or cancers.403 The remarkable success of these endeavors is due to the efforts of generations of virologists, biochemists, molecular biologists, epidemiologists, and clinicians, and to the favorable properties of the viruses themselves.
Cell Transformation
Early studies of tumor viruses focused on isolation of viruses from naturally occurring tumors in animals and description of the effects of these viruses in experimental animals. These studies revealed that a wide variety of viruses can induce tumors (Table 7.1). Diverse taxonomic groups of viruses with DNA genomes can cause tumors: polyomaviruses, papillomaviruses, adenoviruses, herpesviruses, hepadnaviruses, and poxviruses. Among the RNA viruses, a subset of retroviruses cause tumors in animals, and Hepatitis C virus has been implicated in human cancer. Thus, the ability to induce tumor formation is not associated with a particular class of virus or mode of virus replication. The types of tumors induced by viruses are also diverse and include sarcomas (tumors of mesenchymal cells), leukemias and lymphomas (tumors of hematopoietic cells), and carcinomas (tumors of epithelial cells).
Although such experimental pathogenesis studies revealed many fascinating and complex features of viral tumorigenesis, experiments in animals are cumbersome, slow, expensive, and often poorly suited for biochemical and mechanistic analysis. Therefore, cultured cell models of tumorigenesis are widely used. Cells explanted from tumors display many properties that
distinguish them from normal cells. Collectively, these properties are referred to as the transformed phenotype (Table 7.2), and cells displaying these properties are called transformed cells. Infection of normal cells in culture with many (but not all) tumor viruses can cause the rapid acquisition of the transformed phenotype, a process called transformation. The most commonly used cell types for transformation studies are rodent fibroblasts, which in general are easier to transform in vitro than human cells.
distinguish them from normal cells. Collectively, these properties are referred to as the transformed phenotype (Table 7.2), and cells displaying these properties are called transformed cells. Infection of normal cells in culture with many (but not all) tumor viruses can cause the rapid acquisition of the transformed phenotype, a process called transformation. The most commonly used cell types for transformation studies are rodent fibroblasts, which in general are easier to transform in vitro than human cells.
Table 7.1 Oncogenic Viruses | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The most striking property of transformed cells is their ability to form tumors when inoculated into animals. Experiments in animals have classically been used to determine whether a virus can cause a tumor in a living organism and to identify the viral gene(s) responsible for this activity. Although mice are used for the vast majority of such experiments, other commonly used hosts are rats, hamsters, rabbits, and monkeys. Immunodeficient animals (e.g., athymic nude mice) are frequently used in order to avoid immune rejection of the inoculated cells.
In addition to tumorigenicity, transformed cells display a variety of features in culture that distinguish them from normal cells. These include changes in growth, morphology, metabolism, and intracellular and cell surface biochemistry. Normal cells have very stringent requirements for growth. To proliferate in culture, normal cells require peptide growth factors, commonly supplied as fetal bovine serum. When plated on plastic, normal adherent cells typically divide to fill the available space on the surface of the culture vessel until they become a confluent monolayer of cells and then cease proliferation, even if supplied with adequate amounts of nutrients and growth factors. The cessation of growth at confluence is known as density-dependent growth inhibition or contact inhibition. Most normal cells also require attachment to a solid substrate and are unable to grow when suspended in semisolid medium such as agarose or methylcellulose, a property known as anchorage dependence. Finally, normal cells usually display a limited life span in culture after isolation from an animal and can be serially passaged for no more than a few months before they
cease proliferation and adopt a permanently growth-arrested state called senescence.
cease proliferation and adopt a permanently growth-arrested state called senescence.
Table 7.2 Properties of Transformed Cells | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Transformed cells display strikingly different growth properties than normal cells (Table 7.2). Transformed cells have minimal requirements for growth factors and can proliferate in low concentrations of serum (or even in its absence). Transformed cells continue to proliferate after reaching confluence and display a multilayered, piled-up appearance and higher saturation densities than normal cells (Fig. 7.1). If a transformed cell is present in an excess of normal cells in a culture, it will override contact inhibition at confluence and continue to proliferate locally, forming a discrete, clonal group of transformed cells known as a focus (Fig. 7.1).198 Because each focus originates from a single transformed cell, the number of foci formed provides a convenient measure of the number of transforming viruses in an inoculum. In addition, because foci are readily apparent on a background monolayer of cells, focus formation provides a simple means of identifying and isolating transformants. Transformed cells also display anchorage independence and can form colonies in semisolid medium without adhering to a solid surface.294 Anchorage independence is the property of transformed cells that is most closely associated with the ability to form tumors in animals. Finally, in contrast to normal cultured cells, transformed cells that divide indefinitely in culture are said to be immortal.
Transformed cells can also display striking morphologic differences from normal cells due to changes in the cellular cytoskeleton, extracellular matrix, and cell surface.226,371 Transformed cells also switch from oxidative respiration to aerobic glycolysis, a phenomenon known as the Warburg effect. The resulting lactic acid production and acidification of the culture medium (and the tell-tale yellow of medium containing phenol red, a pH indicator) allows the rapid identification of transformed cultures.
It should be noted that transformation is not an all-or-nothing phenomenon. Some transformed cells display only a subset of these properties. For example, in some cases cells can form foci but fail to display anchorage independence or tumorigenicity, or they can display reduced serum requirements but not form foci. It is therefore important to specify the assay used to assess transformation. “Normal” established cell lines, such as murine NIH3T3 cells, are immortal but do not display other features of transformed cells and thus can be regarded as being minimally transformed.
Identification of Viral Oncogenes
To determine whether a virus has tumorigenic or transforming activity, host animals or cells are infected and the appearance of tumors or the acquisition of a transformed phenotype in culture is measured. Acute transforming retroviruses cause the rapid formation of tumors in animals and rapidly transform cells in culture because they carry genetic information known as oncogenes. In contrast, retroviruses that lack oncogenes cause tumors in animals many months after infection and do not transform cells in culture. Most DNA tumor viruses contain oncogenes and can transform cells in culture and induce tumors in animals.
For viruses that contain oncogenes, cell transformation typically occurs within a few days or weeks of infection, although some measures of transformation, such as cell immortalization, take months to assess. When infected at high multiplicity of infection (moi), the entire culture of cells may undergo transformation, which can allow biochemical characterization of cell transformation. Infections at low moi require the use of quantal measures of transformation, such as focus formation or colony formation in agarose, to score transformation because most cells in the culture remain normal.
Although acute properties of transformation are fairly easy to elicit, for some viruses stable transformation that persists in culture is a very rare event. In addition, for lytic DNA viruses, where replication is cytocidal, transformation can only occur when the virus life cycle cannot go to completion. Transformation by these viruses is commonly studied following infection of nonpermissive cells that cannot support a full virus life cycle. In these cells, the initial steps (the early phase) of virus infection take place—virus attachment to cells, penetration into the cells, virion uncoating, and expression of the early proteins—but then
productive infection is aborted before the onset of vegetative viral DNA replication. For example, rat and mouse cells are nonpermissive for replication by simian virus 40 (SV40), a monkey virus, so many transformation assays are conducted in these cells. Alternatively, permissive cells, in which the wild-type virus can complete its life cycle, can be transformed if viral replication is blocked. Infection with replication-defective viral mutants or transfection with nonreplicating, subgenomic viral DNA fragments that contain viral oncogenes can transform permissive cells.299 In addition, some tumor viruses (such as herpesviruses) can establish latency in permissive cells, and in some cases expression of a subset of viral genes in the latently infected cells can lead to transformation. Infection by acute transforming retroviruses usually does not result in cell death, and stable transformation is very efficient, with virtually every infected cell becoming transformed.
productive infection is aborted before the onset of vegetative viral DNA replication. For example, rat and mouse cells are nonpermissive for replication by simian virus 40 (SV40), a monkey virus, so many transformation assays are conducted in these cells. Alternatively, permissive cells, in which the wild-type virus can complete its life cycle, can be transformed if viral replication is blocked. Infection with replication-defective viral mutants or transfection with nonreplicating, subgenomic viral DNA fragments that contain viral oncogenes can transform permissive cells.299 In addition, some tumor viruses (such as herpesviruses) can establish latency in permissive cells, and in some cases expression of a subset of viral genes in the latently infected cells can lead to transformation. Infection by acute transforming retroviruses usually does not result in cell death, and stable transformation is very efficient, with virtually every infected cell becoming transformed.
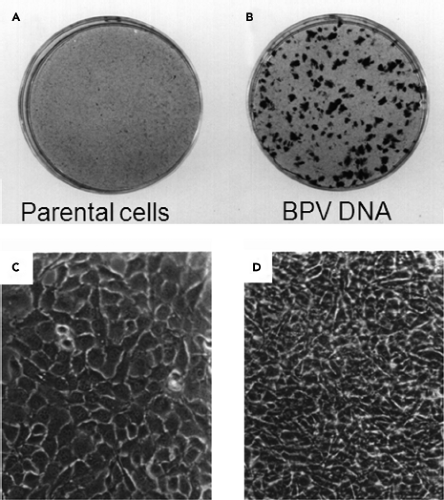 Figure 7.1. Appearance of transformed cells. A: Plate showing a monolayer of normal murine C127 fibroblasts. B: Plate of fibroblasts showing transformed foci induced by transfection with bovine papillomavirus DNA. A and B were stained 12 days after transfection. C: Micrograph of a monolayer of normal murine fibroblasts, showing flat, spread-out morphology. D: Culture of murine fibroblasts transformed by the bovine papillomavirus E5 protein. Note the piled-up, refractile appearance of small, transformed cells. |
An oncogene can display potent activity in one transformation assay but be totally devoid of activity in another assay, even in the same host cell, because it may modulate multiple independent cellular signaling pathways. Host-specific differences can also greatly influence transforming activity, so a variety of host animals or cells are commonly tested to assess transforming activity. Some oncogenes have potent transforming activity in numerous assays but are unable to cause cell immortalization. Therefore, transformation is often studied in established cell lines that are already immortal.
Viral mutants can be used to identify specific viral genes with transforming activity. In early experiments, mutagenized viral stocks were tested for loss of transforming activity or for the acquisition of temperature-sensitive transforming activity. The generation and analysis of conditional mutants were particularly useful for identifying and studying transforming genes, especially those that are essential for virus replication.380,467 In these situations, the virus is first propagated under permissive conditions, and then the virus is used to infect cells under nonpermissive conditions and transformation is assessed. Genetic mapping approaches can then be used to identify the mutant viral gene responsible for the transformation defect. More recently, specific mutations were constructed in individual viral open reading frames, and the resulting virus mutants were assayed for transforming activity. Alternatively, individual viral genes or subgenomic segments can be introduced into cells or transgenic animals and assessed for transforming activity or tumorigenicity.192,420 This approach is complicated when more than one viral oncogene is required for transformation, because no single gene displays
activity. In these situations, combinations of viral genes can be transferred into cells, or the ability of viral genes to cooperate with cellular oncogenes can be tested.
activity. In these situations, combinations of viral genes can be transferred into cells, or the ability of viral genes to cooperate with cellular oncogenes can be tested.
The analysis of murine polyomavirus transforming functions illustrates some of the power and pitfalls of these approaches. Murine polyomavirus (hereafter called polyomavirus) is related to SV40. SV40 and polyomavirus express the early protein, large T antigen (LT), which is required for viral DNA replication. Certain mutations in the SV40 LT gene caused temperature-sensitive defects in cell transformation as well as viral DNA replication, identifying the LT gene as the major oncogene of SV40.2 However, the equivalent polyomavirus LT mutants were temperature sensitive for virus replication but, at least under some conditions, able to transform cells at the nonpermissive temperature. In addition, the polyomavirus LT gene was often disrupted in transformed cells. These findings suggested that, in contrast to SV40, LT was not the major transforming protein of polyomavirus. Indeed, Benjamin26 isolated transformation-defective polyomavirus mutants that could replicate in mouse cells that had been transformed by wild-type polyomavirus, but not in normal mouse cells, so-called host-range–restricted and transformation-defective (hr-T) mutants. Genetic mapping revealed that the hr-T mutant phenotype resulted not from mutations in the LT gene but rather from mutations in an alternative open reading frame in the early region of polyomavirus, which encoded a distinct protein, middle T antigen (mT) (e-Fig. 7.1). SV40 does not encode mT. These findings indicated that SV40 and polyomavirus, though genetically similar, used different mechanisms to transform cells.
To determine the activities of the individual early proteins of polyomavirus, a panel of complementary DNA (cDNA) clones that each expressed a single viral protein was assayed in rat1 cells, an established cell line. The clone expressing polyomavirus mT antigen alone was the only one that displayed transforming activity: cells expressing mT antigen formed foci, appeared morphologically transformed, and formed tumors in animals.468 These results implied that mT is the major transforming protein of polyomavirus. However, cells transformed by mT antigen were unable to grow in low serum and were thus only partially transformed. On the other hand, cells expressing polyomavirus LT alone were able to grow in low serum, even though they did not display other properties of transformed cells.373 By using a different assay to study polyomavirus transformation, namely, the ability of the viral genes to convert primary rat embryo fibroblasts into immortalized cells, Rassoulzadegan et al374 found that LT was active while mT was not. Furthermore, sequential transfer of the LT and mT genes could convert primary cells into immortal, fully transformed cells. Thus, the individual oncogenes of polyomavirus displayed different transforming activities. Together, these important experiments identified the polyomavirus oncogenes, revealed that they had different effects on cell growth, demonstrated that oncogenes can cooperate to generate fully transformed cells, and highlighted the critical importance of the particular assays used to assess transformation.
Another approach to identify viral oncogenes rests on the finding that genes responsible for transformation are frequently retained in transformed cells, whereas the other viral genes can be lost. Thus, DNA virus genes that are uniformly expressed in transformed cells are likely to be viral oncogenes. Experiments searching for viral DNA and RNA in transformed cells led to the presumptive identification of E1A and E1B as the primary adenovirus oncogenes.123,167 As the roster of viral and cellular oncogenes has expanded, it is now often possible to identify viral oncogenes by comparing the sequences of viral genes to sequence databases, because novel viral oncogenes are often homologous to validated oncogenes.
The mapping experiments outlined previously demonstrated that DNA tumor viruses typically contain multiple oncogenes. In contrast, acute transforming retroviruses usually contain one oncogene, although there are examples of retroviruses with two (e.g., avian erythroblastosis virus [AEV] ES4 contains both ErbA and ErbB oncogenes). Nonacute retroviruses lack oncogenes.
Maintenance of Transformation
Is continuous expression of viral oncogenes required to maintain the transformed phenotype, or do tumor virus oncogenes only initiate cell transformation, after which they are not required (a hit-and-run–type mechanism)? If viral genes are required to maintain transformation, then they will be expressed in stably transformed cells. Biochemical and immune-based analysis can detect the presence of viral genes and proteins in cells transformed by many viruses, but often only a subset of viral genes is expressed in these cells. The production of infectious virus by transformed cells would provide definitive evidence for the presence of an intact, functional viral genome in the cells, but one of the striking properties of virally transformed cells is that they often do not produce infectious virus. This is not surprising for viruses, including many of the DNA tumor viruses, whose transforming activity is typically studied in nonpermissive cells unable to support virus replication. In contrast, cells transformed by Rous sarcoma virus or some other retroviruses produce infectious virus.
The presence of wild-type virus genomes in transformed cells was demonstrated by virus rescue experiments. In a typical experiment, SV40-transformed mouse cells (which are nonpermissive for replication of SV40) were fused to nontransformed, permissive monkey cells (e-Fig. 7.2). The fused cells produced infectious wild-type SV40, which could be detected by plaque formation assays in permissive cells.266 This demonstrated that transformed cells could contain full-length, infectious virus genomes in a latent state, which could be activated by replication factors present in permissive cells. We now know that the inability of mouse cells to support SV40 replication is due to the inability of murine DNA polymerase-α/primase to interact with SV40 LT.408
The study of conditional viral mutants and expression constructs proved that viral genes expressed in transformed cells are required to maintain the transformed state. In these experiments, cells transformed under the permissive condition when the viral oncoprotein was active often reverted to normal under nonpermissive conditions that inactivated the oncoprotein. This was initially shown with temperature-sensitive mutants of viral oncogenes, where transformation was extinguished at the higher, nonpermissive temperature.27,302,416 More recently, individual oncogenes driven by inducible/repressible promoters or maneuvers such as RNA interference were used to show that viral oncogene expression was required to maintain the transformed phenotype. Ongoing oncogene expression is not required in cells transformed by all viruses, but it is commonly
observed for cells transformed by a variety of DNA viruses and acute transforming retroviruses.
observed for cells transformed by a variety of DNA viruses and acute transforming retroviruses.
Continuous oncogene expression is also required for cells derived from some virally induced human tumors. For example, repression of human papillomavirus (HPV) oncogene expression in cervical cancer cells causes the rapid cessation of proliferation or the induction of apoptosis (e.g., reference [225]). Thus, even though mutations presumably accumulated in these cells during the extended period of human carcinogenesis, these mutations are not sufficient to maintain the transformed phenotype in the absence of viral oncogene expression. This finding raises the hope that it will be possible to treat virally induced cancers by interfering with the expression or action of viral oncogenes.
To continuously provide viral oncogene products in stably transformed cells, the viral genome must persist. Most commonly, the viral DNA is covalently integrated into the cellular DNA and replicates passively with the host genome. For retroviruses, integration of an essentially intact viral genome is an integral step in the viral life cycle, but for most DNA viruses, integration is incidental (and indeed inimical) to normal viral DNA replication. For these viruses, integration does not occur at specific sites in the cellular genome, and often only portions of the viral genome containing the viral oncogenes are present in the cell. Integration of DNA virus genomes is a rare event, which may explain at least in part why stable transformation by these viruses is so inefficient. Papillomavirus and herpesvirus genomes stably replicate as plasmids in transformed cells, but the DNA of these viruses can be integrated in cancer cells.
Origin of Viral Oncogenes
Where do viral oncogenes come from? Initial studies with retroviruses were conducted with Rous sarcoma virus (RSV). Cells transformed by infection with a mutant RSV carrying a temperature-sensitive src gene rapidly reverted to a nontransformed phenotype when shifted to the nonpermissive temperature, but the mutant was replication competent at both high and low temperatures.74,302 Similarly, transformation-defective RSV deletion mutants were isolated that retained the ability to replicate.27 These important results indicated that a specific gene (src) was responsible for RSV transformation but not for virus replication. Stehelin, Varmus, and Bishop441 later used molecular hybridization to show that src sequences were actually derived from chicken cell DNA. We now know that acute transforming retroviruses such as RSV incorporate altered versions of cellular genes into the viral genomes, a process known as transduction. These transduced genes typically encode constitutively active versions of cellular signal transduction components, such as growth factor receptors and other signaling proteins (see next section). The cellular versions of these genes (designated by the prefix c, e.g., c-src) are known as the proto-oncogenes, whereas the active viral versions (e.g., v-src) are called oncogenes.86 In contrast, the nonacute retroviruses induce tumors by integrating near-cellular proto-oncogenes and activating their expression.
The discovery that cellular proto-oncogenes can be activated by viruses to generate cancer suggested that nonviral cancers might also arise by activation of proto-oncogenes. Indeed, spontaneously arising human tumors frequently contain altered or overexpressed versions of the proto-oncogenes targeted by oncogenic retroviruses. In several cases, drugs have been developed that specifically inhibit activated proto-oncogene products and provide substantial clinical benefit to cancer patients (e.g., in chronic myelogenous leukemia and lung cancer). Thus, the practical legacy of tumor virology extends far beyond virally induced tumors.
In contrast to most retroviral oncogenes, the oncogenes of the small DNA tumor viruses are intrinsic viral genes required for virus replication, and mutations that interfere with transforming activity usually also inhibit viral replication. In some cases, phylogenetic analysis shows that these genes have been essential for virus replication for millions of years. DNA virus oncogenes usually encode proteins that bind and modulate cellular proteins that regulate cell growth. Most commonly, nuclear oncoproteins of a number of DNA tumor viruses bind to the p53 and retinoblastoma (Rb) tumor suppressor proteins and neutralize their growth inhibitory activity, but the oncoproteins of DNA tumor viruses can modulate other cellular targets as well. Some DNA virus oncoproteins mimic constitutively active forms of a cellular regulatory protein.
p53 and Rb are not only targets of viral tumorigenesis. Most sporadic human cancers, as well as some inherited cancers, contain inactivating mutations in the p53 and Rb genes and in genes in the pathways they control.284 As was the case with the discovery of cellular proto-oncogenes, studies of DNA tumor viruses identified cellular proteins and pathways crucial for understanding the genetic basis of naturally occurring, nonviral tumors.
Although retroviral oncogenes usually have cellular origins and DNA tumor virus oncogenes typically have viral origins, these distinctions are not absolute. The Jaagsiekte sheep retrovirus envelope protein is required for both virus replication and cell transformation, and the gp55P oncoprotein of the Friend erythroleukemia retrovirus is a mutant retroviral envelope protein that contributes to erythroleukemia in mice.285,296 On the other hand, the genome of Kaposi sarcoma herpesvirus (KSHV), a large DNA-containing virus, contains a number of genes essential for viral replication and cell transformation that appear to have been acquired from the cellular genome in relatively recent evolutionary times.328,389
Cellular Targets of Viral Oncogenes
The great majority of viral oncogenes stimulate cell proliferation (most of the others inhibit cell death). In untransformed cells, the cell division cycle is regulated by intrinsic oscillators, which ensure the orderly progression of the cell cycle, and by external growth factors, which indirectly affect the activity of the cell cycle machinery. Like serum growth factors, many viral oncogenes can stimulate DNA synthesis in normal cells made quiescent by serum starvation. Thus, the signal transduction pathways activated by growth factors provide a useful framework to understand the function of viral oncogenes (Fig. 7.2).
Serum contains polypeptide growth factors such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF), which bind to specific cell surface receptors to initiate mitogenic signaling. These growth factor receptors are often transmembrane proteins with an extracellular ligand binding domain, a membrane-spanning domain, and an intracellular signaling domain. Ligand binding induces dimerization and/or reorientation of the receptors, initiating intracellular signaling cascades.407 Many of these receptors are tyrosine kinases or associate with tyrosine kinases. The molecular events induced by ligand binding result in activation of the intrinsic tyrosine kinase activity of the receptors (or of the associated kinases) and in tyrosine phosphorylation of the receptor itself. Phosphorylation on
tyrosine in turn generates specific binding sites for a variety of signaling proteins that contain SH2 (src homology region 2) or other protein–protein interaction domains, which themselves may be tyrosine phosphorylated after binding the activated receptor.386 Among the targets of these signaling proteins are intracellular guanine nucleotide exchange factors (GEFs) that activate signal transducing molecules such as p21ras.39 This then initiates a cytoplasmic signaling cascade involving a series of serine/threonine kinases (the mitogen-activated protein [MAP] kinase cascade) that ultimately results in the phosphorylation and activation of nuclear transcription factors, such as AP1 (composed of jun and fos subunits) and c-myc, allowing them to stimulate the expression of various cellular genes.34,36,80 Growth factor treatment also activates phosphotidylinositol 3-kinase (PI3K) signaling, which generates antiapoptotic and pro-growth signals mediated by the downstream kinase, akt.
tyrosine in turn generates specific binding sites for a variety of signaling proteins that contain SH2 (src homology region 2) or other protein–protein interaction domains, which themselves may be tyrosine phosphorylated after binding the activated receptor.386 Among the targets of these signaling proteins are intracellular guanine nucleotide exchange factors (GEFs) that activate signal transducing molecules such as p21ras.39 This then initiates a cytoplasmic signaling cascade involving a series of serine/threonine kinases (the mitogen-activated protein [MAP] kinase cascade) that ultimately results in the phosphorylation and activation of nuclear transcription factors, such as AP1 (composed of jun and fos subunits) and c-myc, allowing them to stimulate the expression of various cellular genes.34,36,80 Growth factor treatment also activates phosphotidylinositol 3-kinase (PI3K) signaling, which generates antiapoptotic and pro-growth signals mediated by the downstream kinase, akt.
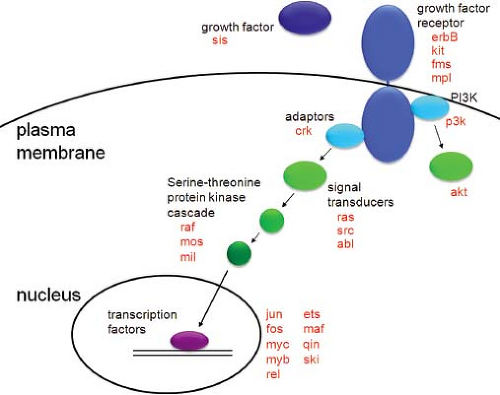 Figure 7.2. Mitogenic signal transduction pathways and retroviral oncogenes. This figure shows a schematic representation of mitogenic signal transduction. Binding of a growth factor to a growth factor receptor initiates signal transduction pathways involving adaptor proteins, signal transducing proteins, and serine/threonine kinases. This results in the phosphorylation of transcription factors to alter gene expression and execute pro-proliferative or antiapoptotic signals. Components of this pathway that were transduced by acute transforming retroviruses are listed in red. |
A major target of transcriptional activation following growth factor treatment is the cyclin D gene.306 Cyclins, including cyclin D, are regulatory subunits of the cyclin-dependent kinases (cdks), which directly regulate the cell cycle by allowing cells to move past specific cell cycle checkpoints (e.g., the G1/S boundary) (Fig. 7.3). Cyclin concentrations vary periodically throughout the cell cycle, and association of cyclins with cdks stimulates the activity of the cdk complexes, which catalyze the phosphorylation of nuclear substrates. For example, association of cyclin D with cdk4 results in phosphorylation of members of the Rb family (Fig. 7.3). Phosphorylation of Rb proteins causes their release from E2F proteins, a family of transcription factors that was first identified because E2F is required for activation of the adenovirus E2 promoter.268 Rb release increases transcription of a set of E2F-dependent cellular genes that encode proteins required for cellular DNA synthesis, including DNA polymerases, topoisomerases, and enzymes involved in nucleotide synthesis.114 Thus, growth factor treatment triggers a signal transduction cascade that results in the inactivation of the Rb family and stimulation of the G1 to S phase transition and cellular DNA synthesis.
Cdks are also regulated by the p53 pathway.284 The p53 protein is a transcription factor that can stimulate or repress the expression of various genes. Typically present at low levels
and inactive, the expression and activity of p53 is induced by a variety of stresses, including DNA damage. An important target of p53-mediated transcriptional activation is p21WAF1/CIP1, an inhibitor of cdk activity and cell cycle progression.139 Therefore, a direct consequence of p53 activation is p21WAF1/CIP1 induction and the inhibition of cdk activity. This causes the retention of the Rb family in the underphosphorylated form, which inhibits cell cycle progression by preventing E2F-mediated gene activation. Conversely, inhibition of p53 reduces the levels of p21WAF1/CIP1, and the resulting increase in cdk activity and Rb phosphorylation dissociates E2F and Rb, allowing DNA synthesis to occur.
and inactive, the expression and activity of p53 is induced by a variety of stresses, including DNA damage. An important target of p53-mediated transcriptional activation is p21WAF1/CIP1, an inhibitor of cdk activity and cell cycle progression.139 Therefore, a direct consequence of p53 activation is p21WAF1/CIP1 induction and the inhibition of cdk activity. This causes the retention of the Rb family in the underphosphorylated form, which inhibits cell cycle progression by preventing E2F-mediated gene activation. Conversely, inhibition of p53 reduces the levels of p21WAF1/CIP1, and the resulting increase in cdk activity and Rb phosphorylation dissociates E2F and Rb, allowing DNA synthesis to occur.
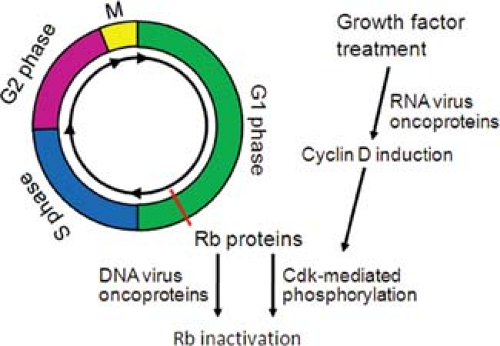 Figure 7.3. The cell cycle. The figure shows a simplified version of the cell cycle. The active form of the retinoblastoma family members prevents progression through the G1 phase. Rb-mediated inhibition is abrogated by growth factor treatment, which results in accumulation of cyclin D, stimulation of cyclin-dependent kinases (cdks), and Rb phosphorylation. Rb can also be inhibited by the expression of several DNA virus oncoproteins, such as adenovirus E1A, human papillomavirus (HPV) E7, or simian virus 40 (SV40) large T antigen (see Fig. 7.12). |
According to this simplified view of cell cycle control, proliferation can be stimulated by activation of mitogenic signaling or by inhibition of tumor suppressor proteins. Not surprisingly, most viral oncogenes function in just this way, either by activating signal transduction cascades or by inhibiting tumor suppressor pathways.31,47 Indeed, much of our molecular understanding of mitogenic signal transduction and the role of tumor suppressor proteins in cell proliferation and cancer is derived from genetic and biochemical analysis of viral oncoproteins.
Cells not only respond to mitogenic signals but also possess intrinsic pathways that limit cell proliferation in response to a variety of stresses. DNA damage elicits cell cycle arrest (via p53 activation) to allow cells time to repair the damage. If the damage is extensive, apoptosis will result. Virus infection or oncogene action can also activate checkpoints that block cell proliferation. For example, replicating viral DNA itself can be recognized as foreign and trigger a DNA damage response.488 In addition, the aberrant activation of mitogenic signaling pathways by viral oncoproteins can cause unscheduled DNA synthesis, which in turn induces apoptosis or senescence.137 Because such premature cell death or inhibition of proliferation will reduce virus yield, these cellular responses can be viewed as antiviral defense mechanisms. In many cases, the p53 and the Rb pathways are required for the execution of these antiviral processes. Neutralization of these tumor suppressor pathways by viral oncoproteins will not only directly stimulate cell proliferation but also interfere with these checkpoints and remove these barriers to transformation and tumorigenesis. Furthermore, when the checkpoint fails, the resulting inaccurate DNA repair or unscheduled DNA synthesis allows the accumulation of mutations in cellular regulatory genes or global genomic instability, which further contribute to the erosion of cell growth control.
Transformation by Retroviruses
Retroviruses were discovered because they caused cancers in animals. The first transmissible tumors associated with retroviruses were avian myeloblastosis in 1908143 and avian sarcomas in 1911,382 caused by avian myeloblastosis virus (AMV) and RSV, respectively. Other early retroviruses associated with cancers include murine mammary tumor virus (MMTV, mammary carcinomas)60 and murine leukemia viruses (MuLVs, lymphomas and leukemias).195 Of the seven retrovirus genera, viruses from five (alpha, beta, gamma, delta and epsilon) are associated with cancers. Only lentiviruses and foamy viruses have not been found to directly induce cancer—and for human immunodeficiency virus type 1 (HIV-1), a lentivirus, a major clinical manifestation of acquired immunodeficiency syndrome (AIDS) is development of cancer.
Most oncogenic retroviruses can be divided into two categories, based on the rapidity with which they induce tumors. Acute transforming retroviruses induce tumors rapidly (often within weeks in experimental animals) and can often transform cells in culture. As mentioned earlier, acute transforming retroviruses carry additional genetic information beyond the standard genes for retroviral replication (gag, pol, and env; see Chapter 47). These additional sequences, the viral oncogenes, are responsible for rapid and efficient tumorigenesis and cell transformation. Nonacute retroviruses induce tumors in animals more slowly, do not carry oncogenes, and usually do not transform cells in culture. Nevertheless, these viruses often induce tumors with high efficiency, typically by causing transcriptional activation of cellular proto-oncogenes.
Acute Transforming Retroviruses and Viral Oncogenes
Acute transforming retroviruses belong to the alpha- and gammaretrovirus genera (Table 7.1). The first acute transforming retroviruses to be studied were avian alpharetroviruses, beginning with RSV and avian acute leukemia viruses such as AMV. Acute transforming gammaretroviruses include murine and rat sarcoma viruses, murine acute leukemia viruses, and feline and simian sarcoma viruses. Study of acute transforming retroviruses and their cognate proto-oncogenes has provided numerous important insights into pathways of growth regulation. Indeed, some of the most important proteins in signal transduction and cancer were first identified through study of retroviral oncogenes, including Ras, Src, Abl, Myc, Fos, and Jun.
RSV and src
Infection of chick embryo fibroblasts with RSV results in cell transformation; a focus-formation assay can be used to quantify the amount of transforming virus present.463 After the original isolation of RSV, several different strains of related transforming viruses were developed. Some of these strains (e.g., Schmidt-Ruppin [SR-RSV] and Prague [Pr-RSV]) are replication competent. Others, such as the Bryan high-titer strain, consist of a replication-defective RSV genome, which carries the viral oncogene, as well as a related replication-competent “helper” virus genome (e.g., Rous-associated virus [RAV]). In a mixed infection, this helper virus encodes viral replication proteins in trans to allow the production of infectious virus.204
Passage of replication-competent RSV occasionally results in loss of transforming capacity without affecting viral replication—such variants are termed transformation-defective RSV (tdRSV).130 Thus, oncogenesis and transformation are not required for RSV replication. The size of the tdRSV genome was smaller than wild-type RSV, suggesting that viral genomic sequences responsible for transforming activity were missing from tdRSV. These deleted sequences were mapped to the 3′ end of the RSV genome,484 and they were eventually termed src,86 providing the first example of a retroviral oncogene (Fig. 7.4). Src is expressed via a spliced messenger RNA (mRNA) from the splice donor site at the 5′ end of the genome and an acceptor site at the 5′ end of the src gene.202
Antisera from RSV-infected rabbits were used to identify the protein product of the src oncogene as a 60-kD phosphoprotein designated pp60v-src.50 When an anti-src antibody was incubated with a cell lysate from RSV-transformed cells and
γ −32P-adenosine triphosphate (ATP) as a phosphate donor, 32P was transferred to the antibody heavy chain, thus identifying pp60v-src as a protein kinase.88 Subsequently it was shown that pp60v-src is a tyrosine-specific protein kinase; that is, it phosphorylates tyrosine residues in its substrate proteins.89,223 Phosphorylation of proteins on tyrosines is a relatively rare modification (compared to phosphorylation on serine and threonine), and pp60v-src was the first tyrosine kinase discovered. Tyrosine phosphorylation is now known to be a major mechanism to regulate protein activity.
γ −32P-adenosine triphosphate (ATP) as a phosphate donor, 32P was transferred to the antibody heavy chain, thus identifying pp60v-src as a protein kinase.88 Subsequently it was shown that pp60v-src is a tyrosine-specific protein kinase; that is, it phosphorylates tyrosine residues in its substrate proteins.89,223 Phosphorylation of proteins on tyrosines is a relatively rare modification (compared to phosphorylation on serine and threonine), and pp60v-src was the first tyrosine kinase discovered. Tyrosine phosphorylation is now known to be a major mechanism to regulate protein activity.
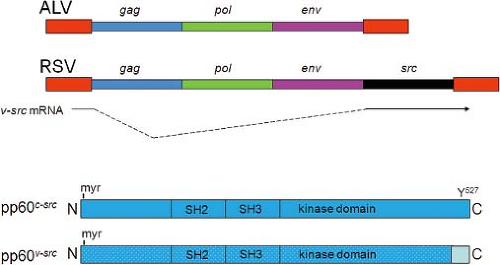 Figure 7.4. Structure of Rous sarcoma virus (RSV) and pp60src. The top part of the figure shows the proviral structure of avian leukosis virus containing the three retroviral genes required for virus replication, gag, pol, and env. The long terminal repeats (LTRs) are shown in red. The second provirus shows RSV with the src gene located at the 3′ end of the viral genome. A schematic picture of spliced v-src messenger RNA (mRNA) is shown below the RSV provirus, with spliced-out introns depicted as dashed lines. The bottom part of the figure shows schematic diagrams of pp60v-src and pp60c-src. Both proteins contain conserved SH2 and SH3 protein–protein interaction domains, tyrosine kinase domains, and an amino-terminal myristal (myr) group. A regulatory tyrosine at position 527 in pp60c-src (Y 527) is also shown. pp60v-src contains several amino acid substitutions compared to pp60c-src, represented by the stippling, as well as a frameshift mutation that replaces the 19 carboxyl-residues of pp60c-src, including the Y527, with 12 unrelated amino acids. |
Another key discovery was the determination of the origins of the RSV src gene. Hybridization experiments with radioactive src-specific DNA probes revealed the presence of homologous sequences in DNA from uninfected chickens.441 The src-specific DNA also hybridized with DNA of other species, and the extent of hybridization correlated with the evolutionary distance from chickens. Thus, the src gene of RSV, v-src, was derived by capture of a normal cell gene, c-src, referred to as a proto-oncogene.
The domains of pp60v-src are shown in Figure 7.4. The protein is modified by myristoylation at the N-terminus, which anchors it to the inner surface of the plasma membrane.52 Membrane attachment is necessary for transformation, although not for kinase activity.243 The kinase domain is located in the internal region of pp60v-src. Two adjacent domains are “src homology” domains—SH2 and SH3. These domains were originally identified as regions of homology between v-src and src-related oncogenes of other avian acute transforming retroviruses.393 The SH2 and SH3 domains mediate the specific interaction of pp60v-src (and other proteins) with cellular proteins, including regulatory proteins and substrates for phosphorylation.261,319 SH2 and SH3 domains are present in many cellular proteins, and they are important protein–protein interaction motifs. SH2 domains bind phosphotyrosine residues in specific sequence contexts in their target proteins, while SH3 domains bind proline-rich regions.261
The c-src proto-oncogene encodes a similar protein, pp60c-src, which differs from pp60v-src by several amino acid substitutions throughout the protein and by a different C-terminal sequence due to a frameshift mutation in v-src (Fig. 7.4).454,460 The C-terminus of pp60c-src is a regulatory domain that contains a crucial tyrosine residue at position 527 (Y 527). When Y 527 is phosphorylated, it binds the SH2 domain on the same molecule of pp60c-src, resulting in a closed conformation in which the kinase is inactive.503 Dephosphorylation of Y 527 displaces the SH2 domain, resulting in activation of src kinase.94 Of note, the viral protein pp60v-src lacks the regulatory Y 527 and is thus constitutively in the open, kinase-active form where it signals for cell transformation by catalyzing high-level phosphorylation of substrates.
Substrates phosphorylated by pp60v-src provide the key to understanding transformation and oncogenesis by RSV. Various substrates have been identified by searching for proteins that show enhanced levels of tyrosine phosphorylation in RSV-transformed cells compared to normal cells.91,97,367,381,398,415 pp60v-src substrates whose phosphorylation is important for RSV transformation include the mitogen-activated protein kinase (MAPK) Erk-1/2,381 focal adhesion kinase (FAK),398 and Connexin43, a tight junction protein.98
Overview of Other Retroviral Oncogenes
The basic principles elucidated by analysis of RSV also apply to the other acute transforming retroviruses and are summarized here. With a few exceptions described below, acute transforming retroviruses contain viral oncogenes resulting from capture of cellular proto-oncogenes (Table 7.3), although the location
of the oncogene in the viral genome can vary (Fig. 7.5). In some cases the same proto-oncogene has been captured independently by several different viruses. For example, the avian retroviruses Fujinami sarcoma virus and PRC-II both captured the c-fps proto-oncogene from the chicken genome, and the Snyder-Theilen strain of feline sarcoma virus captured the homologous gene (c-fes) from the cat genome.194 Proto-oncogene proteins, which generally play positive roles in stimulation of cell growth or division, are typically tightly regulated. The viral oncogene proteins often differ from their parent proteins by amino acid substitutions, deletions, and/or fusion to an intrinsic viral replication protein, most commonly gag. These differences lead to activation of the viral oncogene proteins compared to the proto-oncogene proteins by allowing the viral proteins to escape from the tight regulatory control imposed on the normal cellular homologs. For example, deletions may lead to loss of regulatory regions present in the normal proto-oncogene protein (e.g., pp60v-src vs. pp60c-src as described earlier). Amino acid substitutions can also result in loss of regulatory mechanisms (e.g., the v-ras proteins), and fusion of viral sequences to the oncogene protein can enhance activity (e.g., the v-abl protein). In other cases, the viral oncogene product may be identical to the proto-oncogene product but delivers a sustained or enhanced signal because it is overexpressed. In either case, the viral oncogene proteins signal for cell proliferation by the same molecular mechanisms as the proto-oncogene proteins, but in a constitutive or uncontrolled manner (see Fig. 7.2).
of the oncogene in the viral genome can vary (Fig. 7.5). In some cases the same proto-oncogene has been captured independently by several different viruses. For example, the avian retroviruses Fujinami sarcoma virus and PRC-II both captured the c-fps proto-oncogene from the chicken genome, and the Snyder-Theilen strain of feline sarcoma virus captured the homologous gene (c-fes) from the cat genome.194 Proto-oncogene proteins, which generally play positive roles in stimulation of cell growth or division, are typically tightly regulated. The viral oncogene proteins often differ from their parent proteins by amino acid substitutions, deletions, and/or fusion to an intrinsic viral replication protein, most commonly gag. These differences lead to activation of the viral oncogene proteins compared to the proto-oncogene proteins by allowing the viral proteins to escape from the tight regulatory control imposed on the normal cellular homologs. For example, deletions may lead to loss of regulatory regions present in the normal proto-oncogene protein (e.g., pp60v-src vs. pp60c-src as described earlier). Amino acid substitutions can also result in loss of regulatory mechanisms (e.g., the v-ras proteins), and fusion of viral sequences to the oncogene protein can enhance activity (e.g., the v-abl protein). In other cases, the viral oncogene product may be identical to the proto-oncogene product but delivers a sustained or enhanced signal because it is overexpressed. In either case, the viral oncogene proteins signal for cell proliferation by the same molecular mechanisms as the proto-oncogene proteins, but in a constitutive or uncontrolled manner (see Fig. 7.2).
Table 7.3 Oncogenes Transduced by Acute Transforming Retroviruses | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Figure 7.5. Genome maps of avian acute transforming retroviruses. The genome structures of the following avian acute transforming viruses are shown: ALV, avian leukosis virus; MC29, avian myelocytoma virus MC29; AMV-BAI, avian myeloblastosis virus BAI; AEV-ES4, avian erythroblastosis virus ES4. The proviral long terminal repeats (LTRs) are shown in red, and the retroviral structural genes gag, pol, and env are shown in blue, green, and lavender, respectively. Transduced oncogenes are shown in black or gray. |
Most acute transforming retroviral genomes are replication defective, because capture of the proto-oncogene sequences generally results in loss of part of the viral genome (Fig. 7.5). Therefore, in order to produce infectious virus, these viruses require co-infection with a helper virus that supplies replication functions. Infection of cells with low concentrations of an acute transforming virus stock can generate foci of transformed cells harboring only the acute transforming viral genome (and not the helper virus genome). Cells from these foci express the viral oncogene, but they do not produce infectious virus—they are called nonproducer cells. Infectious transforming virus can be recovered from these nonproducer cells by infecting them with a replication-competent helper virus to provide replication functions.
Retroviral oncogenes also differ from their corresponding proto-oncogenes because they lack introns. This likely reflects the mechanism of oncogene capture, which is thought to be initiated by integration of a retroviral provirus upstream from a cellular proto-oncogene.454 Read-through transcription from the retrovirus into the proto-oncogene causes the production of a fusion transcript containing 5′ retroviral sequences and 3′ proto-oncogene sequences. The fusion transcript is then processed by mRNA splicing to remove the proto-oncogene introns, followed by packaging into virions. In a subsequent infection, recombination during reverse transcription presumably results in recovery of the 3′ end of the viral genome and the generation of an acute transforming retroviral genome containing a captured oncogene lacking introns.
Mechanism of Action of Retroviral Cytoplasmic and Membrane-Associated Oncogene Products (see Fig. 7.2)
Many oncogenes encode membrane-associated proteins that are derived from RTKs and other growth factor receptors. For example, the v-erbB oncogene of AEV ES4 is a transduced version of the EGF receptor.125 In comparison to the wild-type EGF receptor, the v-erbB protein of AEV lacks the extracellular growth factor binding domain, as well as a tyrosine-containing inhibitory autoregulatory domain at the C-terminus (analogous to Y 527 of pp60c-src).210 As a result, the truncated v-erbB protein delivers downstream signals in an EGF-independent manner. Other RTKs captured and constitutively activated by acute transforming retroviruses include the stem cell factor receptor (v-kit) and the colony-stimulating factor 1 receptor (v-fms).219,425,517 The v-mpl oncogene encodes a constitutively active version of the thrombopoietin receptor, which lacks intrinsic kinase activity but associates with intracellular tyrosine kinases.109,249,291 The v-cbl oncogene encodes a ubiquitin ligase that can regulate receptor abundance and activity.239 On the other hand, the v-sis oncogene of simian sarcoma virus is a transduced version of a growth factor itself, the platelet-derived growth factor B chain (PDGF-B) gene.79,338 Thus, v-sis can transform only cells that express the PDGF receptor.
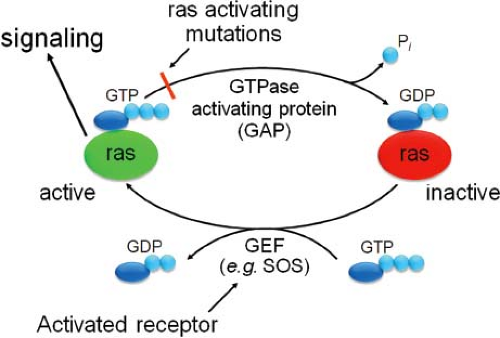 Figure 7.6. The ras cycle. Cellular ras proteins exist in two different forms, active guanosine triphosphate (GTP)-bound ras (green) and inactive GDP-bound ras (red). Inactive ras is activated by guanine nucleotide exchange factors (GEFs, such as SOS) that load GTP onto ras; GEFs are activated by growth factor receptor signaling. Active ras is inactivated by its intrinsic GTPase activity, which hydrolyzes bound GTP to guanosine diphosphate (GDP). GTPase activity is stimulated by GTPase activating proteins (GAPs). Activating point mutations at particular positions in v-ras cause the accumulation of ras in the active form, usually by preventing GAP action. Active ras induces signaling by binding and activating effector proteins such as raf. |
The src family constitutes a class of membrane-associated tyrosine kinases that function in the cytoplasm and lack a ligand binding domain. Thus, src family members are not RTKs. In addition to v-src itself, other avian acute transforming retroviruses contain the v-fps/v-fes oncogene and the v-yes oncogene.194 These oncogene proteins share substantial homology with pp60v-src in the kinase domain as well as in other domains.261,393 There are other cellular tyrosine protein kinases in the src family that function in different signal transduction pathways (e.g., lck, whose protein is important for signaling in B lymphocytes478), but they have not been captured by acute transforming retroviruses. The genes encoding other cytoplasmic tyrosine kinases unrelated to src have been transduced and activated by other acute transforming retroviruses, such as the v-abl oncogene of Abelson murine leukemia virus.496,497
The cytoplasmic products of other retroviral oncogenes are kinases that phosphorylate protein substrates on serines or threonines, rather than tyrosines. Three oncogenes in this class are v-raf (of murine sarcoma virus 3611 [and the homologous v-mil from an avian virus]),234,372 v-mos (of Moloney murine sarcoma virus),413 and v-akt (of Akt murine leukemia virus).24 The corresponding cellular proto-oncogene proteins function in different signaling pathways. Cellular Raf protein is a mediator in signal transduction from RTKs and Ras (see later), initiating a cascade of protein phosphorylations that ultimately leads to phosphorylation and nuclear translocation of MAPKs of the extracellular signal-regulated kinase (ERK) family.262,514 Cellular Mos protein is required for meiosis and is a component of cytostatic factor (CSF).508 v-mos is activated primarily by overexpression from the viral long terminal repeat (LTR).33 Cellular Akt protein is a key intermediate in downstream signaling from PI3 kinase, which is also coupled to RTK signaling (see Fig. 7.2). Akt activation delivers survival and proliferative signals to cells. In addition, PI3 kinase itself has been captured by an avian acute transforming retrovirus (AS-16) as the v-p3 k oncogene.64 For the viral oncogenes that encode protein kinases, constitutive, unregulated enzyme activity causes overactive signaling through the corresponding signal transduction pathways, leading to transformation and tumorigenesis.
Other important cytoplasmic oncogenes are the ras oncogenes, v-Hras and v-Kras, identified in Harvey and Kirsten murine sarcoma viruses, respectively.86,145 These viruses were generated by the capture of two closely related cellular proto-oncogenes, c-Hras and c-Kras, when MuLVs were passaged through rats. The cellular Hras and Kras proteins, which play critical roles in signal transduction, are guanosine triphosphatases (GTPases) (known as G proteins) that bind guanine nucleotides (guanosine diphosphate [GDP] or guanosine triphosphate [GTP]). In the GTP-bound form, Ras proteins induce signals that result in cell proliferation (e.g., by binding and activating Raf kinase), but they are inactive when bound to GDP (Fig. 7.6).39,44 Guanine nucleotide exchange factors (GEFs), which stimulate replacement of GDP with GTP, cause inactive Ras to cycle to GTP-bound, active Ras. GEFs include adaptor proteins such as Grb-2 or Sos, which are mobilized by RTK activation.136,292 The active GTP-bound Ras cycles back to the inactive form by hydrolysis of the bound GTP to GDP by the intrinsic GTPase activity of the Ras protein, which is stimulated by GTPase activating proteins (GAPs).35 The v-Hras and v-Kras proteins contain missense mutations at key residues (e.g., amino acids 12 and 59 for v-Hras)459 that reduce the ability of GAP to stimulate GTP hydrolysis.1 Therefore, the v-Ras proteins accumulate in the active, GTP-bound form because they are unable to cycle to the inactive form, and they signal constitutively for cell growth.
The avian sarcoma virus CT10 and its transduced oncogene, v-crk,307 have provided insight into a family of cellular proteins known as adaptor proteins. The proto-oncogene protein c-crk (crkI), along with proteins from related proto-oncogenes crkII and crkL, contain SH2 and SH3 domains, but they do not have enzymatic activity. Rather, they function as adaptor proteins that assemble proteins into signal transduction complexes by way of interactions via the SH2 and SH3 domains. Cellular signaling proteins that interact with Crk family proteins include Abl, Sos, and FAK.304
Mechanism of Action of Retroviral Nuclear Oncogene Products (see Fig. 7.2)
The nuclear oncogenes of retroviruses are largely derived from proto-oncogenes that encode DNA binding proteins, many of which act as transcription factors. The v-myc oncogene, originally identified in the avian acute leukemia virus MC29 (but also captured by other avian acute transforming viruses), was one of the first studied.421,476 The c-myc protein (Myc) is involved in early responses to mitogenic stimuli and binds E-box motifs in promoters/enhancers of target genes.138 Myc exists as a homodimer or as a heterodimer with a related protein, Max. The Myc-Myc homodimers bind E-boxes with low affinity, while Myc-Max heterodimers bind with high affinity.32 Early after a mitogenic stimulus, levels of Myc transiently rise, leading to activation of E-box–containing genes such as those encoding E2F and cyclin D and repression of genes including those encoding cdk inhibitors.138 v-myc appears to transform cells because of its constitutive overexpression of the retroviral LTR.
The v-myb oncogene was also discovered in two different avian acute leukemia viruses (AMV BAI A and AMV E26).258 It is derived from c-myb, which encodes a cellular DNA binding protein that acts as a transcription factor important in hematopoietic cell development.28 Structural alterations in v-myb appear to be responsible for activation.28,193
The oncogene of acute transforming viruses of the Finkel- Biskis-Jinkins (FBJ) murine osteosarcoma virus complex is v-fos,472 and v-jun is the oncogene of avian sarcoma virus S17 (“jun” is 17 in Japanese).297 The c-fos and c-jun proto-oncogenes encode nuclear DNA binding proteins that are subunits of the transcription factor, activator protein-1 (AP1).375 Jun can homodimerize, or it can heterodimerize with Fos. Jun-Jun homodimers bind AP1 sites in DNA with low affinity, while Fos-Jun heterodimers bind these sites with high affinity. Activation of Fos transcription from undetectable levels is one the earliest events after mitogenic stimulation—levels of Fos protein rise within 15 minutes of bombesin stimulation in fibroblasts.324 In contrast, Jun protein is present in the absence of mitogenic stimulation, and Jun levels do not change substantially in response to growth factor treatment. As a result, early after mitogenic stimulation, there is a shift from Jun-Jun homodimers to Fos-Jun heterodimers, resulting in activation of promoters/enhancers containing AP1 sites.329 Expression of these target genes (e.g., c-myc)227 is important for cell division and growth. Both the v-jun and the v-fos proteins contain structural alterations responsible for oncogenic activation. For example, v-jun contains missense mutations and a deletion that removes an auto-inhibitory segment, resulting in constitutive activation.41
Another nuclear oncogene is v-erbA of AEV. The c-erbA proto-oncogene encodes the thyroid hormone receptor-α (THR-α), a sequence-specific DNA binding protein.395,487 In contrast to wild-type THR-α, which binds DNA and activates transcription of target genes, the v-erbA protein consists largely of the DNA binding domain.105,395 As a result, it binds to DNA nonproductively and acts as a dominant negative form by inhibiting transcription of THR-α–responsive genes. This leads to a block in erythroid cell differentiation, which in turn results in enhanced proliferation of undifferentiated erythroid progenitors.
The v-rel oncogene was initially identified in avian reticuloendotheliosis virus.73,443 The c-rel proto-oncogene encodes a member of the nuclear factor-κB (NFκB) transcription factor family and is important for B-cell development.180 C-Rel can homodimerize or heterodimerize with the p50 NFκB protein and bind NFκB sites in target genes. Point mutations and amino acids derived from env are required for v-rel activation.172,457
Tumorigenesis by Nonacute Retroviruses
Oncogenic nonacute retroviruses belong to the alpha-, beta-, and gammaretrovirus genera. Prototypic nonacute retroviruses include avian leukosis virus (ALV), MMTV, and various MuLVs. These viruses are replication competent, lack viral oncogenes, and do not transform cultured cells. In contrast to tumors induced by acute transforming retroviruses, which are often polyclonal, tumors induced by nonacute retroviruses are monoclonal or oligoclonal (i.e., derived from one or a few transformed cells). Another feature of these viruses is that high-level infection in animals typically occurs many months before development of tumors. Thus, infection is not synonymous with oncogenic transformation for nonacute viruses. The common mechanism by which most nonacute retroviruses induce tumors is insertional activation of cellular proto-oncogenes by the integrated retroviral genome.
B Lymphomas Induced by ALV and Activation of c-myc
Pioneering experiments investigating tumorigenesis by nonacute retroviruses were conducted on chicken B-cell lymphomas induced by ALV. Neel et al331 analyzed DNA from these tumors and found that many of them did not contain intact integrated ALV proviruses, although each tumor contained at least some segment of viral DNA. They studied tumor RNA by northern blot analysis by using two different radioactive viral cDNA hybridization probes, one representing the entire viral coding region and another representing only the ends of the viral RNA including portions of the viral LTR. The full-length probe did not detect viral RNAs in all tumors; however, the LTR probe invariably detected transcripts in tumors, and different tumors gave transcripts of similar sizes. This led Neel et al331 to propose the promoter insertion model of ALV tumorigenesis, in which transcription from the downstream ALV LTR continues into adjacent cellular sequences (Fig. 7.7). Furthermore, the similar sizes of the transcripts suggested that the ALV proviruses were inserted into the same chromosomal locus in different tumors. Further experiments showed that the ALV LTR was indeed inserted into the same chromosomal region in independent tumors, adjacent to the c-myc proto-oncogene.211 This placed c-myc under control of the strong ALV promoter/enhancer, resulting in read-through transcription into the proto-oncogene. Thus, c-myc can be activated either by transduction by the acute transforming retroviruses or by proviral insertion by the nonacute retroviruses. Many ALV-induced tumors contain a deletion of a portion of integrated proviral ALV DNA, resulting in loss of the upstream LTR, so that most of the viral DNA is not transcribed. Deletion of the upstream LTR likely stimulates the activity of the downstream LTR, which drives enhanced transcription of c-myc.
In some ALV-induced B lymphomas overexpressing c-myc, the relative transcriptional orientations of ALV DNA and c-myc were not consistent with the promoter insertion model.352 For example, some tumors contained proviral DNA
inserted upstream of c-myc but in the opposite orientation, whereas in other cases the provirus was inserted downstream of c-myc (Fig. 7.7). Activation of c-myc expression in these cases can be explained by activation of the endogenous c-myc promoter by the enhancer sequences in the ALV LTR, which can activate adjacent promoters in an orientation- and position-independent manner. Thus, activation of proto-oncogenes by nonacute retroviruses can be considered as LTR activation, either by promoter insertion or by enhancer activation. Insertional activation of proto-oncogenes has proven to be a common mechanism of tumorigenesis by nonacute retroviruses. Other variations of these mechanisms are described in the next sections.
inserted upstream of c-myc but in the opposite orientation, whereas in other cases the provirus was inserted downstream of c-myc (Fig. 7.7). Activation of c-myc expression in these cases can be explained by activation of the endogenous c-myc promoter by the enhancer sequences in the ALV LTR, which can activate adjacent promoters in an orientation- and position-independent manner. Thus, activation of proto-oncogenes by nonacute retroviruses can be considered as LTR activation, either by promoter insertion or by enhancer activation. Insertional activation of proto-oncogenes has proven to be a common mechanism of tumorigenesis by nonacute retroviruses. Other variations of these mechanisms are described in the next sections.
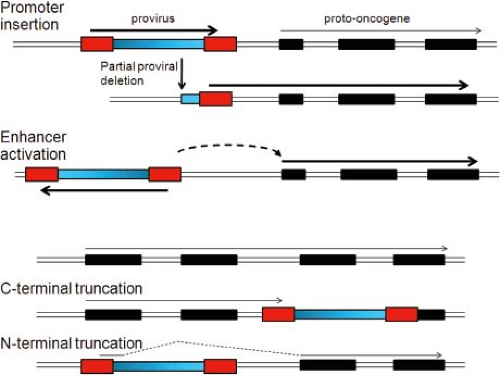 Figure 7.7. Insertional activation of cellular proto-oncogenes by retroviruses. The top two diagrams show proto-oncogene activation by promoter insertion. The top diagram shows a provirus integrated upstream of a proto-oncogene in the same transcriptional orientation. The thickness of the horizontal arrows indicates the relative level of expression. Partial deletion of the provirus activates the downstream long terminal repeat (LTR), which results in read-through transcription into the proto-oncogene. The third diagram shows proto-oncogene activation by enhancer activation. In the example shown, the provirus integrates farther upstream from the proto-oncogene in the opposite transcriptional orientation, and the enhancer in the viral LTR activates expression from the proto-oncogene promoter (dashed arrow). Proviruses inserted downstream of a proto-oncogene can also enhance expression. The bottom three diagrams show a proto-oncogene with four exons. Proviral insertion in the same transcriptional orientation in the 3′ portion of the proto-oncogene could result in synthesis of a C-terminally truncated protein product due to cleavage/polyadenylation sequences in the LTR. A provirus inserted in the 5′ portion of the proto-oncogene could be transcribed into downstream proto-oncogene exons followed by splicing from a viral splice donor site to a proto-oncogene splice acceptor site. This could result in the deletion or splicing out of proto-oncogene exons and expression of an N-terminally truncated protein product. If the deleted portions regulate the activity of the wild-type protein, these deletions might result in proto-oncogene activation. |
MMTV Tumorigenesis
MMTV (a betaretrovirus) also employs insertional activation. Common proviral insertion sites (CISs) were identified340 in independently arising MMTV-induced mammary tumors. For MMTV, the first CIS did not correspond to any proto-oncogene known at the time, but by analogy to ALV-induced B lymphomas, it was hypothesized that the CIS marked a cellular gene that was activated by the MMTV provirus. This gene was named int-1 (integration site-1), and int-1 RNA was overexpressed in tumors. Subsequently int-1 was renamed wnt-1 because the Drosophila homolog had been identified by the developmental mutation wingless.378 Wnt-1 is an important molecule in the Wnt-β-catenin signal transduction pathway, which is mutated in a wide variety of human cancers, most notably colorectal cancer.254 Thus, CISs initially identified in nonacute retroviral tumors led to discovery of new proto-oncogenes (Table 7.4). In contrast to ALV-induced B lymphomas where the provirus was integrated quite near c-myc, the MMTV provirus in most cases was inserted farther from Wnt-1.340 Activation of Wnt-1 likely occurs by enhancer activation, because transcriptional enhancers can work over relatively long distances (kilobases). Other CISs in MMTV-induced tumors include int-2 (FGF-3),120 int-3, and int-4.247
MuLV Leukemogenesis
MuLVs are gammaretroviruses and include Moloney MuLV (M-MuLV), SL3-3 MuLV, and radiation leukemia virus (RadLV), which induce T lymphoma, and Friend MuLV (F-MuLV), which induces erythroid or myeloid leukemia. Activation of c-myc commonly occurs in leukemias induced by M-MuLV.417,440 Other M-MuLV
tumors contained novel CISs that led to the discovery of new proto-oncogenes such as the cytoplasmic serine/threonine protein kinases, pim-1 and pim-248,99 (Table 7.4). A number of other CISs have been identified in T lymphomas induced by M-MuLV or RadLV, some of which correspond to known cellular genes such as cyclin D2.205 F-MuLV–induced tumors show activation of a different set of proto-oncogenes than those activated by M-MuLV. Proto-oncogenes commonly activated by F-MuLV include fli-1 (a nuclear DNA binding protein of the ets family), fim-1,435 and fim-2 (M-CSF).182 In rapidly arising erythroid leukemias induced by the Friend virus complex, a provirus activates the spi-1 proto-oncogene, which encodes another ets family transcription factor.320,350 In addition, an acute transforming retrovirus has also been identified in the Friend virus complex: spleen focus-forming virus (SFFV), which contains an oncogene encoding gp55. Gp55 is unusual in that it is not derived from a cellular proto-oncogene, but rather is a deleted form of an endogenous (genetically transmitted) MuLV env gene that recombined into the F-MuLV genome.498
tumors contained novel CISs that led to the discovery of new proto-oncogenes such as the cytoplasmic serine/threonine protein kinases, pim-1 and pim-248,99 (Table 7.4). A number of other CISs have been identified in T lymphomas induced by M-MuLV or RadLV, some of which correspond to known cellular genes such as cyclin D2.205 F-MuLV–induced tumors show activation of a different set of proto-oncogenes than those activated by M-MuLV. Proto-oncogenes commonly activated by F-MuLV include fli-1 (a nuclear DNA binding protein of the ets family), fim-1,435 and fim-2 (M-CSF).182 In rapidly arising erythroid leukemias induced by the Friend virus complex, a provirus activates the spi-1 proto-oncogene, which encodes another ets family transcription factor.320,350 In addition, an acute transforming retrovirus has also been identified in the Friend virus complex: spleen focus-forming virus (SFFV), which contains an oncogene encoding gp55. Gp55 is unusual in that it is not derived from a cellular proto-oncogene, but rather is a deleted form of an endogenous (genetically transmitted) MuLV env gene that recombined into the F-MuLV genome.498
Table 7.4 Proto-Oncogenes Identified As Common Insertion Sites | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The different activated proto-oncogenes in leukemias induced by M-MuLV and F-MuLV likely reflect the fact that different signaling pathways are important for transformation of lymphoid versus erythroid/myeloid cells. Studies of MuLV leukemogenesis revealed that the viral LTRs played critical roles in leukemogenicity. First, exchange of the U3 regions of the LTRs between an oncogenic MuLV (e.g., M-MuLV or SL3-3) and a nononcogenic virus (e.g., an endogenous MuLV) demonstrated that leukemogenicity was lost when the U3 region of the LTR was derived from the nononcogenic MuLV.118,281 Furthermore, substitution of the enhancer region of the F-MuLV LTR into M-MuLV shifted the disease spectrum from T-lymphoid to erythroid/myeloid tumors.68 This finding reflects the fact that enhancers are often tissue specific; that is, they specifically bind transcription factors present in a subset of differentiated cells. Indeed, in reporter assays, the M-MuLV LTR is most active in T-lymphoid cell lines and less so in erythroid/myeloid cells, while F-MuLV LTR is most active in erythroid/myeloid cells.428 Taken together, these results indicate that in order for an MuLV to induce tumors, it must have LTRs that efficiently activate cellular proto-oncogenes in the target cells.
Recombinant inbred strains of mice have also been used to study MuLV leukemogenesis. Certain strains of mice genetically transmit replication-competent endogenous MuLVs to their offspring. Crossing such strains with other mouse strains, followed by in-crossing, yields recombinant inbred lines. Some of these lines have high rates of leukemia development due to activation of the endogenous virus, which in turn infects cells and activates cellular proto-oncogenes. Leukemias in such lines have been used to identify new proto-oncogenes that are activated in the tumors, such as evi-1 (endogenous virus insertion site-1) and evi-2, associated with myeloid leukemias.92
Another feature of leukemogenesis by MuLVs is the formation of recombinants between these viruses and endogenous MuLV proviruses present in the mouse germline. The genomes of most higher eukaryotes carry retroviral DNA that was introduced by germline infection by retroviruses sometime during evolution. This endogenous retroviral DNA is transmitted vertically from parents to offspring, and multiple endogenous proviruses have accumulated over time. Indeed, approximately 8%
of the human genome is derived from endogenous retroviruses. Many endogenous retroviruses are replication defective, perhaps reflecting evolutionary selection against genetically transmitted, replication-competent viruses, but some can still be expressed. When mice are infected with MuLVs, recombination between the incoming MuLV and endogenous MuLVs can occur to generate mink cell focus-inducing (MCF) recombinant viruses.207 MCFs express an endogenous MuLV-derived envelope protein that allows them to infect cells by using a different cellular receptor than the original MuLV. Most MuLVs that infect mice or mouse cells are ecotropic and bind to the ecotropic receptor murine cationic amino acid transporter 1 (MCAT-1) on mouse cells; in contrast, MCFs infect via the xenotropic and polytropic retrovirus receptor (XPR-1) receptor. MCFs have been suggested to be the proximal leukemogens for some MuLVs (e.g., in AKR mice that develop spontaneous leukemia by activation of a ecotropic endogenous MuLV [Akv-MuLV]),207 although for other MuLVs MCF recombinants may play early roles in disease.148 For MuLVs with weak LTR enhancer activity (e.g., Akv-MuLV), MCF recombinants also acquire a stronger LTR by recombination with a second endogenous provirus.448
of the human genome is derived from endogenous retroviruses. Many endogenous retroviruses are replication defective, perhaps reflecting evolutionary selection against genetically transmitted, replication-competent viruses, but some can still be expressed. When mice are infected with MuLVs, recombination between the incoming MuLV and endogenous MuLVs can occur to generate mink cell focus-inducing (MCF) recombinant viruses.207 MCFs express an endogenous MuLV-derived envelope protein that allows them to infect cells by using a different cellular receptor than the original MuLV. Most MuLVs that infect mice or mouse cells are ecotropic and bind to the ecotropic receptor murine cationic amino acid transporter 1 (MCAT-1) on mouse cells; in contrast, MCFs infect via the xenotropic and polytropic retrovirus receptor (XPR-1) receptor. MCFs have been suggested to be the proximal leukemogens for some MuLVs (e.g., in AKR mice that develop spontaneous leukemia by activation of a ecotropic endogenous MuLV [Akv-MuLV]),207 although for other MuLVs MCF recombinants may play early roles in disease.148 For MuLVs with weak LTR enhancer activity (e.g., Akv-MuLV), MCF recombinants also acquire a stronger LTR by recombination with a second endogenous provirus.448
Other Mechanisms of Insertional Oncogenesis
While the predominant mechanisms of oncogenesis by nonacute retroviruses are LTR-mediated activation of proto-oncogenes by promoter insertion or enhancer activation, several related mechanisms have also been identified, all of which involve integration of proviral DNA into the genome at specific places, leading to changes in cellular gene expression and development of tumors.
In one such mechanism, a provirus is inserted upstream of a proto-oncogene or within a proto-oncogene in the same transcriptional orientation as the cellular gene. The viral genome is transcribed beginning in the upstream LTR, but transcription continues past the poly(A) signal in the downstream LTR into the proto-oncogene (Fig. 7.7). Splicing of the fusion transcript from the viral splice donor into downstream proto-oncogene splice acceptor sites can generate mRNAs that encode truncated proto-oncogene proteins. This has been observed in erythroid leukemias induced by ALV in line 151 chickens, where a truncated c-ErbB (EGF receptor) protein is produced,164 and in myeloid leukemias induced by M-MuLV in pristane-primed Balb/c mice, where a truncated c-Myb protein is generated.330 Similarly, transcription of a proto-oncogene could terminate at the LTR of a provirus inserted into a downstream portion of the proto-oncogene, resulting in a C-terminal truncation (Fig. 7.7). In these cases, the truncations remove regulatory domains from the proto-oncogene proteins, leading to the synthesis of unregulated, growth stimulatory proteins.
As mentioned earlier, inoculation of chicken embryos with ALV leads to rapid development of B lymphomas harboring proviral activation of c-myc. In addition, some ALV-induced lymphomas contain proviruses at both c-myc and a novel CIS, bic-1.84 Although bic-1 RNA was overexpressed in these tumors, presumably due to provirus insertion, bic-1 does not contain protein-coding sequences. Rather, bic-1 encodes the precursor for microRNA (miRNA) miR155—a so-called onco-miR that is overexpressed in a variety of human cancers.149 miR155 causes the down-regulation of growth inhibitory genes, which presumably accounts for its oncogenic potential.38 Insertional activation of other miRNAs has also been observed in retrovirally induced tumors. The miR-103-363 cluster is activated in RadLV-induced T lymphomas,275 and miR-106 a is activated in SL3-3 MuLV-induced T lymphomas.293
Another mechanism of insertional oncogenesis involving miRNAs has been described for T lymphomas induced by SL3-3 MuLV. In these tumors, the provirus is inserted into downstream noncoding sequences of the gfi-1 proto-oncogene.100 These sequences contain binding sites for several miRNAs, including miRNA-155. This results in overexpression of a truncated gfi-1 mRNA, presumably because the inserted provirus eliminates binding sites for inhibitory miRNAs.
Finally, insertions of the SFFV provirus into the p53 tumor suppressor gene occur in erythroleukemia cell lines established from tumors induced by the acute transforming Friend SFFV/F-MuLV complex.25,323 The insertions inactivate p53, and in these tumors the normal p53 gene is generally lost. Similarly, in BXH2 recombinant inbred mice, proviral insertion occurs in evi-2, which encodes the NF-1 tumor suppressor.277 In these cases, then, rather than activating proto-oncogenes, proviral insertion inactivates tumor suppressor genes.
Multiple Changes in Nonacute Retrovirus-Induced Tumors
Multiple genetic changes may be involved in some tumors induced by nonacute retroviruses. Proviral insertions next to more than one proto-oncogene have been observed in some MMTV-induced mammary tumors and M-MuLV–induced T lymphomas (e.g., wnt-1 and int-2 for MMTV and c-myc and pim-1 for M-MuLV).311,417 Although in some cases this reflects two independent tumors, each containing an insertion event near a different proto-oncogene, in other cases the same tumor cell harbors both of the activated proto-oncogenes, suggesting that the proto-oncogenes cooperate in tumorigenesis. This notion has been further developed by studying leukemogenesis in transgenic mice. For example, mice with a pim-1 transgene under expression of a T-cell–specific promoter develop lymphomas very inefficiently. However, if these mice are infected with M-MuLV, tumors develop extremely rapidly, and analysis of the tumors revealed proviral activations of both c-myc and n-myc.473 This result provided direct evidence that overexpressed pim family members and myc family members can cooperate in T lymphomagenesis. In later experiments, several additional pairs of cooperating proto-oncogenes have been identified.474
Secondary events may also be important in tumors induced by acute transforming retroviruses. Abelson MuLV (carrying the v-abl gene) induces B-cell lymphomas when it is co-infected with an M-MuLV helper virus but not with other helper MuLVs, suggesting that M-MuLV also contributes to tumor development. The critical region for this activity was localized to the M-MuLV LTR, and a CIS for the M-MuLV helper in Abelson MuLV tumors was identified adjacent to the ahi-1 gene.361 Thus, expression of the v-abl oncogene along with ahi-1 protein238 is required for efficient lymphomagenesis.
Another approach to identify activated proto-oncogenes that contribute to later steps in tumorigenesis involves in vitro or in vivo passage of tumors. For example, in vitro passage of rat T lymphomas induced by M-MuLV results in acquisition of additional proviral CISs, which have been termed tumor progression loci (e.g., tpl-1 and tpl-2).20,349 Likewise, in vitro infection of growth factor–dependent M-MuLV–induced
lymphomas with a second MuLV (MCF-MuLV) resulted in the generation of factor-independent cells, which contained proviral activation of new proto-oncogenes (e.g., gfi-1).159,179 Finally, as described earlier, insertional inactivation of p53 occurs during in vitro passage of erythroleukemia cell lines from Friend virus complex–infected mice, so it is a late step in the development of these lines.25
lymphomas with a second MuLV (MCF-MuLV) resulted in the generation of factor-independent cells, which contained proviral activation of new proto-oncogenes (e.g., gfi-1).159,179 Finally, as described earlier, insertional inactivation of p53 occurs during in vitro passage of erythroleukemia cell lines from Friend virus complex–infected mice, so it is a late step in the development of these lines.25
 Figure 7.8. Proto-oncogene activation in nonviral tumors. The figure shows three mechanisms of proto-oncogene activation in tumors of nonviral origin: tandem amplification of the oncogene and flanking cellular DNA, resulting in overexpression of the oncogene; chromosomal translocation resulting in overexpression and/or structural alteration of the oncogene, leading to its constitutive activation; and point mutations in the coding region of an oncogene, resulting in constitutive activation. The thickness of the horizontal arrows indicates the relative level of expression. Black, proto-oncogene; blue, flanking normal cell DNA; yellow, translocated chromosome. Red X indicates mutation. |
Proto-Oncogene Discovery in the Age of Genomics
The initial discovery of CISs and activated proto-oncogenes was very labor intensive and typically required identifying virus-induced tumors harboring a small number of proviruses, followed by generating a lambda phage library of tumor DNA and cloning all of the proviruses. Adjacent cell sequences from each provirus were then used as hybridization probes on Southern blots of DNA from other virus-induced tumors to identify additional tumors that harbored proviral insertions in the same cellular site.211,352 The availability of whole genome sequences substantially increased the pace of CIS discovery, particularly in mice.422 New cloning techniques such as inverse polymerase chain reaction (PCR) facilitated the cloning of proviral DNA and viral–host junctions.422 Sequencing of the adjacent cellular DNA in these clones, followed by alignment of the cellular DNA sequence with the host genome, allowed rapid localization of each proviral insertion site within host cell DNA. Common insertion sites could be readily identified by insertions into similar chromosomal locations in different tumors.
The advent of high-throughput DNA sequencing has further enhanced the efficiency of CIS discovery. In a recent approach, linker-ligated PCR techniques are used to amplify viral–cellular junctions from tumor samples, and then the entire reactions are analyzed by deep sequencing.265 This provides high-resolution analysis of all of the proviral insertions in a tumor, and then comparison with other tumors induced by the same virus is used to identify CISs. In a recently published study, over 9,000 insertion sites were analyzed in a collection of 476 murine tumors, and multiple new CISs were identified.265
Activation of Proto-Oncogenes in Nonviral Tumors
Studies of retroviral oncogenes and their corresponding proto-oncogenes led to the first discoveries of molecular genetic alterations in human cancers, namely, activation of proto-oncogenes. Genetic alterations in cancer are now major subjects of study in cancer research. The basic mechanisms of proto-oncogene activation in tumor cells—gene amplification, chromosomal translocation, and missense mutations—mirror the consequences of retroviral activation of oncogenes (Fig. 7.8).
Gene amplification in tumor cells is manifested by tandem head-to-tail duplication of chromosomal regions (up to several hundred kilobases in length), resulting in as many as ∼10 additional copies of the amplified region of the chromosome. Amplified regions typically contain a proto-oncogene (as well as flanking genes) and were originally detected on Southern blots by cross-hybridization with viral oncogene probes. For example, c-myc is amplified in several kinds of tumors such as lung cancer and neuroblastoma.410,411 Amplification leads to overexpression of the protein product of the proto-oncogene and excessive stimulation of growth. Early studies on c-myc amplification led to the discovery of c-myc–related genes that were also amplified in tumors (e.g., n-myc in neuroblastomas and l-myc in lung cancers). These findings expanded the concept of proto-oncogenes beyond those that had been captured by acute transforming viruses: proto-oncogenes are cellular genes that are activated in tumors and stimulate growth or division.
Chromosomal translocations have also been observed in tumors; for example, the 9:22 translocation (the Philadelphia chromosome) is pathognomonic for chronic myelogenous leukemia (CML).385 The first molecular description of such translocations was in Burkitt lymphoma (of B-cell origin), where c-myc is characteristically present at an 8:14 translocation breakpoint,103 which moves c-myc on chromosome 8 next to the immunoglobulin heavy chain gene. In other cases, Burkitt lymphoma cells harbor a translocation of the c-myc gene into the immunoglobulin light chain locus on chromosome 22. In B lymphocytes, whose function is to produce antibodies, the immunoglobulin genes are highly expressed. Therefore, translocation
of c-myc into these regions leads to c-myc overexpression and growth stimulation. Similarly, the 9:22 translocation in CML juxtaposed c-abl from chromosome 9 next to the Bcr (breakpoint cluster region) on chromosome 22.108 This translocation results in a novel Bcr-abl fusion protein, which exhibits elevated tyrosine kinase activity compared to the normal Abl protein.264 Thus, chromosomal translocations can have the same consequences as proviral insertion and proto-oncogene transduction. Characteristic translocations have been detected in various additional tumors (particularly of hematopoietic lineage, but also in prostate and lung carcinoma), and genes at the translocation junctions either are overexpressed or have altered signaling properties.257 These genes are now considered proto-oncogenes as well.
of c-myc into these regions leads to c-myc overexpression and growth stimulation. Similarly, the 9:22 translocation in CML juxtaposed c-abl from chromosome 9 next to the Bcr (breakpoint cluster region) on chromosome 22.108 This translocation results in a novel Bcr-abl fusion protein, which exhibits elevated tyrosine kinase activity compared to the normal Abl protein.264 Thus, chromosomal translocations can have the same consequences as proviral insertion and proto-oncogene transduction. Characteristic translocations have been detected in various additional tumors (particularly of hematopoietic lineage, but also in prostate and lung carcinoma), and genes at the translocation junctions either are overexpressed or have altered signaling properties.257 These genes are now considered proto-oncogenes as well.
Missense mutations in proto-oncogenes in tumors were first detected by DNA transfection experiments. When naked DNA from certain tumor cell lines or primary tumors was introduced into mouse NIH-3T3 fibroblasts, foci of transformed cells appeared, whereas DNA from normal cells was, in general, devoid of transforming activity.426 Thus, these tumor cells contained a genetic change that could be transferred to other cells to cause transformation. Molecular cloning from the EJ bladder carcinoma cell line identified the transforming gene as cellular H-ras.346 The H-ras gene in the EJ cells had a missense mutation at residue 12 (G12V) of the protein,458 resulting in inhibition of GTPase activity and constitutive signaling, similar to changes in the v-ras proteins described earlier. Other tumors had missense mutations in the K-ras gene117 or in a third homolog, N-ras.168 Ras mutations are some of the most frequent genetic alterations in human tumors (e.g., they are present in greater than 90% of pancreatic cancers), but this assay has identified other transforming genes in human tumor DNA as well. Activating mutations have also been found by direct sequencing of cellular proto-oncogenes (e.g., EGF receptor mutations in lung cancer, c-kit mutations in gastrointestinal stromal tumors, and B-raf mutations in melanoma).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


