Cytomegaloviruses
Edward S. Mocarski Jr
Thomas Shenk
Paul D. Griffiths
Robert F. Pass
Introduction
Human cytomegalovirus (HCMV) is a ubiquitous virus infection with worldwide distribution. The virus is the most significant infectious cause of congenital disease, an important opportunist in the immunocompromised host and an occasional cause of febrile illness as well as infectious mononucleosis in the general population. HCMV infects an overwhelming majority of the population, spreading efficiently throughout life and all over the world through direct contact with bodily secretions. Like other human herpesviruses, HCMV is never completely cleared and remains latent for the life of the host. Persistently and sporadically shed virus is an important recurrent source of virus for transmission. Susceptibility to HCMV disease is associated with a compromised immune system, particularly related to defects in cell-mediated CD4 and CD8 T-cell function. During pregnancy, intrauterine transmission to the fetus results in sensorineural damage. In immunocompromised
individuals (following solid organ transplantation and hematopoietic cell allografting, immunosuppressive therapies, and genetic or acquired immunodeficiency), this virus remains clinically important despite available antiviral therapies aimed at reducing the overall disease burden. More effective and safe, orally bioavailable antiviral drugs are needed.
individuals (following solid organ transplantation and hematopoietic cell allografting, immunosuppressive therapies, and genetic or acquired immunodeficiency), this virus remains clinically important despite available antiviral therapies aimed at reducing the overall disease burden. More effective and safe, orally bioavailable antiviral drugs are needed.
Transplacental transmission during pregnancy, rare with other human herpesviruses, underscores the medical importance of HCMV as well as the motivation for universal vaccination.481 Virus transmits during primary maternal infection (in HCMV-seronegative women) in a pattern reminiscent of rubella; however, HCMV causes recurrent maternal infection (in HCMV-seropositive women) following either reinfection with additional viral strain or reemergence of persistent/latent infection. It is now recognized that this virus is transmitted in at least 1% of pregnancies worldwide regardless of population serostatus. Sensorineural damage (hearing loss, eye sight compromise, and learning disabilities) impacts roughly 12% to 25% of infected newborns,162 with half developing disease over the first few years of life. A small proportion of congenital infections cause severe systemic, life-threatening cytomegalic inclusion disease (CID). Overall awareness of disease risk is poor, both in the general population and among physicians, possibly owing to the limited options that are available to treat congenital HCMV disease. Because person-to-person transmission depends on direct contact with infected bodily secretions, viral shedding patterns in urine, saliva, breast milk, and genital secretions mediate exposure. Good hygiene practices (e.g., handwashing) can reduce rates, although widespread shedding and transmission results in universal transmission in populations worldwide. Transmission is most frequent in childhood and is mostly asymptomatic. A varying proportion of the population, ranging as high as 50% to 60% in areas of North America and Europe, escapes infection early in life and remains susceptible as adults. Primary infection during pregnancy in HCMV-naïve women is associated with an average 33% risk of transplacental transmission,289 whereas roughly 1% of recurrent infections result in transmission.95 Therefore, HCMV universal vaccination is highly desirable29 and includes the challenge of preventing primary as well as recurrent intrauterine maternal transmission.562
Space limitations dictate emphasis on HCMV, with some reference to key observations in animal models, in this chapter. A growing number of publications and reviews are available. The reader is strongly advised to consult earlier reviews and chapters focusing on cytomegaloviruses to obtain a more complete understanding of this field. In particular, Cytomegalovirus: Biology and Infection,247 the last version of this chapter404 as well as earlier versions18,397,402,404,446,627 and the reference book, Human Herpesviruses: Biology, Therapy and Immunoprophylaxis,28 provide more extensive information. Given the limited number of references that can be included in the print version of this chapter, we generally reference a key recent report or review, expecting that this will lead the reader to relevant earlier work. The online materials provide more extensive references.
History
HCMV has been recognized as an opportunistic pathogen in immunocompromised hosts as well as an important infectious cause of birth defects for half a century.704,705 Observations of cytopathology associated with HCMV disease in the immunocompromised host and newborns extends back nearly 100 years before cell culture or virus isolation methods were available. Severe HCMV-associated CID was first recognized by in association with “owl’s eye” cytopathology in autopsy specimens prior to any knowledge of viral etiology. Surrogate animal models employing natural cytomegaloviruses of mice, rats, and guinea pigs played important roles in early appreciation of pathogenesis.247 More recent studies using rhesus macaque cytomegalovirus (rhCMV) have been possible due to isolation of specific pathogen-free monkeys.41,490 By the early 1950s, diagnosis of HCMV disease was facilitated by the identification of inclusion-bearing cells in urine along with the demonstration of virus-like particles by low-resolution electron microscopy (EM). One investigator in particular, Margaret Smith, pursued studies on salivary gland virus of mice, which today is called murine CMV (MCMV), for at least two decades before the mid-1950s, when human salivary gland virus was isolated from urine of congenitally damaged newborns by her, at Washington University, as well as by Thomas Weller at Harvard and Wallace Rowe at National Institutes of Health (NIH). MCMV and HCMV replicated and produced a similar cytopathology, but only on fibroblasts from the homologous species. Species-specificity is now recognized as a key characteristic of all cytomegaloviruses. In the period from 1956 through 1970, the viral etiology of HCMV disease in newborns and immunocompromised transplant recipients was elucidated.704,705 Biologically similar viruses associated with a characteristic tissue and cell type distribution were isolated from many mammals. By the early 1970s, diagnosis of HCMV by virus isolation was established as a gold standard. By the turn of the 21st century, quantitative polymerase chain reaction (PCR) detection of viral DNA in blood and secretions became the standard for diagnosis of infection.164
As the health economic and societal consequences of HCMV damage, including lifelong sensorineural hearing damage and neurological impairment, became fully appreciated, this earned a high priority status as a target for universal vaccination.29,643 Primary maternal infection leads to frequent transplacental transmission and is tied to severe disease in newborns.65 Viral transmission from primary and recurrent maternal infection both contribute to the overall congenital disease burden in the United States695 and other developed countries, with recurrent infection predominating in other areas of the world. In the overall population, transplacental transmission is infrequent, and ranges broadly from roughly 0.3% to 2% of newborns, depending on many factors. Transmission is more frequent in younger women as well as during the third trimester, although transmission during the first or second trimester is associated with greater risk of congenital disease. Placental infection122,469 may be much more frequent and may be a barrier to intrauterine transmission, although this area needs more systematic study. Perinatal and postnatal infection of full-term newborns, often acquired from breastfeeding,232 is of little disease consequence. Premature or immunodeficient infants risk HCMV disease during delivery, from blood transfusion, or from breast milk,232 and sometimes results in a systemic disease constellation.52 Lactating HCMV seropositive mothers commonly shed virus in milk along with HCMV-specific antibody that may neutralize virus, thereby dampening transmission.173 Once infected, infants and children shed virus in saliva and urine for months to years and remain an important source of virus infecting parents as well as other childcare providers.95 Remarkably low awareness of HCMV in the general population
as well as among physicians536 poses a challenge to efforts in dealing with congenital disease.
as well as among physicians536 poses a challenge to efforts in dealing with congenital disease.
HCMV exhibits remarkable genome variation and heterogeneity during natural infection,139,216,217,520 a characteristic that has been documented in virus shed by congenitally infected infants.520 The consequences of this variation are presently unknown. Because transmission occurs commonly through contact with bodily secretions and fomites, handwashing is an effective intervention to prevent primary infection in pregnant women.11 Development of a vaccine to prevent congenital HCMV remains an area of unmet medical need and potential promise.50
HCMV remains an important etiologic agent of opportunistic infections and disease in immunocompromised hosts, generally emerging in the face of poor T-cell function. Infection in immunocompetent adults is clinically benign, although febrile illness and heterophile-negative infectious mononucleosis may be common.136 The T-cell response to HCMV is remarkable. High frequencies of antigen-specific major histocompatibility complex (MHC) class II–restricted CD4 and MHC class I–restricted CD8 T-cell responses develop over the course of life,290,644 increasing in frequency with age.691 T-cell immunity is important for the lifelong suppression of virus replication, as well as for maintaining latency. Helper CD4 and, in particular, cytotoxic CD8 T-cell response levels are recognized as central to the effective host control of viral infection and disease.489,530,672,691 HCMV infection and disease predominate during immunosuppressive therapy, particularly in settings of allograft rejection following solid organ transplant (SOT) and allogeneic hematopoietic cell transplant (HCT) recipients, as well as in acquired immunodeficiency disease syndrome (AIDS). Given that settings where this virus causes disease can sometimes be predicted because they result from medical interventions, preemptive therapy and prophylaxis strategies are commonly employed to suppress infection before disease appears.190,302 The currently licensed antiviral drugs, ganciclovir/valganciclovir, are used to treat clinically evident disease as well as for prophylaxis and preemptive therapy strategies, with foscarnet and cidofovir as back-up drugs.505 Although highly beneficial, these antiviral drugs have been recognized as inadequate in many settings that affect outcomes in transplantation as well as in congenital disease.
General Characteristics
All beta herpesviruses share common characteristics, including appearance in electron micrographs (Fig. 62.1) a prolonged replication cycle in cell culture, species specificity, and a tropism for differentiated hematopoietic and epithelial cell types. Cytomegaloviruses isolated from a mammalian species are most readily propagated in cultured fibroblasts from the homologous host species. During natural infection, HCMV engages epithelial and myeloid (monocyte/macrophage and dendritic) cells, as well as fibroblasts and endothelial cells.523 HCMV-infected cells develop characteristic cytopathology, exhibiting both nuclear and cytoplasmic inclusions. The latter is associated with a distinct cytoplasmic viral assembly compartment (AC) composed of cellular membranes and organelles that support viral final steps in maturation and release.144 When propagated in human fibroblasts, HCMV clinical isolates acquire mutations141 in a manner that suggests a process of adaptation.139,619 A comparison of laboratory-propagated strain AD169 and wild-type strain Merlin is depicted in Figure 62.1. Mutation of RL13 and UL128 (Table 62.1) occurs rapidly, even after few passages, suggesting that these membrane impede viral replication in fibroblasts.619 Although the negative impact of either gene product in fibroblasts remains elusive, pUL128, along with pUL130 and pUL131A (any of which mutates during virus passage in fibroblasts), forms a pentameric envelope complex that contains glycoprotein (g)H and gL and facilitates entry into epithelial and endothelial cells.523,539,699 TB40/E is a low passage endotheliotropic strain used for experimental investigations.584
Human fibroblasts (e.g., MRC5) are commonly employed for isolation as well as propagation of HCMV, and these cells have been key to understanding how viral gene functions control the various steps in replication (Fig. 62.2). Fibroblasts retain full permissiveness when immortalized with either telomerase or human papillomavirus E6/E7 oncoproteins. Retinal pigment epithelial (e.g., ARPE-19) and astrocytoma (e.g., U373-MG) cancer cell lines are susceptible to HCMV, but other transformed cell lines are typically nonpermissive. Although replication stalls, cytomegaloviruses can enter and proceed through the early stages of the viral replication cycle in many cell types, including cells from other animal species. When exposed to HCMV fibroblasts from nonhomologous animal species such as mouse, attachment, entry, uncoating, and viral IE gene expression proceed (see Fig. 62.2), but viral DNA replication fails to occur. Studies on MCMV infection of human fibroblasts suggest roles for virus-encoded cell death suppressors377 as well as other viral gene controlling early steps568 as critical determinants to overcome the species barrier.
HCMV DNA or antigens in peripheral blood (PB) are important and specific markers of infection and disease,207 particularly in at-risk immunocompromised transplant recipients.190,302 Two common methods, quantitative DNA PCR amplification, which can be applied to whole blood, tissue, and body fluids, and detection of viral antigen, applied to PB cells as an antigemia assay, are complementary methods for diagnosis of infection.207 Viral antigens and nucleic acids may also be assessed directly in affected tissues. These indicators of active HCMV infection are used in combination with clinical diagnosis to guide antiviral therapy.505 Primary infection in immunologically normal children or adults, including pregnant women and newborns, is readily diagnosed by detection of virus or viral DNA in urine or saliva. These body fluids may contain virus continuously for months to years, and virus reappears in these fluids sporadically during life. Serologic assessment of HCMV-specific antibodies is a more common means of diagnosing previous infection, and is a highly specific and accurate indicator of long-term infection. Natural latency of HCMV occurs in blood marrow (BM)–derived hematopoietic cells580 where viral DNA is present at very low frequencies (10−5) and low copy number (2 to 10 genome equivalents per cell)581,589 and is associated with limited viral transcription.47,515 The precise status and regulation of HCMV during life-long infection remains a topic of active investigation.581
Viral Strains and Cell Tropism
HCMV strains accumulate deletion and point mutations when propagated in cell culture.139,141,619 Replication and release of progeny virus improves with adaptation through repeated passage on fibroblasts, while, at the same time, ability to infect endothelial and epithelial cells is compromised.139,523,619 As a result, laboratory strains such as AD169112 and Towne167,417 carry substantive mutations and rearrangements compared to
a wild-type HCMV prototype106,139,141,149,150 (see Fig. 62.1). Adapted strains replicate very well in fibroblasts but poorly on cultured macrophages, dendritic cells, endothelial cells, or epithelial cells. In addition to adaptive mutations, it has become clear that viral stocks often consist of multiple strain variants and that laboratory adapted viral strains in common use around the world differ markedly.77,138 Serial propagation of common laboratory strains results in stable strain variants77,138 that exhibit unique biological properties such as upregulation of cell cyclin–dependent kinases resulting in pseudomitosis246 as well as other alterations in host cell response to infection.399 Comparison of viral genomes from low passage or even nonpropagated wild-type strains in clinical samples facilitated current estimate of HCMV genome coding capacity106,139,141,149,150 and led to the derivation of bacmid clones of low-passage strains for experimental use.584,619 Recent deep DNA sequencing has revealed considerable heterogeneity as well as the presence of multiple viral strains within clinical samples.139,216,217,520 Deep sequencing approaches have also facilitated assembly of a high-resolution transcriptome204 and unveiled a level of messenger RNA (mRNA) greater than previously appreciated for this virus, two features that still need to be integrated into the growing understanding of HCMV biology.
a wild-type HCMV prototype106,139,141,149,150 (see Fig. 62.1). Adapted strains replicate very well in fibroblasts but poorly on cultured macrophages, dendritic cells, endothelial cells, or epithelial cells. In addition to adaptive mutations, it has become clear that viral stocks often consist of multiple strain variants and that laboratory adapted viral strains in common use around the world differ markedly.77,138 Serial propagation of common laboratory strains results in stable strain variants77,138 that exhibit unique biological properties such as upregulation of cell cyclin–dependent kinases resulting in pseudomitosis246 as well as other alterations in host cell response to infection.399 Comparison of viral genomes from low passage or even nonpropagated wild-type strains in clinical samples facilitated current estimate of HCMV genome coding capacity106,139,141,149,150 and led to the derivation of bacmid clones of low-passage strains for experimental use.584,619 Recent deep DNA sequencing has revealed considerable heterogeneity as well as the presence of multiple viral strains within clinical samples.139,216,217,520 Deep sequencing approaches have also facilitated assembly of a high-resolution transcriptome204 and unveiled a level of messenger RNA (mRNA) greater than previously appreciated for this virus, two features that still need to be integrated into the growing understanding of HCMV biology.
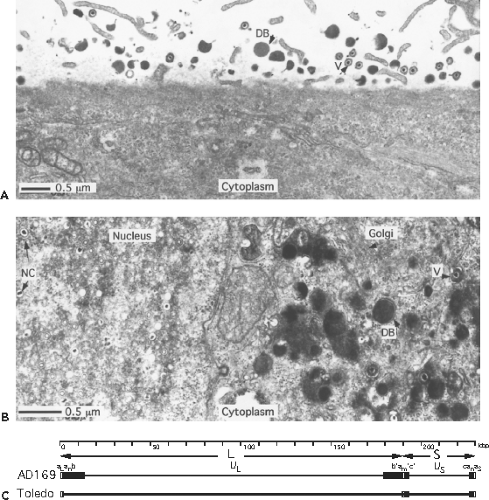 Figure 62.1. Virus particles, replication, and genome structure. A: Surface of a cell onto which virions (V) and dense bodies (DB) of human cytomegalovirus (HCMV) (Towne strain) have attached. This illustrates the size and approximate ratio of these two types of particle in a virus preparation and the ability of both to attach to the cell surface. B: Productively infected cell with nucleocapsids (NC) in the nucleus and maturing virus (V) and dense bodies (DB) in the enlarged Golgi region of the cytoplasm, which forms the characteristic cytoplasmic inclusion of HCMV. Many capsids lacking a dense core of nucleic acid can be observed in the nucleus where, along with nucleocapsids, they form a characteristic inclusion within a kidney-shaped nucleus that is displaced by a cytoplasmic inclusion, a compartment where final steps in maturation occur (giving an “owl’s eye” appearance to cells).247 Envelopment of nucleocapsids (and capsids) occurs at the inner nuclear membrane, where thickened patches of modified membrane develop, followed by final envelopment in the cytoplasm. Dense body envelopment occurs exclusively at cytoplasmic membranes. The bar represents 0.5 um. C: HCMV genome structure. The top line is a size scale in kbp (kilobase pairs = 1000 bp). The second line depicts the L and S components of the genome by arrows. The complete sequenced AD169var UK strain genome structure112,159 is shown on the third line with unique sequences (thin lines) flanked by inverted repeats (boxed areas). The genome structure of a low passage strain Merlin as an example of a wild strain is depicted on the fourth line.106,159 Note that the region shown as b sequence repeat on the left end of the L component in the AD169 strain is retained in wild strains as unique sequence; the b sequence repeat on the right is replaced by additional unique (UL) sequence. The lettering above the genome depicts the following features. The L-terminal a sequence repeat (a L), zero to several (n) additional copies of the a sequence (an), L-terminal b sequence repeat of the L component, the unique sequences of the L component (UL), the L-S junction b sequence inverted repeat (b’), one to several (m) additional copies of the a sequence inverted repeats at the L-S junction (a’m), the L-S junction c sequence inverted repeat (c’), the unique sequences of the S component (US), S-terminal c sequence repeat, and the S-terminal a sequence repeat (aS) with a variable number of additional copies of the a sequence (an). The b and b’ repeats are also called TRL and IRL, respectively; and c’ and c repeats are also called IRS and TRS, respectively. In different strains of CMV, including clinical isolates, the a sequence ranges in size from 700 to 900 bp. |
 |
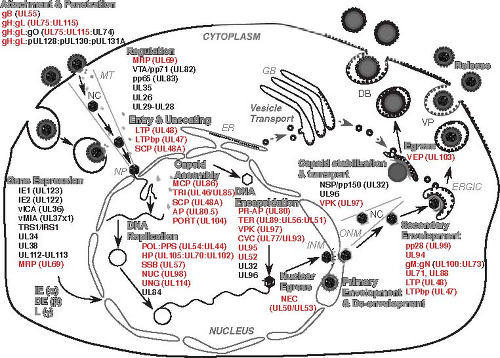 Figure 62.2. Summary of the human cytomegalovirus (HCMV) replication pathway. Major steps in productive replication are indicated in large gray font with outline, black arrows indicate the progression of steps and viral functions (see Table 62.1 for summary descriptions). Individual gene products listed under each step are identified by provisional abbreviated names399,400 as either herpesvirus core (red text) or beta herpesvirus conserved (black text). Viral attachment and penetration occur either via direct fusion at the cell surface (fibroblasts), dependent on gB, gH:gL and gH:gL:gO, or via endocytosis into other cell types (endothelial and epithelial cells) where the pentameric complex, gH:gL:p128:p130:p131A, also facilitates entry (see text). In addition to the interferon (IFN)–like activation of cells by the process of attachment and penetration, input virion tegument proteins (UL69/MRP, pp71/VTA, pp65, UL35, UL26, and UL29-UL28) regulate cellular pathways. NC-associated UL47, UL48, and smallest capsid protein (SCP) are predicted to facilitate the final steps in entry and uncoating that deliver input nucleocapsid (NC) via microtubules (MT) to nuclear pore (NP) complexes where the viral genome is released into the nucleus. Transcriptional regulation of viral and host cell gene expression is mediated by IE genes (IE1, IE2) or DE genes (UL34, UL35, UL112-UL113, and UL69); cell death suppression is mediated by IE gene products vICA and vMIA, and other regulatory processes are facilitated by UL34, UL38, and UL112-UL113 proteins. DNA replication depends on core proteins (POL:PPS, HP, SSB, NUC, and UNG) as well as one beta herpesvirus-specific protein (UL84 gene product) that facilitates initiation of DNA synthesis. Capsids assemble from MCP, TRI, SCP, PORT, and AP. Preformed capsids process PR-AP (UL80) and AP as viral DNA is encapsidated by the TER complex (UL89, UL56, UL51) through a PORT (UL104) pentamer, followed by predicted addition of the CVC complex (UL77, UL93) onto NC pentamers, with UL95, UL52, UL32, and UL96 added for NC stabilization. Nuclear egress of the NC is mediated by the NEC (UL50, UL53). Following primary envelopment at the inner nuclear membrane (INM), and de-envelopment at the outer nuclear membrane (OMN), capsid stabilization is ensured by the function of NSP/pp150 (UL32) and UL96, with nuclear egress and transport facilitated by VPK (UL97). Glycoproteins incorporated into the envelope are synthesized in the endoplasmic reticulum (ER), glycosylated in the Golgi body (GB), and delivered by vesicle transport (dashed gray arrow) to join NC at sights of secondary envelopment on ER Golgi intermediate compartment (ERGIC). Secondary envelopment requires UL99, UL94, gM/UL100:gN/UL73, UL71, UL88, UL47, and UL48, acting together with VPK. Following the acquisition of an envelope, virus particle (VP) as well as capsidless dense body (DB) egress is facilitated by VEP/UL103 for release into the extracellular space. |
The AD169 and Towne stocks distributed by the American Type Culture Collection (ATCC) include a mixture of genomes. Replication-competent variants with substantive genome rearrangements and deletions have been independently propagated from various AD169 and Towne preparations.77,138 Cell tropism factors have come from such studies538 and have opened the way to a more complete understanding of HCMV biology. Two examples include the role that virion envelope glycoprotein gpRL13 plays in suppressing replication in fibroblasts619 and the role that the pentameric complex composed of gH:gL:pUL128:pUL130:pUL131A plays in facilitating entry into epithelial and endothelial cells.523,539,699
Classification
The starting point for classification of cytomegaloviruses infecting humans and other animals is comparative biology, physicochemical characteristics, and virion morphology. Over the last 20 years, genome sequence analysis has eclipsed other approaches in viral taxonomy.146,465 Cytomegaloviruses have been isolated from a wide variety of mammalian species, including dogs, horses, bats, cows and pigs, although many have not been fully characterized.247 Beta herpesviruses, in general, are associated with universal infection in their natural host species. Four groups of beta herpesviruses have been officially recognized465: (1) known human and primate cytomegaloviruses; (2) muromegaloviruses, including MCMV and rat CMV; (3) beta herpesvirus causing roseola (human herpesvirus type 6B [HHV-6B]) as well as two close relatives (HHV-6A and HHV-7); and, (4) proboscivirus, endotheliotropic elephant herpesvirus. Other beta herpesviruses, such as guinea pig CMV and porcine CMV, as well as Tupaia herpesvirus, remain unclassified.465 The beta herpesvirus subfamily exhibits a greater level of evolutionarily and genetic divergence than either alpha herpesviruses or gamma herpesviruses. This situation poses a challenge to investigators trying to understand virus biology by studying these surrogate animal models. MCMV and HCMV exhibit many common biological attributes in pathogenesis, immunomodulation, and latency, but appear to achieve these ends via evolutionarily divergent mechanisms and gene products.348,394,398,401,405 RhCMV has been championed as more HCMV-like41,490 and has raised interest in cytomegaloviruses as vaccine vectors in the face of preexisting immunity.237 Importantly, nonhuman primate surrogates have obvious limitations in that the host animals are limited, plus RhCMV is evolutionarily divergent. There remains a need for intensive investigation of natural HCMV infection and immunity, as well as for further optimization of humanized small animal models593,675 to facilitate direct study of HCMV.
The HCMV genome is the largest of any characterized herpesvirus, at 236,000 bp (HCMV), with an annotated capacity to encode at least 167 protein-coding gene products139 (Table 62.1 and Fig. 62.3), with extensive alternate mRNA splicing in certain regions204 (Table 62.2), plus noncoding RNAs as well as micro (mi)RNAs156,182,223,623 (Fig. 62.3). Other cytomegaloviruses exhibit a level of genome complexity similar to HCMV, whereas roseoloviruses have smaller genomes encoding approximately 85 genes. Alternative annotation methods82,473 and sensitive experimental detection156,182,204,223,623 have complemented each other in the annotation of HCMV genome coding potential.
Neither human roseoloviruses nor nonhuman cytomegaloviruses share significant DNA sequence identity with HCMV. Evolutionary relationships between beta herpesviruses emerge from comparisons of predicted protein coding sequences and appear to follow relationships of host animal species consistent with longstanding pathogen–host co-evolution.405 Independent coevolution belies the biological similarity of cytomegaloviruses infecting diverse mammalian hosts. Only 75 of the estimated 167 HCMV protein–coding genes are conserved across this group, including 40 core herpesvirus and seven (UL49, UL79, UL87, UL88, UL91, UL92, and UL95) betagamma conserved genes (Table 62.1). On one end of this spectrum, 163 of the predicted 168 chimpanzee CMV proteins are homologous and colinear with HCMV.149 Only about 111 RhCMV genes are homologous to HCMV genes531; in guinea pig CMV, relatedness falls off to 84 homologs,282 and in MCMV or rat CMV genomes 75 sequence homologs are present.282 These comparisons have consistently shown that beta herpesviruses are remarkably diverse in genetic composition. As information accumulates, it appears that the immune modulators in these viruses evolved to target a common set of host immune control pathways from evolutionarily distinct origins and using distinct mechanisms. Because of the smaller number of total genes, a high proportion of human roseoloviruses genes have homologs in HCMV,149 and these overlap with the genes retained in all animal cytomegaloviruses. With few exceptions, both beta herpesvirus-conserved genes and herpesvirus core genes are concentrated between UL23 to UL123, with additional US22 family members flanking this region. Many genes within as well as outside this central region are involved in modulation of the host response to virus infection. Genes near the ends of viral genomes represent the most recent evolutionary acquisitions and provide evidence of evolutionary adaptation to host defense pathways.405
Like all herpesviruses, beta herpesviruses have linear DNA genomes with direct terminal repeats containing the cis-acting signals (pac1 and pac2) recognized by the encapsidation machinery to initiate packaging and direct genome cleavage.702 HCMV and guinea pig CMV have large (approaching 1,000 bp) terminal repeats, whereas MCMVs has a small (<50 bp)
terminal repeat that suffices for genome cleavage and packaging.385 Similar to human alpha herpesviruses, primate cytomegaloviruses have an internal inverted copy of the a sequence terminal repeat that supports homologous recombination-driven genome rearrangement during replication.424 As a result, HCMV and chimpanzee CMV have class E genomes (Fig. 62.1; see Chapter 59, Herpesviridae) generating four isomers that appear to package with equal efficiency, whereas RhCMV, roseoloviruses, muromegalovirus, and guinea pig CMV have linear DNA genomes that do not rearrange at all (referred to as class A genomes). Genome isomerization is dispensable for HCMV replication555 as it is in other herpesviruses. The importance of genome isomerization in the biology of any herpesviruses remains a complete mystery.
terminal repeat that suffices for genome cleavage and packaging.385 Similar to human alpha herpesviruses, primate cytomegaloviruses have an internal inverted copy of the a sequence terminal repeat that supports homologous recombination-driven genome rearrangement during replication.424 As a result, HCMV and chimpanzee CMV have class E genomes (Fig. 62.1; see Chapter 59, Herpesviridae) generating four isomers that appear to package with equal efficiency, whereas RhCMV, roseoloviruses, muromegalovirus, and guinea pig CMV have linear DNA genomes that do not rearrange at all (referred to as class A genomes). Genome isomerization is dispensable for HCMV replication555 as it is in other herpesviruses. The importance of genome isomerization in the biology of any herpesviruses remains a complete mystery.
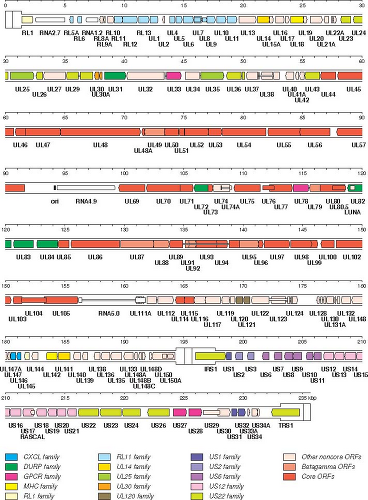 Figure 62.3. Genetic organization and content of wild-type human cytomegalovirus HCMV, based on an updated interpretation668 of consensus sequences. 145,418 The inverted repeats TRL/IRL and TRS/IRS including a sequences are shown in a thicker format than UL and US. Protein-coding regions are indicated by arrows, and gene names are listed below. Introns are shown as narrow white bars. Genes corresponding to those in TRL/IRL and TRS/IRS of strain AD169 are given their full nomenclature, but UL and US prefixes have been omitted from UL1-UL150 and US1-US34A, respectively. Herpesvirus core genes and gene families are color-coded as indicated in the legend. White genes are unique. |
Table 62.2 Summary Information on HCMV miRNAs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Consistent with the conserved set of proteins and despite variation in genome structure and existence of genome isomers, all beta herpesviruses, follow a replication pathway (Fig. 62.2) from DNA replication21,424 through capsid formation and DNA encapsidation,84,99 nuclear egress, and final virion maturation steps80,653 common to all herpesviruses.
Virion Structure
HCMV has a structure characteristic of all herpesviruses,80,84,99 with a DNA core inside of a highly stable icosahedral capsid made up of 162 capsomeres surrounded by an envelope derived from host cell membrane containing viral glycoproteins to
control attachment and entry into cells. Herpesviruses have a particularly thick tegument (or matrix) layer of virus-encoded proteins between the capsid and envelope. Cryoelectron microscopy in combination with computer-assisted tomographic image reconstruction (cryoEM or cryoET)84,342 has provided accurate nucleocapsid dimensions as well as other structural details of the 200 to 230 nm virion particle. HCMV has a 130-nm icosahedral nucleocapsid, somewhat larger than other herpesviruses, that accommodates a large genome. Overall, the virion is the most structurally complex of the characterized herpesviruses. Whether observed in thin sections of infected cells (Fig. 62.1) or as purified virion and dense body preparations resolved either by transmission EM or cryoEM, virus particles have a pleomorphic appearance with a voluminous tegument layer that is asymmetric.729
control attachment and entry into cells. Herpesviruses have a particularly thick tegument (or matrix) layer of virus-encoded proteins between the capsid and envelope. Cryoelectron microscopy in combination with computer-assisted tomographic image reconstruction (cryoEM or cryoET)84,342 has provided accurate nucleocapsid dimensions as well as other structural details of the 200 to 230 nm virion particle. HCMV has a 130-nm icosahedral nucleocapsid, somewhat larger than other herpesviruses, that accommodates a large genome. Overall, the virion is the most structurally complex of the characterized herpesviruses. Whether observed in thin sections of infected cells (Fig. 62.1) or as purified virion and dense body preparations resolved either by transmission EM or cryoEM, virus particles have a pleomorphic appearance with a voluminous tegument layer that is asymmetric.729
HCMV capsids are composed of four herpesvirus core proteins, major capsid protein (MCP, the UL86 gene product) comprising hexons and most pentons, triplexes composed of two subunits, triplex subunit 1 (TRI1, the UL46 gene product or minor capsid protein) and triplex subunit 2 (TRI2, the UL85 gene product or minor capsid protein binding protein), and the smallest capsid protein (SCP, the UL48A gene product). Unlike the situation in HSV-1 where SCP is dispensable, all capsid proteins are essential for HCMV replication.72,80,315 By analogy with HSV-1,84,99 one specialized penton composed of the portal protein (PORT, the UL104 gene product) acts as a channel for both encapsidation and release of viral DNA together with two principal subunits of the terminase, subunit 1 (TER1, the UL89 gene product) and subunit 2 (TER2, the UL56 gene product). A capsid vertex-capping (CVC) complex composed of UL77 and UL93 proteins decorates all pentons133 and the proteins encoded by UL51 and UL52 likely provide stability. Inside the HCMV nucleocapsid is a ∼236-kb linear DNA genome together with two virion (v) RNAs, ∼300 nucleotide vRNA-1, and ∼500 nucleotide vRNA-2, embedded as an RNA–DNA hybrid in an essential region the lytic origin of DNA synthesis (oriLyt).21,443 The nucleocapsid is enclosed in a tegument (or matrix) composed of at least 32 virus-encoded proteins, many of which are phosphorylated. Small amounts of cytoplasmic proteins681 as well as RNAs657 are captured into mature virus particles, likely passively. The tegument is surrounded by a lipid bilayer envelope that is derived from the ER-Golgi intermediate compartment (ERGIC) or recycling endosomal compartment modified by insertion of approximately 23 virus-encoded glycoproteins. Five envelope glycoproteins (gB, gH:gL, gM:gN) provide essential replication functions and are targets of neutralizing antibody. In total, the virion is composed of at least 66 virus-encoded proteins that play diverse roles during infection (Table 62.1 and Fig. 62.2). Many virion proteins mediate and modulate entry and egress, influence cell tropism, and interface with the host response to infection. Although additional minor tegument and envelope components are likely to be recognized given the large coding capacity and truly extensive mRNA splicing,204 HCMV has the most complex of herpesvirus virions.
Purified HCMV preparations contain an abundance of noninfectious defective particles in addition to virions. Infected cells release both capsidless and capsid-containing particles such that defective particles constitute roughly 99 percent of material in highly purified virus preparations. The most carefully purified HCMV preparations exhibit particle-to-PFU (particle forming unit) ratios exceeding 100, and high multiplicity propagation can result in particle-to-PFU ratios approaching 10,000 due to enrichment of defective interfering particles.630 Capsidless dense bodies constitute approximately 50% of particles collected from culture supernatants (Fig. 62.1). Dense body formation depends upon pp65 tegument protein and takes place in the same cytoplasmic AC using the same cytoplasmic membranes as virions (Fig. 62.2). Noninfectious enveloped particles (NIEPs) form when genomeless capsids mature through the cytoplasm and are released.210 Even carefully executed studies seeking to identify virion and dense body structural components657,681 have encountered heterogeneity, cross-contamination, and difficulty in removing host contaminants even though these virus particles have been enriched by differential sedimentation. Furthermore, virus strain-to-strain variability may also influence protein composition of virus particles (Plachter, personal communication). An abundance of DNA-containing defective particles are produced during infection but cannot be distinguished or separated from infectious virions by physicochemical properties.
Capsid
CryoEM analyses342 revealed unique characteristics of the inner capsid surface of HCMV and simian CMV nucleocapsids, in addition to the larger volume to accommodate these large genomes. The icosahedral nucleocapsid exhibits icosahedral T = 16 symmetry assembled from 162 capsomeres like all herpesviruses and the HK97 group of tailed bacteriophages.84,99 One hundred fifty hexons, each consisting of six molecules of MCP, make up the triangular faces of the capsid. Eleven pentons, each consisting of five molecules of MCP together, plus one specialized penton made of PORT complete the capsid wall. Hexons and pentons together form the bulk of the 15-nm–thick capsid walls. Therefore, like all herpesviruses,84,99 the HCMV capsid comprises 955 molecules of MCP, 12 molecules of PORT, 320 copies of a 2:1 complex consisting of TR2 and TR1 making contact with three capsomers just above the capsid floor, and 900 molecules of SCP, forming six member rings on MCP at hexon tips. The importance of UL77 and UL93 as a CVC complex or the precise role of UL51 and UL52 in nucleocapsid stabilization, have not yet been resolved in structural studies. The organized layer of material on the outer surface of the capsid has been ascribed to one of the major tegument proteins, pp150,729 a structural detail that is unique to HCMV.
Based on studies with HSV-1, an HCMV procapsid shell is likely formed when MCP is translated and imported into the nucleus together with scaffold subunits comprised of assembly protein (AP, UL80.5 gene product) and maturational protease (PR), a PR-AP fusion protein (the UL80 gene product).210 After chaperoning subunits to the nucleus where maturation proceeds, the scaffold is replaced as viral DNA is packaged by encapsidation machinery. The capsid assembly process is also common to other herpesviruses84,99 and yields three distinct nuclear capsid forms, termed A, B, and C capsids. C capsids represent DNA-containing nucleocapsids that appear to be in the process of maturing; whereas, A and B capsids represent aberrant particles that appear to have failed to complete encapsidation. Viral DNA is arranged in three-dimensional hexagonally packed arrays within the interior of the nucleocapsid together with polyamines and oriLyt RNA but without additional virus or host proteins. Some HCMV B capsids complete maturation to become NIEPs. In addition, B capsids accumulate when
maturation is blocked, either using HCMV-specific encapsidation inhibitors,158,215 by inhibiting expression of the PR-AP730 or by employing other viral mutants that fail to encapsidate viral DNA.73,80 Therefore, there is strong evidence that viral DNA encapsidation drives the generation of C capsids (nucleocapsids) and that these translocate to the cytoplasmic AC where maturation continues and final envelopment takes place.
maturation is blocked, either using HCMV-specific encapsidation inhibitors,158,215 by inhibiting expression of the PR-AP730 or by employing other viral mutants that fail to encapsidate viral DNA.73,80 Therefore, there is strong evidence that viral DNA encapsidation drives the generation of C capsids (nucleocapsids) and that these translocate to the cytoplasmic AC where maturation continues and final envelopment takes place.
Tegument
The 32 known tegument proteins (Table 62.1) carry out diverse activities, from conditioning the host cell at the beginning of infection to orchestrating the final stages of virion assembly. They are added to the maturing nucleocapsid in sequential layers, beginning in the nucleus and continuing in the cytoplasmic AC. At the start of infection, these proteins, located entirely within the virion between the nucleocapsid and the lipid bilateral envelope, direct nucleocapsid translocation on microtubules to nuclear pore complexes, delivering the viral genome to the nucleus while also overtaking the host cell machinery (Fig. 62.2). During the final stages of maturation, tegument proteins control nucleocapsid stability, trafficking, and envelopment from the nucleus through to final steps in egress (Fig. 62.2). Many tegument proteins are conserved across herpesviruses. The small amounts of viral and cellular RNAs as well as host proteins that appear to be passively acquired during envelopment reside in the tegument. Most tegument proteins are phosphorylated and many are highly immunogenic.80,262
The most abundant tegument protein in virions is pp65 (lower matrix protein, UL83 gene product). This protein is the major constituent of capsidless dense bodies and is acquired during envelopment in the AC. Despite its abundance and potential importance during natural infection in humans, UL83 is dispensable for replication in cultured cells.564 pp65 is highly immunogenic and has proven very useful for monitoring virus-specific immunity in the population because it is a target of MHC class I–restricted CD8 and MHC class II CD4 T-cell responses.644 This tegument protein is also the most abundant viral protein in virus-infected cells and may be transferred to neutrophils that come into contact with virus-infected cells during natural infection. Detection of pp65 in PB neutrophils has been the basis of the antigenemia diagnostic assay.336 Immediately following viral entry, pp65 localizes to the nucleus of infected cells where it has an immunomodulatory role dampening the interferon-like cellular response to infection.262 Late in infection, pp65 is associated with the AC where virion and dense body envelopment occur.
The HCMV UL32, UL48, and UL82 genes encode abundant tegument proteins that play crucial roles during infection. The virion transactivator (VTA) pp71 (upper matrix protein, UL82 gene product), a sequence homolog of UL83, localizes to the nucleus following entry and recruits cellular transcription machinery to activate immediate early (IE) gene transcription.472,631 The pp150 (large matrix phosphoprotein, UL32 gene product) is capsid-proximal in virions729 and plays an essential role sustaining stability of maturing nucleocapsids during translocation from the nucleus to the cytoplasmic AC.651,653 This phosphoprotein is recognized by more than 80% of HCMV-seropositive sera and is also an important target of cellular immunity. The largest tegument protein (LTP, UL48 gene product) is capsid proximal,729 and is expected to stabilize nucleocapsids, like its HSV-1 homolog.560,645 An important enzyme, the viral protein kinase (VPK, the UL97 gene product),494 is incorporated into virions as a tegument constituent and may impact early stages of infection,366 even though its major role is later in facilitating a number of steps during maturation.494
Many protein–protein contacts are involved in the organization of the tegument layer. A number of approaches have been employed to recapitulate these interactions, most recently focused on building a systems-level framework of binary relationships between structural proteins.474,662 Interactions between pp150 tegument protein and MCP, pUL47/pUL48, and MCP as well as pUL48 and pp28 may help direct envelopment in the AC.80 It is likely that many additional contacts serve to establish and maintain the structure of the tegument and provide continuity between the nucleocapsid and the envelope.
The remaining tegument proteins account for a small percentage of virion or dense body proteins,681 but contribute to entry and maturation (Table 62.1 and Fig. 62.2). One class is involved in replication steps, including disassembly of virions and release of the viral genome into the nucleus, transcriptional regulation, or virion assembly. Other tegument proteins modulate or modify the host cell response to infection, inactivating host cell transcriptional repression mechanisms, blocking intrinsic host defenses, altering the host cell cycle, and optimizing the intracellular environment for virus replication. US22 family members (UL23, UL24, UL25, UL26, UL29-28, UL36, UL43, IRS1, US22, US23, US24, US26, and TRS1 gene products) modulate host cell signaling and cell death pathways.
Envelope
Virions, dense bodies and other noninfectious virus particles are enclosed in a lipid bilayer envelope derived from cytoplasmic ERGIC or endosomal membranes80,653 as depicted in Figure 62.2. Remarkably, as many as 23 different viral glycoproteins have been associated with purified virion and dense body preparations.681 Some of these contribute to attachment and entry, but most are more likely involved in modulation of the host cell response to infection. In contrast to the alpha- and gamma herpesviruses where a subfamily-specific envelope glycoprotein such as gD (herpes simplex virus type 1 [HSV-1]) and gp350 (Epstein-Barr virus [EBV]) dictates attachment and entry, HCMV relies on homologs of the three major conserved glycoprotein complexes (gcI, gcII, and gcIII, comprised of gB, gM:gN, and gH:gL, respectively). gB and gH:gL are key to attachment and entry, whereas gM:gN is involved in maturation (Table 62.1, Figs. 62.2 and 62.3). These glycoproteins accumulate on internal infected cell membranes as well as on the plasma membrane during infection, and they are the principal targets of antibodies that neutralize virus.80
UL55-coded HCMV gB forms a trimer (gcI) on the envelope to mediate membrane fusion in attachment and entry. This is a major target of neutralizing antibody and, like other viral fusion proteins, undergoes a conformational change to fuse the virion envelope and target cell membranes during entry. Based largely on structural comparisons to truncated gB from HSV-1 or EBV,132 which are closely related sequence homologs, HCMV gB is a class III fusion protein related to rhabdovirus G and baculovirus gp64. gB mediates binding to heparan sulfate proteoglycan, an initial step in attachment, as well as either pH-independent entry directly at the plasma membrane, as occurs in fibroblasts, or pH-independent entry via the endocytic route, as occurs in endothelial and epithelial
cells.262 gB is not involved in maturation or release of progeny virions, but is important for both cell-to-cell spread and cell–cell fusion leading to syncytia, both of which involve membrane fusion. Cellular receptor(s) for gB are still being investigated. Candidates include cell surface integrins α2β1, α6β1, and αVβ3 on all cells,262 and particularly β1 integrin via a disintegrin-like domain,186 epidermal growth factor receptor (EGFR) on monocytes,108 and platelet-derived growth factor receptor α (PDGFαR) on endothelial, epithelial, and fibroblast cells.602 HCMV receptors (also called entry mediators) may enhance early viral gene expression through signaling pathways, but the role of signaling in entry remains unclear.262,434
cells.262 gB is not involved in maturation or release of progeny virions, but is important for both cell-to-cell spread and cell–cell fusion leading to syncytia, both of which involve membrane fusion. Cellular receptor(s) for gB are still being investigated. Candidates include cell surface integrins α2β1, α6β1, and αVβ3 on all cells,262 and particularly β1 integrin via a disintegrin-like domain,186 epidermal growth factor receptor (EGFR) on monocytes,108 and platelet-derived growth factor receptor α (PDGFαR) on endothelial, epithelial, and fibroblast cells.602 HCMV receptors (also called entry mediators) may enhance early viral gene expression through signaling pathways, but the role of signaling in entry remains unclear.262,434
Late in replication, as virion maturation proceeds, gB is cleaved by a cellular furin-like protease to generate a 116-kD surface component linked by disulfide bonds to a 55-kD transmembrane component. Unlike the situation with many RNA viruses, proteolytic cleavage is not a requisite for gB function under any conditions that have been studied.262 HCMV neutralizing antibody in convalescent sera recognizes gB as well as other envelope glycoproteins. A soluble form of gB has been shown to elicit protective immunity as an oil-in-water (MF59) adjuvanted subunit vaccine,228,456 suggesting that this strategy may lead to a safe vaccine to prevent infection and disease. When administered prophylactically, pooled human gammaglobulin with high gB-specific binding antibody titer has been reported to benefit SOT recipients595 and congenitally infected infants.359,431 Antibodies that recognize different antigenic domains can inhibit viral attachment or prevent fusion,488 reinforcing both aspects of gB function during entry.262
A second envelope glycoprotein complex (gcII) includes UL100-coded gM and UL73-coded gN. gM may be the most abundant envelope glycoprotein.681 gM:gN is clearly important during maturation307,308 and has not been implicated in entry. gM is an eight-membrane spanning protein that binds heparan sulfate. Experimentally, only a small portion of gM is required for complex formation with gN.357 gN is one of the most highly variable gene products and has been used to track viral strains in disease settings. One particular variant (gN4) has been associated with an increased congenital disease risk.479
The third envelope glycoprotein complex is composed of gH (UL75 gene product) and gL (UL115 gene product) that may be modified with additional proteins to control infection in different cell types. The main function of gH:gL is to influence attachment and gB-mediated fusion. HCMV gH:gL and gB together mediate cell fusion more efficiently than gB alone,262 similar to the situation in other herpesviruses.132 Therefore, most evidence indicates gH, in particular, controls postattachment enhancement of fusion.262 Detailed crystallographic structural studies completed on HSV-1 and EBV gH:gL132 have not provided insight into the gH:gL control of gB activity. gH is a transmembrane protein that requires gL as a chaperone to properly mature.80 gH alone, as a component of gH:gL or in other higher order complexes, behaves in ways that suggest it controls the interaction with a cellular receptor,679 and although integrin αvβ3 may be engaged,703 this area remains elusive.
As first shown with EBV, subpopulations of envelope gH:gL associate with additional proteins that act as tropism determinants and dictate entry efficiency for particular cell types. A majority of gH:gL exists as the unmodified heterodimer glycoprotein complex. One additional HCMV component, the UL74-encoded gO, may either be a chaperone540,710 or a structural component,679 and apparently facilitates entry.262,569 In addition to the trimeric gH:gL:gO complex, a pentameric gH:gL complex containing UL128, UL130, and UL131A gene products541 enhances infection in epithelial and endothelial cells539,699 as well as virus interactions with neutrophils, dendritic cells, and many other cell types.523 This complex may recognize novel receptors to initiate an endocytic entry pathway or facilitate fusion at a postattachment step that follows endocytosis.523,539 Viruses lacking components of the pentameric gH:gL:pUL128:pUL130:pUL131A complex enter and replicate without compromise in fibroblasts. Attachment and endocytosis into endothelial and epithelial cells is also independent of the pentameric complex, but the final step of entry into the cytosol is inefficient in its absence.539,541 Evidence is accumulating around the pentameric complex that virus particles produced in one cell type differ in biological characteristics from particles produced in another cell type.569 For reasons that remain largely unexplained, mutations in UL128, UL130, or UL131A accumulate rapidly as HCMV is propagated in fibroblasts, where entry occurs directly at the cell surface, have given the impression that the pentameric complex expression is deleterious to replication in this cell type.262,539,569
Many of the HCMV envelope glycoproteins that are not directly involved in entry or egress have immunomodulatory potential. The virion contains glycoproteins that bind to IgG encoded by RL11 and UL119-118609 as well as secreted glycoproteins, such as the UL22A gene product, the RANTES (Regulated and Normal T Cell Expressed and Secreted) chemokine decoy receptor (Table 62.1) that impact the host response to infection. G protein–coupled receptor (GPCR) homologs encoded by UL33, UL78, US27, and, particularly, US28, have attracted the most attention as immunomodulators. US28 encodes a constitutively strong and ligand-inducible CC/CX3C chemokine receptor,202,298,426 long suggesting a role in infection or behavior of virus-infected cells as well as pathogenesis. US28 has been associated with signaling via phosphorylation-, beta-arrestin-, Gα12- and RhoA-dependent activity, and induction of apoptosis and cell migration.575,686,690 US27 does not signal but influences viral spread,436 and virion-associated UL33 and UL78 gene products have biological roles that remain unclear.575,686,690 Receptor heterodimerization may contribute to the activities of viral GPCRs.670
Viral Genome
The genomes of HCMV and closely related chimpanzee CMV align closely, retaining a class E genome arrangement with unique long (UL) region and unique short (US) regions flanked by terminal and internal repeats in an arrangement first described for HSV-1 (ab-UL–b´a´c´-US–ca) (see Chapter 59, Herpesviridae) and shown in Figure 62.1. This arrangement of terminal and internal inverted a sequences, which contain the genome cleavage/packaging signals, promotes genome isomerization during replication. Such genome inversion generates four equimolar and independently infectious isomers with regard to the orientation of the short (S) and long (L) genome components. This process is dispensable for replication555 and the artificial creation of an inverted copy of the terminal repeat in a viral genome that naturally lacks such repeats, triggers isomerization.386 The c´ and c repeats (also called IRS and TRS) flanking the S genome component have a partially duplicated
IE gene set (IRS1 and TRS1) that are conserved in HCMV strains. In contrast, the large b and b´ repeats (also called TRL and IRL) flanking the L genome component originally characterized in the AD169 strain, are apparently the result of extensive cell culture propagation.149 The RhCMV genome239,531 has direct terminal repeats but lacks internal repeats or genome isomerization. Directly repeated a sequences of variable length and copy number are found at genome termini and include two herpesvirus-conserved cis-acting cleavage/packaging signals, pac1 and pac2. These elements are recognized by the encapsidation machinery to initiate genome packaging and feed the DNA into a capsid as well as to signal cleavage once a genome-length is reached. The cleavage event produces ends with single overhanging 3´-nucleotides, with the location determined by the location of pac1 and pac2 motifs.
IE gene set (IRS1 and TRS1) that are conserved in HCMV strains. In contrast, the large b and b´ repeats (also called TRL and IRL) flanking the L genome component originally characterized in the AD169 strain, are apparently the result of extensive cell culture propagation.149 The RhCMV genome239,531 has direct terminal repeats but lacks internal repeats or genome isomerization. Directly repeated a sequences of variable length and copy number are found at genome termini and include two herpesvirus-conserved cis-acting cleavage/packaging signals, pac1 and pac2. These elements are recognized by the encapsidation machinery to initiate genome packaging and feed the DNA into a capsid as well as to signal cleavage once a genome-length is reached. The cleavage event produces ends with single overhanging 3´-nucleotides, with the location determined by the location of pac1 and pac2 motifs.
The viral genome contains an origin of DNA synthesis (oriLyt), a second cis-acting function, located between the divergent UL57 and UL69 genes in the middle of the UL region. This position is conserved in all characterized beta herpesviruses. oriLyt is required to support viral DNA replication. The oriLyt in cytomegaloviruses is large (∼1,500 bp) and structurally complex, as assayed in a transient replication assay or within the context of the viral genome.21,443 In HCMV, it includes a pyrimidine-rich sequence, reiterated elements, direct and inverted repeat sequences, transcription factor-binding sites, and RNA–DNA hybrids.21,443,495
The HCMV genome has two transcription enhancers controlling IE promoters, a third type of cis-acting element, as well numerous629 transcriptional promoters that become active at different times during infection through a regulatory cascade that controls the behavior of host cell RNA polymerase (RNA pol) II machinery. The best studied is the major IE promoter (MIEP) enhancer, a large genomic region that activates transcription of IE1 (p72) and IE2 (p86). A second complex promoter–enhancer controls US3 IE gene expression. When isolated from HCMV and linked to heterologous genes, the MIEP-enhancer directs efficient transcription in a wide-range of mammalian expression vectors and cell lines. In the context of the viral genome, expression is influenced by cell type and, importantly, is repressed in undifferentiated cells and at late times during productive replication.628,629 Therefore, the MIEP enhancer plays a pivotal role in activating viral gene expression immediately following entry into permissive cells, in repressing viral gene expression during latency and in activation of viral gene expression during reactivation.510,512,515,628 The enhancer may also function as a latent origin of DNA replication to maintain the viral genome in dividing hematopoietic progenitor cells.403 The MIEP enhancer of HCMV, as well as the analogous enhancer found in other cytomegalovirus genomes, is composed of a dense assembly of transcription factor binding sites that extend over several hundred base pairs of DNA.628,629 A cis-acting repression signal (crs) located between the MIEP TATA box and transcription initiation site interacts with IE2 gene products and shuts down MIEP expression as productive infection proceeds. The different cis-acting elements act on host RNA pol II transcription initiation628,629 to control the cascade of both protein-coding and noncoding gene products that are made during replication.
Genome Organization and Expression
The conventional depiction of the organization of the HCMV genome (Fig. 62.3), as established in initial published work,112 depicts one of the four natural isomers with the L component to the left and S component in a multiline layout. Additional considerations and modifications have emerged from genome comparisons,145 including the current arrangement of protein coding genes RL1-13-UL1-150-IRS1-US1-US34-TRS1 (Table 62.1). The numbering of genes has resulted in the UL148 to UL133 segment being inverted due to the unusual organization of the Toledo strain genome where this region was first characterized.106 The current consensus genome map omits some potential ORFs and emphasizes a uniform nomenclature.399 A few genes have been added as a result of strain comparisons and empirical mapping, resulting in the current estimate of a minimum of 167 protein-coding genes, plus genes for four large noncoding RNAs, two oriLyt RNAs, and at least 23 miRNAs (Table 62.2). The genome includes cis-acting signals for replicative DNA synthesis (oriLyt), cleavage/packaging (pac1 and pac2 within terminal a sequences) and the RNA transcription, including the MIEP-enhancer within the UL (located between UL124 and UL128) and the US3 promoter-enhancer within the US region (located between US3 and US6). As in all herpesviruses, genes encoding different HCMV temporal classes of protein-coding and noncoding RNAs are interspersed across the viral genome. For example, the first set of genes to be expressed during infection, IE genes, include the IE1-IE2 locus and the UL36-UL37 locus within UL, US3 within US and IRS1/TRS1 within the S component inverted repeats (Table 62.1). These features are conserved in chimpanzee CMV but diverge in other primate and nonprimate cytomegaloviruses.41,369,490,531
During passage in fibroblasts,141,619 both HCMV (AD169, Towne) and RhCMV (68.1) strains accumulate point mutations, deletions, and duplications such that common laboratory strains lack as many as 20 genes while retaining a consistent genome size through sequence duplication.106 A common duplication affects a large segment of the RL region,106 which is naturally part of UL (Figs. 62.1 and 62.3). It should be noted that HCMV strains do not spontaneously lose any og the many genes that are dispensable for replication. Despite spontaneous deletions, HCMV maintains a consistent, 236 kbp genome size.145,166,399,727
HCMV genome annotation remains provisional. Protein-coding regions shorter than about 80 codons or regions overlapping larger ORFs have typically not been included unless verified as biologically functional.145,399 The refinement of HCMV annotation has included many methods, including comparisons of strains to unpropagated HCMV, chimpanzee CMV, alternative algorithms, and relating protein composition to coding capacity. Liberal estimates of coding capacity,418 improved annotation methods,82 transcriptome analysis,204 and ribosomal profiling741 all contribute to the current estimate. The functional analysis of HCMV genes has generally followed one of two strategies. Many viral genes have yielded biological activity when studied in isolation or when mutated in the virus. Many have revealed their function through a specific phenotype in viral replication, pathogenesis, immune modulation, or latency. Alternatively, directed or random mutagenesis of viral genomes has yielded phenotypes that define biological function during replication or pathogenesis. The virus strain, host cell type, precision of the mutagenesis, and approaches to evaluation may all impact the physiological relevance of the data that is generated by either approach. A wider
variety of viral gene products have been studied in isolation than have been studied in the context of viral infection, which leaves the physiological function of many gene products in need of further study. The genome complexity of HCMV, together with the range of settings where this virus interfaces with host cell and host defense pathways for replication, persistence, pathogenesis, and latency provides opportunity for additional insights to emerge.
variety of viral gene products have been studied in isolation than have been studied in the context of viral infection, which leaves the physiological function of many gene products in need of further study. The genome complexity of HCMV, together with the range of settings where this virus interfaces with host cell and host defense pathways for replication, persistence, pathogenesis, and latency provides opportunity for additional insights to emerge.
Although different strains of HCMV exhibit >95% DNA sequence homology, certain regions exhibit a high interstrain variability. UL73, UL74, UL144, and UL146 glycoprotein genes, in addition to the terminal repeat a sequence, are the most polymorphic (Table 62.1), variability that has been very useful in tracking viral genome transmission in clinical settings. These polymorphic genes have been used in attempts to correlate particular genotypes with disease as well as for assessing geographic distribution of strains and to establish the level of recombination between strains in reinfected individuals. Recent deep sequence evaluation of viral genomes in clinical samples has revealed a truly immense level of variation that occurs within infected individuals139 and has suggested that HCMV undergoes error-prone replication with sequence variation levels as high as some RNA viruses.520
HCMV encodes 23 miRNAs (Table 62.2) during productive viral replication.168,221,473,623 In contrast to alpha herpesviruses and gamma herpesviruses, where miRNAs are clustered, HCMV miRNA genes are dispersed across the genome. All but miR-UL70-1-5p, which has not been tested, associate with Argonaute protein silencing complexes623 and initiate a diversity of biological consequences.156,674 Most mature miRNAs that have been assayed221 show DE kinetics, except miR-UL70–1, which shows an IE pattern. Many of the miRNAs are in regions of the genome known to be dispensable for replication in fibroblasts (e.g., miRNAs mapping to US4, US5, and US22), such that no single virus-encoded miRNA is crucial for replication. Several play accessory roles modulating gene expression that influences cell behavior or host response, and may be important in pathogenesis. The sequences of miRNAs are not conserved between herpesvirus subfamilies or even between human beta herpesviruses HCMV and HHV-6B.673 Although conservation between HCMV and chimpanzee CMV221 limited (miR-US5-2) conservation with RhCMV have been noted,235 MCMV carries a completely unique set of miRNAs.88,160 More work will be needed to determine whether the lack of sequence conservation reflects a type of evolutionary divergence that immunomodulatory proteins are already known to exhibit. HCMV encoded miRNAs have not yet been demonstrated during the latent phase of infection, although cellular miRNA has been implicated in a CD34+ cell model of latency.485
Replication
HCMV pathogenesis involves productive replication in a variety of cell types,81,523 including epithelial, endothelial, neuronal, myeloid, and fibroblast cells. Most studies aimed at understanding viral replication properties have been carried out in fibroblasts, a stromal cell present in all tissues of the host. The ease of studying HCMV replication in these cells, together with significantly greater yields of virus and gene products,668 has made it possible to establish the main themes of viral gene expression and regulation, DNA synthesis and maturation (Fig. 62.2). Viral tropism differences for various cell types were initially recognized through studies employing clinical strains propagated on either cultured endothelial or myeloid cells, including vascular endothelial cells, macrophages, and dendritic cells.583 Viral strains propagated in this manner retained tropism characteristics that are lost during propagation on fibroblasts, but still accumulate mutations.141 Cell lines such as ARPE-19 epithelial cells proved useful in studies of viral tropism determinants. Clinical isolates must undergo adaptation to replicate efficiently in fibroblasts, overcoming poorly understood negative impacts of RL13 and UL128-UL131A141 that drive genome mutations in fibroblasts.619 Evidence clearly shows that loss of any one of three components (pUL128, pUL130, and pUL131A) of the gH:gL pentameric complex compromises replication efficiency on epithelial and endothelial cells.523,539,541,698
Following entry, three kinetic classes of genes, IE (or α), delayed early (DE or β) and late (L or γ), are expressed sequentially in a coordinately regulated manner over the course of a 48 to 72 hour productive replication cycle. Expression of major IE gene transcription is controlled by upstream enhancers on the viral genome as well as a tegument protein (pp71) that acts as the virion transactivator to increase expression. IE gene expression depends upon preformed host and viral machinery and is independent of de novo viral or host expression. Host cell RNA pol II and related transcription machinery controls transcription of the viral genome throughout infection, although transcription is regulated via virus-encoded transactivators. Translation depends entirely on host cell ribosomes. Expression of DE genes is required for viral DNA synthesis and requires expression of functional IE gene products. The DE class of β genes may be further divided into early–late subclasses, β1 and β2, which differ in the pattern of expression.399 Expression of L genes is dependent on expression of DE genes and includes gene products that form virions and control maturation. Two distinct categories of L genes are recognized as leaky late, or γ1 and true late, or γ2, based on the pattern of expression and dependence on viral DNA synthesis.399 Similar to other herpesviruses, the expression of most HCMV L genes is leaky, and occurs independent of viral DNA synthesis though dependent on DE gene expression. Typically, viral DNA synthesis inhibitors such as foscarnet (phosphonoacetic acid) have been used to block viral DNA synthesis to assist in dissecting DE and L patterns of gene expression. The leakiness of late gene expression and the recognition of kinetic subclasses spawned the alternate terminology—α, β, and γ— first used to describe HSV-1 patterns. Although the replication cycle of HCMV is slow, requiring 48 to 72 hours to reach final stages of maturation and release of progeny, the expression of IE gene products starts within minutes of infection. The switch from early phase to late phase is prolonged, from 24 to 36 hours postinfection (hpi) and may be even longer dependent on cell type. Initiation of IE gene expression is sensitive to cell cycle status, linked to regulation of p53,739 such that cells in S, G2, or M phases do not produce IE proteins until cells return to G1. Maximum levels of virus progeny are released from fibroblasts starting around 5 days postinfection and continue for several days before cells die, succumbing to a serine protease-dependent programmed death pathway the timing of which is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA).377
Attachment and Entry
Entry occurs in distinct steps399: (a) binding to specific cell surface receptors, (b) viral envelope fusion with cellular membranes to release nucleocapsids into the cytoplasm, either directly at the plasma membrane (as occurs in fibroblasts) or after endocytosis into cells (as occurs in endothelial and epithelial cells), (c) nucleocapsid translocation toward the nucleus on cytoskeletal filaments, (d) nucleocapsid interaction with nuclear pores, and (e) release of the viral genome into the nucleus (see Fig. 62.2). At the same time, independent of nucleocapsid translocation to the nucleus, tegument proteins are released into the cytosol and traffic to sites where they function in diverse ways, modulating the initial host response to infection and orchestrating the transcription of IE genes. This process has common characteristics among all herpesviruses, so understanding of how these steps are controlled comes from direct study of HCMV as well as from surrogates such as MCMV, guinea pig CMV, and simian CMV, as well as comparisons to other herpesviruses. Initial contact with cells is via cell surface heparan sulfate, a feature common to many herpesviruses. Investigations have shown that envelope glycoproteins gB and gM bind heparan sulfate, and may overlap to make initial contact with cells.262 HCMV attachment does not require dedicated subgroup-specific receptor-recognizing envelope glycoprotein, such as gD (HSV-1/HSV-2) or gp350 (EBV).132,285 Instead, HCMV completes attachment as well as fusion steps with herpesvirus-conserved gB and gH:gL. Initial binding of virions to cells leads to a cascade where gB trimers and gH:gL heterodimers orchestrate events through sequential cellular receptors that ultimately results in fusion between the viral envelope and cell membrane,262 and release of the nucleocapsid and tegument proteins into the cytoplasm. This entry step is disrupted when cholesterol is reduced.275 In addition to the major envelope glycoproteins controlling entry into any cell type, a trimeric complex consisting of gH:gL:gO and a pentameric complex comprised of gH:gL:pUL128;pUL130:pUL131A facilitate entry in certain settings.523,582 The pentameric complex facilitates attachment and entry into epithelial and endothelial cell types,523,541,698 suggesting that this complex may play an important role in HCMV pathogenesis. Additional entry options have come to light, such as the requirement for pUS16 to mediate efficient viral infection of endothelial and epithelial cells when the pentameric gH:gL complex is absent.83
Considerable attention has been focused on gB because of its common role in attachment and fusion of all herpesviruses, mediating delivery nucleocapsids into the cytosol. Several receptor and entry mediator candidates have been shown to interact with gB. Some of these have turned out to be dispensable for infection of susceptible cells.131 In this regard, β2 microglobulin, annexin II, and aminopeptidase N (CD13) are unlikely receptors for viral entry. More recent identification of cell surface integrins α2β1, α6β1, and αVβ3,262 as well as EGFR on monocytes108 and PDGFαR on endothelial, epithelial, and fibroblast cells,602 remain under evaluation. Furthermore, host membrane characteristics conferred by tetherin expression enhance entry.687 In addition to mediating the delivery of the nucleocapsid to the cytoplasm, the interaction of HCMV envelope glycoproteins with cell surface proteins may trigger cellular signaling pathways to enhance viral and cellular gene expression to facilitate infection.262 Toll-like receptor 2 (TLR2) is one such candidate on monocytes. Candidate receptors have also been found to interact with gH, including αvβ3 integrin.131 Although there is little question that HCMV enters cells through specific interactions between major envelope glycoproteins and cellular plasma membrane receptors, much remains to be learned about the pathways that are involved.131,262,399,523
Intracellular Trafficking and Uncoating
Once the HCMV nucleocapsid is deposited into the cytoplasm, cytoplasmic microtubules are predicted to facilitate translocation to the nucleus where viral DNA is released.399 In HCMV, the large tegument protein (LTP; UL48), as well as a binding protein (LTPbp; UL47) play essential roles in replication,167,727 potentially analogous to the HSV-1 UL36 gene product, controlling uncoating and release of viral DNA from nucleocapsids. Both HCMV UL47 and UL48 are likely to be important for replication,80,400 but so far only viral mutants in a dispensable deubiquitinylating function of UL48 have been studied.293 Greater focus on uncoating and nucleocapsid translocation to nuclear pores is needed.
IE Gene Regulation and Function
Once the HCMV genome is delivered to the nucleus, IE gene expression ensues. RNA pol II transcription machinery transcribes IE as well as all other protein-coding and noncoding RNAs made from the HCMV genome. Regulation of viral gene expression occurs via two broad strategies: (1) viral as well as cellular factors that directly influence the transcription machinery by binding to promoter/enhancer elements directly (transcription factors) or through interactions with other proteins (adaptors) and (2) viral factors that alter chromatin remodeling by regulating the opposing activities of histone acetyl transferases (HATs) acting together with demethylases and histone deacetylases (HDACs) and methylases. HDAC-dependent repression of viral IE gene expression, in particular, is a cell-intrinsic host defense mechanism that must be defused before productive replication can ensue. Epigenetic regulation is important in permissive cells, even though the viral genome does not take on a recognizable chromatin structure, and also during latency, where viral genomes take on an organized chromatin arrangement and viral HDAC inhibitors can drive reactivation.510,512,579
The regulation and activities of IE gene products have been a focus of considerable review.399,510,512,579,628,629,631,707 Five loci distributed across the viral genome give rise to IE gene transcription (Table 62.1), UL36 and UL37, IE1 and IE2 (UL122 and UL123), TRS1, IRS1, and US3. IE1/IE2 and US3 gene expression is under control of independent transcriptional enhancers. The major IE (MIE or IE1/IE2) gene is transcribed from the MIE promoter (MIEP) into alternatively spliced and polyadenylated mRNAs encoding two predominant nuclear phosphoproteins, IE1-p72 and IE2-p86, along with minor IE proteins. Although IE2-p86 is recognized as the most important viral transactivator and key to viral gene expression during productive replication, IE1-p72 and IE2-p86 together defuse epigenetic repression, activate DE and L classes of viral genes during productive replication, autoregulate IE gene expression, and establish nuclear sites where lytic viral DNA synthesis proceeds. In addition, these major IE proteins contribute in crucial ways to the switch between latency and reactivation.
MIEP expression is dependent on a transcriptional enhancer (also called the HCMV enhancer) that is subject to positive- and negative-acting viral and cellular chromatin
remodeling pathways controlled by HATs and HDACs acting together with methyltransferases and demethylases.510,512,579 This locus is the only region of any cytomegalovirus genome known to exhibit CpG suppression, consistent with a need for the region to remain unmethylated during latency and reactivation.629 No matter the cell type, epigenetic repression of the MIEP must be relieved by the concerted and sequential activity of tegument proteins such as pp71 and ppUL69 and the major IE proteins IE1-p72 and IE2-p86, acting with other viral proteins and host transcription machinery to sustain viral gene expression over the very long HCMV replication cycle.323,374,654 The failure of HCMV to replicate in cells that have entered S phase is due to the inability of these viral functions to overcome epigenetic repression740 and is associated with the failure to encode IE proteins.550
remodeling pathways controlled by HATs and HDACs acting together with methyltransferases and demethylases.510,512,579 This locus is the only region of any cytomegalovirus genome known to exhibit CpG suppression, consistent with a need for the region to remain unmethylated during latency and reactivation.629 No matter the cell type, epigenetic repression of the MIEP must be relieved by the concerted and sequential activity of tegument proteins such as pp71 and ppUL69 and the major IE proteins IE1-p72 and IE2-p86, acting with other viral proteins and host transcription machinery to sustain viral gene expression over the very long HCMV replication cycle.323,374,654 The failure of HCMV to replicate in cells that have entered S phase is due to the inability of these viral functions to overcome epigenetic repression740 and is associated with the failure to encode IE proteins.550
Individual MIE proteins counteract cell-intrinsic resistance mechanisms triggered by viral infection.427,512 MIE proteins IE2-p86 and IE1-p72 induce cell cycle arrest with prodeath or prosurvival impact depending on the setting.377,427,631 MIE protein IE1-p72 binds chromatin,516,576 orchestrates STAT (Signal Transducer and Activator of Transcription) signaling,427 disrupts nuclear domain 10 (ND10; also called promyelocytic leukemia protein [PML] bodies or PML oncogenic domains [PODs]),323,374,654 in addition to binding HDACs576 and Daxx511,512 and contributing to chromatin remodeling and direct regulation of gene expression.
Three additional IE regions (UL36/UL37, TRS1, and IRS1) encode functions that modulate other aspects of the cell-intrinsic response to viral infection in addition to their direct impact on gene expression, and one IE gene (US3) is involved in posttranslational down modulation of MHC class I gene levels on the host cell to evade cytotoxic T-cell surveillance. UL36, UL37 × 1, UL37 × 3, TRS1, and IRS1 are each individually dispensable for viral replication in fibroblasts; each carries out modulatory activities that contribute to viral pathogenesis. The US3 protein is the earliest viral protein disrupting MHC class I antigen presentation to make infected cells less attractive targets for cytotoxic T-cell immune surveillance480 and will be discussed together with other US2 and US6 family members.
UL37 × 1 encodes the very potent mitochondrial cell death suppressor, vMIA. This small protein localizes to the outer mitochondrial membrane and sequesters Bax and Bak, but does not prevent oligomerization of these Bcl2 family members or mitochondrial fission that results from this association.286,435 Through interactions with Bax, Bak, BclxL, and GADD45 family members, vMIA prevents cytochrome c release, the key step in the activation of executioner caspases driving apoptosis.377 vMIA associates with endoplasmic reticulum (ER)–derived mitochondrial-associated membranes,75,711,713 increases ER release of calcium stores, and enhances cell rounding574 and, as a consequence, vMIA has been associated with HCMV cytopathic effects mediated through viperin.573 Although vMIA is a very potent inhibitor of apoptosis,377 its role in HCMV infection is to prevent a novel mitochondrial HtrA2 serine protease-dependent death pathway that terminates the viral replication cycle in fibroblasts.380 Because of its role in titrating out mitochondrial proteins that antagonize infection, vMIA may appear essential for replication (e.g., AD169) or dispensable for replication (e.g., Towne). Small mitochondrial Bax and Bak inhibitors with sequence characteristics of UL37 × 1 are evolutionarily conserved in beta herpesviruses.377 MCMV encodes two mitochondrial-localized cell death suppressors with related function, m38.5/vMIA and m41.1/vIBO, that independently target Bax26,435 and Bak,91 respectively.
UL36 is a US22 family member that encodes the viral inhibitor of caspase 8 activation (vICA).377 vICA suppression of extrinsic apoptosis is crucial for viral infection of macrophages,123,379 but apparently dispensable for replication in other cell types. HCMV UL36, as well as the RhCMV homolog are mutated in commonly used strains.585 UL36-encoded vICA binds procaspase 8 and prevents cleavage/activation, behaving in a manner that is mechanistically similar to the long form of cellular FLICE (another name for caspase 8) inhibitory protein (cFLIPL). vICA is highly conserved, with a homolog encoded by all beta herpesviruses. The contribution of vICA-associated caspase 8 suppression during infection and pathogenesis comes from studies using MCMV mutant viruses in mice.123 Viruses that lack vICA replicate fine in fibroblasts but are attenuated due to susceptibility to extrinsic apoptosis in macrophages.123,377,405 The evolutionary acquisition of multiple cell death suppressors in HCMV and MCMV has facilitated identification of programmed necrosis as a bona fide host defense pathway that evolved in mammals to defend against viruses.405 Necrotic death is triggered by vICA suppression of caspase 8, causing release of receptor interacting protein (RIP) kinase 3 to form a complex with DNA-dependent activator of interferon signaling (DAI)677 and is inhibited by MCMV M45-encoded viral inhibitor of RIP activation (vIRA).678 An HCMV RIP inhibitor has not been identified.
IRS1 and TRS1 are highly homologous US22 family members that inhibit interferon-inducible protein kinase R (PKR).371 Either must be retained in order for viral replication to proceed.370 Independent of PKR, TRS1 inhibits macroautophagy,111 and TRS1, more than IRS1, may influence DE gene expression, DNA synthesis,278,634 and capsid assembly,371 all of which may ultimately be related to inhibition of PKR.370
Regulation of MIEP Enhancer
The MIEP-enhancer is a common tool for eukaryotic expression vectors in experiments and commercial production of biologics because it is constitutively active and strong.561 In the context of the viral genome, the MIEP-enhancer is epigenetically silenced and requires the activity of input viral tegument proteins pp71 and ppUL69 as well as IE and DE proteins that are made during infection to remain active.579,628,629 Regulation focused on cellular chromatin remodeling machinery bears on models of viral latency and reactivation where MIEP expression is key to this switch.512 Mutations within the MIEP enhancer result in viruses that exhibit MOI-dependent growth properties and that are compromised in models of latency and reactivation.512,628,629 Once released from nucleocapsids following entry, HCMV genomes delivered to the nucleus associate with ND10, in a manner that has been observed in many herpesviruses and other DNA viruses.374 Initiation of IE gene transcription, modulation of cell intrinsic signaling, and inhibition of the DNA damage response all take place in association with ND10,179 which becomes disrupted by IE1-p72, even though this disruption is dispensable for viral replication.576 Relief from HDAC-mediated repression is initially carried out predominantly by the tegument protein pp71, encoded by UL82, is the principal virion transactivator (VTA) of HCMV. VTA relieves HDAC repression511,512,628 by localizing to ND10, degrading Daxx to
prevent Daxx/ATRX-mediated repression of the MIEP, which is mediated via HDACs.258,352,430,492 These properties of pp71 make it useful in experimental settings to increase production of infectious virus following transfection of viral DNA or bacmids. pp71 associates with tegument protein ppUL35,559 an HCMV DNA damage response modifier.545 Upon entry, the pp71:ppUL35 tegument complex more efficiently stimulates MIEP expression than pp71 alone, whether surveyed in reporter assays or in the context of virus infection.559 Viral mutants lacking UL82 or UL35 exhibit MOI-dependent replication similar to that observed with IE1-p72, MIEP enhancer, and crs mutant viruses.628,629 Following the initiation of IE gene expression, HDAC suppression is sustained by IE1-p72.
prevent Daxx/ATRX-mediated repression of the MIEP, which is mediated via HDACs.258,352,430,492 These properties of pp71 make it useful in experimental settings to increase production of infectious virus following transfection of viral DNA or bacmids. pp71 associates with tegument protein ppUL35,559 an HCMV DNA damage response modifier.545 Upon entry, the pp71:ppUL35 tegument complex more efficiently stimulates MIEP expression than pp71 alone, whether surveyed in reporter assays or in the context of virus infection.559 Viral mutants lacking UL82 or UL35 exhibit MOI-dependent replication similar to that observed with IE1-p72, MIEP enhancer, and crs mutant viruses.628,629 Following the initiation of IE gene expression, HDAC suppression is sustained by IE1-p72.
Other tegument proteins contribute to optimal MIEP expression. Virion ppUL69 enhances the initial stages of viral infection, although it has its major impact later in infection. ppUL26 is a transcriptional activator of the MIEP that probably regulates phosphorylation levels of other tegument proteins,413 and pp65 activates the MIEP possibly by recruiting the cellular IFI16 protein to the control region.137 ppUL29/28, encoded from a spliced mRNA, stimulates MIE gene expression by modifying the HDAC1-containing nucleosome remodeling and deacetylase protein (NuRD) complex, handing off to pUL38 in association with the MTA2 component of NuRD at early times in infection.396,656 As infection proceeds, MIE gene products impose additional regulation on the MIEP enhancer, as described in the next section. In addition to the viral proteins, cellular signal transduction pathways induced by virus particles or other factors such as serum growth factors as well as cell type and cell cycle state, impact MIEP expression. This is particularly evident at low MOIs as well as in particular cell types.579
Mutation and substitution of enhancer components have reinforced the role of the MIEP enhancer in the initiation of viral gene expression during replication. The MIEP enhancer is composed of an upstream region (−580 to −300 relative to the transcription start site) that is less important in most assays than the proximal region (−300 to −39 relative to the transcription start site).628,629 The multitude of transcription factor binding sites contributing to enhancer activity during infection628,629 includes multiple cyclic AMP response element binding protein ATF/CREB and NF-kB binding sites. Investigations have shown that many can be disrupted without compromising replication, consistent with a highly redundant role for this complex transcriptional control region. Substitution experiments replacing MIEP enhancers have shown that MCMV carrying a HCMV enhancer replicates normally in mouse fibroblasts, but not in mice. The MCMV enhancer does not work in HCMV, but the simian CMV enhancer confers normal growth levels and plaquing efficiency on HCMV.628,629 These experiments reinforce the role of the enhancer as a critical cis-acting signal for MIE expression as well as some level of evolutionary adaptation to the homologous host species within the context of the viral genome.
IE1 and IE2
The principal products of the MIE gene, IE1-p72 and IE2-p86, result from alternative mRNA splicing that places 85 amino terminal amino acids together with carboxyl terminus derived from UL123 and UL122, respectively. Both of these proteins are primarily involved in regulating viral transcription to initiate productive replication as well as the latency-reactivation axis, although expression from this locus is not sufficient to drive reactivation.512,628,629,707 These proteins modulate the host cell response to infection,367,654 acting in concert with virion tegument as well as other IE gene products to establish a cellular environment to support replication. IE2-p86 is both a crucial activator of later kinetic classes (DE and L) of viral genes as well as a repressor of MIE expression,631 whereas IE1-p72 functions primarily as a coactivator together with IE2-p86.205 IE1-p72 disrupts ND10374,629 and sustains an open chromatin configuration initiated by the VTA, and probably acts independent of VTA in reactivation from latency.511 In addition, less abundant IE proteins IE1-p38, IE2-p55, and IE2-p18, have no known function.629 Later during infection, abundant leaky L (p60) and true L (p40) proteins are made from independent promoters embedded within UL122, and these are colinear with the carboxyl terminus of IE2-p86. Although both of these have the ability to repress MIE expression by binding to the crs, neither plays a crucial role in replication.629,706
IE2 proteins that share the carboxyl-terminus of IE2-p86 all negatively autoregulate expression from the MIEP by binding to the minor groove of the crs between −13 and −1 relative to the transcription start site (CGTTTAGTGAACC−1), thereby blocking transcription by inducing a repressive chromatin state509,510 rather than by disrupting transcription by RNA pol II more directly. This was the first IE2-p86 binding site identified in the HCMV genome, but there are many others within the promoters of DE and L genes that enhance transcription. A sequence related to the crs in the US3 gene (CGTG/TCAGTCCACACG+1) is repressed by ppUL34 rather than IE2-p86.55,629 The highly abundant leaky L 60-kD and very abundant L 40-kD (L40) IE2 proteins, once thought to maintain repression throughout infection, are largely accessory. Only IE2-p86 appears crucial for the activation and repression necessary to complete viral replication.706
The major function of IE1-p72 and IE2-p86 is to mediate activation of viral genes expressed through the remainder of infection.629 IE1-p72 and IE2-p86 associate with ND10. IE1-p72 disrupts these domains and undermines ND10-dependent activation of interferon (IFN), but does not alter their organizational role in viral gene expression or in initiation of viral DNA synthesis. Although an independent role of IE1-p72 in countering IFN activation is established,459 infection with IE1-p72–deficient virus at an MOI greater than 1 leaves ND10 intact and replication at levels similar to that of wild-type virus. The theme established by pp71 continues with the MIE genes, and IE1-p72 inactivates HDACs associated with ND10 to facilitate efficient, IE2-p86-dependent DE and L gene expression.511,512,631 Although normally disrupted by IE1-p72, the ND10 areas become sites of DNA synthesis independent of IE1-p72 function, starting with ppUL84 associating with IE2-p86, attracting UL112-113 gene products and viral replication complex proteins to initiate viral DNA replication.21,278,295,443 The replication defect of IE1-p72-deficient virus at low MOI likely results from failure to inactivate HDACs and allow for adequate activation of DE gene expression rather than either a failure to disrupt ND10, suppress the IFN response, or initiate DNA replication.511,512 It should be noted that the UL97 kinase667 and other gene products expressed later in infection (UL30, UL35, and US32) alter PML morphology or disrupt ND10.546
HCMV IE2-p86, a major nuclear phosphoprotein, is crucial for replication, although exact mechanisms have proven difficult to establish because of technical difficulties with conditional
mutant viruses that have been made. This gene has been subjected to extensive mutagenesis and evaluation by transient assay and many mutants have been built into virus constructs that fail to replicate. Most attention has been focused on the full-length MIE product, IE2-p86, the only UL122 gene product that is absolutely necessary for replication. The additional IE, DE, and L gene products encoded in the UL122 region are completely dispensable.90,551,552,553,631,706,707 The MCMV homolog (called ie3) seems to function analogously to HCMV IE2-p86,314,372 although the MCMV homolog of HCMV IE1 (called ie1) and a novel MCMV ie2 gene are dispensable for all aspects of viral infection708 and fail to contribute to transcriptional patterns during infection.314 HCMV IE2-p86 transactivates promoters through direct interaction with DNA (the MIEP crs and many other crs-like sites distributed throughout the genome), influencing RNA pol II transcription machinery directly by interacting with p53, cdk9 and cdk7, cycinT1, Bed4, HDAC1 and HDAC2 in a viral transcriptosome283,284 as well as via epigenetic regulation, and employing functional domains that are located in both amino-terminal and carboxyl-terminal regions of the protein.631,707 In the absence of IE2 mutant viruses to reveal core aspects of IE2-p86 function, transient assays defined IE2-p86 domains that are important for positive or negative regulatory impact, and these have proven useful. The regions contributing to transactivation include two domains located between aa 25 and aa 85 as well as between aa 544 and aa 579, at the extreme carboxyl terminus, as well as dimerization (aa 388 to 542) and helix-loop-helix aa (463 to 513) regions that overlap a core region (aa 450 and 544) important for most associated activities.631,707 The 450 to 544 region directs the interaction with a number of cellular transcription factors that have been implicated in regulation by IE2-p86,283,284 as well as for interaction with the viral UL84 protein, which counteracts IE2-p86 transactivation function but is crucial for initiation of DNA replication. IE2-p86 also interacts with a broad number cell cycle regulated proteins, sustaining the unique cell cycle block instituted during infection. Transactivation involves an adapter-like protein stabilizing RNA pol II transcription factor D TFIID-associated factors on the promoters, interacting with the carboxyl terminus of TATA element binding protein TBP and TFIIB.631,707
mutant viruses that have been made. This gene has been subjected to extensive mutagenesis and evaluation by transient assay and many mutants have been built into virus constructs that fail to replicate. Most attention has been focused on the full-length MIE product, IE2-p86, the only UL122 gene product that is absolutely necessary for replication. The additional IE, DE, and L gene products encoded in the UL122 region are completely dispensable.90,551,552,553,631,706,707 The MCMV homolog (called ie3) seems to function analogously to HCMV IE2-p86,314,372 although the MCMV homolog of HCMV IE1 (called ie1) and a novel MCMV ie2 gene are dispensable for all aspects of viral infection708 and fail to contribute to transcriptional patterns during infection.314 HCMV IE2-p86 transactivates promoters through direct interaction with DNA (the MIEP crs and many other crs-like sites distributed throughout the genome), influencing RNA pol II transcription machinery directly by interacting with p53, cdk9 and cdk7, cycinT1, Bed4, HDAC1 and HDAC2 in a viral transcriptosome283,284 as well as via epigenetic regulation, and employing functional domains that are located in both amino-terminal and carboxyl-terminal regions of the protein.631,707 In the absence of IE2 mutant viruses to reveal core aspects of IE2-p86 function, transient assays defined IE2-p86 domains that are important for positive or negative regulatory impact, and these have proven useful. The regions contributing to transactivation include two domains located between aa 25 and aa 85 as well as between aa 544 and aa 579, at the extreme carboxyl terminus, as well as dimerization (aa 388 to 542) and helix-loop-helix aa (463 to 513) regions that overlap a core region (aa 450 and 544) important for most associated activities.631,707 The 450 to 544 region directs the interaction with a number of cellular transcription factors that have been implicated in regulation by IE2-p86,283,284 as well as for interaction with the viral UL84 protein, which counteracts IE2-p86 transactivation function but is crucial for initiation of DNA replication. IE2-p86 also interacts with a broad number cell cycle regulated proteins, sustaining the unique cell cycle block instituted during infection. Transactivation involves an adapter-like protein stabilizing RNA pol II transcription factor D TFIID-associated factors on the promoters, interacting with the carboxyl terminus of TATA element binding protein TBP and TFIIB.631,707
DE Gene Regulation and Function
Following peak expression of MIE regulatory proteins, between 8 and 12 hpi, the second class of early genes, DE, become transcriptionally active independent of the host cell type.668 Although some DE gene products are produced in abundance from the start, most of the 65 proteins (Table 62.1) and assorted miRNAs (Table 62.2) accumulate gradually. The DE period of viral replication continues through 18 to 24 hpi when viral DNA synthesis initiates. DE genes are crucial for viral DNA synthesis and include several functions that become important later in infection for maturation and egress. Many DE genes have a substantial impact on replication when disrupted.167,727 DE gene products dispensable for replication in fibroblasts may contribute to modulation of the host cell and host animal response to infection. Several HCMV DE genes switch or add transcriptional start sites later in infection, which means that transcript levels represent the combined products of different kinetic classes of gene expression.
The principles of DE gene regulation707 are intertwined with subversion of host cell cycle regulated pathways550 and both depend on the key regulatory protein IE2-p86 functioning together with a number of collaborating factors.629,631 The promoter regions of three DE genes (UL112-UL113, UL54, and UL4) have been studied in detail. Although transient cotransfection assays first revealed candidate transcriptional control elements in the promoters for these genes, it was only after mutations were introduced into the viral genome and evaluated in the context of infection that concrete information emerged. The activation of these three genes illustrates the subpatterns of early gene expression that is dependent on IE gene expression as well as the differential regulation by host and viral functions as infection proceeds. Expression of the UL112-113 transcripts707 that are differentially spliced to yield four unique proteins (pp34, pp43, pp50, pp84), starts by 8 hpi and remains constant through L times (β1 pattern). Transcription shifts to a different start site at L times but the same mRNA splicing pattern and a single polyadenylation site is used throughout infection. Promoter-proximal ATF/CREB and IE2-p86 binding sites are important for sustained transcription throughout infection, with the ATF/CREB site more important early and the IE2-p86 binding site more important at L times. Expression of UL54,707 encoding DNA POL, starts by 8 hpi and increases steadily through L times (β2 pattern), relying on a single transcription start site but two polyadenylation sites. Unlike L genes discussed in the next section, expression of β2 genes is not reduced in the presence of viral DNA synthesis inhibitors. Promoter-proximal SP1 and IE2-p86 binding sites are more important early, and an ATF/CREB site is crucial for the increase at L times during infection, although the transcription start site remains unchanged. Finally, UL4,707 which encodes a minor virion envelope glycoprotein (gp48), shows a transcription pattern during infection that is similar to UL54 (β2 pattern), but this gene is predominantly regulated at the translational level by ribosome stalling.54,543 Transcriptional activation is dependent on Elk-1 and IE2-p86 binding sites in the promoter plus some consequence of MAPK signaling that acts via the TATA element. Translational stalling is dependent on a carboxyl-terminal Pro within a 22 aa ORF (uORF2) within the transcript upstream UL4, thereby preventing full expression of gp48. Translation stops because uORF2 fails to release peptidyl tRNA dependent on the release factor eRF1,543 and this blocks the large ribosomal subunit exit tunnel and halts the ribosome.54 Although elegant studies have deduced that this form of regulation is common across eukaryotes and prokaryotes, the natural context for this exquisite level of regulation as well as a mechanism to relieve inhibition in HCMV remain to be elaborated.
A number of DE gene products regulate gene expression. pUL34 is a specific repressor of US3 expression, acting on a novel crs.328 Four UL112-113 gene products (pp34, pp43, pp50, pp84), function together with IE1-p72, IE2-p86, and TRS/IRS to modulate DE gene expression, UL21A enhances replication at early times,184,185 and DE proteins contribute to continued suppression of epigenetic repression by HDACs. UL26 activates the MIEP,349,413 pUL38 antagonizes the tuberous sclerosis complex to prevent ER stress (407 Qian, 2011 #7341), and pUL29-28 stimulates gene expression by interfering with HDACs, modifying the nucleosome remodeling and deacetylase (NuRD) complex396,656 to sustain derepression of the MIE gene throughout infection. In addition, UL97 promotes KAT5/Tip60 HAT phosphorylation to increase expression330 and UL27 inhibits KAT5/Tip60 HAT and suppresses
expression of cyclin-dependent kinases.518 Finally several abundant tegument proteins, including pp65, pp71, pp150, and LTP/ppUL48 are transcriptionally active as DE genes, although their activities support virion maturation or steps during infection of subsequent cells.
expression of cyclin-dependent kinases.518 Finally several abundant tegument proteins, including pp65, pp71, pp150, and LTP/ppUL48 are transcriptionally active as DE genes, although their activities support virion maturation or steps during infection of subsequent cells.
Cellular gene expression is activated during HCMV infection. Global transcription204,246,434,623,668 and, most recently, global proteomic/metabolic analyses,412,549 has shown the extensive stimulation of cellular lipid metabolism and energy pathways to favor HCMV replication, contrasting with the extensive shut-off of the host cell by HSV.682 Viral replication and maturation follow stimulation and parallel accumulation of viral DNA synthesis functions, giving the impression that the prolonged replication cycle of this virus may require sustained cellular functions and proceeds only as they become available.550 HCMV infection stimulates cellular RNA as well as protein synthesis while completely dysregulating the cell cycle550 such that infected cells appear to be at G1/S, G2, or even M. Some of this comes as a result of dysregulation of cellular cyclin-dependent kinases246 and mislocalization of proteins normally associated with checkpoint control to sites of viral maturation in the cytoplasm.203 In fibroblasts, cellular DNA synthesis is usually held in check as a pseudo-G1 block,246 with contributions from tegument proteins, pp71 and ppUL69, as well as IE2-p86 in maintaining this cellular environment.550 Systems level analyses have repeatedly confirmed more focused earlier assessments showing that glucose metabolism,731,732 mitochondrial energy production,377,711 and cell cycle–regulated expression550 are all broadly activated, whereas cytoskeletal, extracellular matrix, adhesion functions, and cellular DNA synthesis550 are broadly down modulated.
One abundant noncoding RNA, the β2.7/RNA2.7, accumulates to represent more than 60% of total viral polyadenylated transcripts at L times of infection.204,668 This most abundant DE gene product is dispensable for replication384 but associates with mitochondrial complex I (nicotinamide adenine dinucleotide-ubiquinone oxidoreductase) to stabilize ATP production and prevents apoptotic cell death when respiration is blocked by rotenone treatment513 as well as necrotic death from ischemia/reperfusion injury.737 Although naturally a noncoding RNA, the β2.7/RNA2.7 promoter allows for high-level expression of heterologous genes, regulated with DE kinetics, within the HCMV genome.605
DNA Synthesis and Nucleotide Metabolism
Lytic HCMV DNA synthesis occurs within the nucleus of infected fibroblasts, starting as early as 14 to 16 hpi, increasing by 24 hpi, and reaching greater than 10,000 viral genome copies per cell668 at the time progeny virions start to form.21,443 In fibroblasts, viral DNA labeled for 1 h at either 19 or 23 hpi quantitatively chases into maturing virions and leaves the cell via the cytoplasm over the next 24 to 48 hours.467 In epithelial and astrocytoma cells, viral replication is more subdued and maximal viral DNA levels do not exceed 1,000 viral genomes per cell.668 DNA synthesis initiates from the complex oriLyt site between the UL57 and UL69 genes (Fig. 62.3), although the mechanism is still unclear. The HCMV, MCMV, or GPCMV oriLyt region is large, almost 3,000 bp, making this replication origin considerably more complex than others, including those from other human beta herpesviruses. This region can be divided into Essential Region I, with a bidirectional ppUL84:IE2-p86-responsive oriLyt promoter and a pyridine-rich Y-block,443 and Essential Region II, which includes an RNA–DNA hybrid structure495 and an adjacent RNA stem-loop structure capable of binding ppUL84.130 This region is also rich in other direct and inverted repeat sequences and transcription factor–binding sites. Viral DNA destined for replication circularizes after being delivered to the nucleus, associating with ND10, sites of early transcription where DNA synthesis initiates following the successive localization of IE1-p72, IE2-p86, and ppUL84, then UL112-113 proteins and, finally, ppUL44, which brings in other replisome components. A distinct subregion containing viral DNA and replisome components eventually dominates the nucleus as a distinct replication compartment. Six herpesvirus core replication fork proteins compose the replisome that synthesizes DNA.424 In HCMV, this replisome composed of the UL54-encoded DNA polymerase catalytic subunit (POL) together with the UL44-encoded polymerase processivity subunit (POL:PPS complex), the UL57-encoded single-strand DNA binding protein (SSB), and the heterotrimeric helicase-primase (HP) consisting of UL105-encoded HP1, UL70-encoded HP2, and UL102-encoded HP3.
Initiation of DNA synthesis requires oriLyt promoter activity, which is dependent on a ppUL84:IE2-p86 complex. ppUL84, a DE gene long known to play a key role in this process,443 forms homomultimers and binds to IE2-p86, thereby antagonizing transactivation by this protein. Its role in viral DNA synthesis is highlighted by the phenotype of mutant viruses.443 In complex with all UL112-113 proteins,295 ppUL84, a putative DExH/D box protein443 recruits ppUL44/PPS.635 Interactions between these components must form to support viral replication.295 As such, oriLyt recognition by ppUL84 bridges to the replisome via PPS. In addition, cellular functions such as C-EBP277 and hnRNP-K278 associate with these complexes and are necessary for viral DNA synthesis. Genetic evidence strongly supports the role of a ppUL84:IE2-p86 complex in initiation of oriLyt-dependent viral DNA synthesis,443 although the lack of a cell-free system leaves other details to be investigated. Other HCMV IE genes pUL36/vICA, pUL37 × 1/vMIA, ppIRS1, and ppTRS1 make the host cell environment more conducive to the process without being actively involved in the replication compartment.443
HCMV DNA synthesis may start as theta form before switching to a rolling circle mode of replication. The architecture of replicating DNA is consistent with concatemeric structures generated by rolling circle form of replication, and this is the likely template for the genome encapsidation. IE2-p86, ppUL84, and four UL112-113-encoded phosphoproteins (pp34, pp43, pp50, pp84) produced via alternative mRNA splicing707 all associate with prereplicative sites near ND10 sites in the nucleus to coordinate assembly of the viral replisome. Based on studies of viral mutants, all of these proteins must be simultaneously coexpressed to form a complex, colocalize with ND10, and recruit ppUL44/PPS to prereplication foci.295 It has long been appreciated that ppUL44/PPS and ppUL57/SSB show distinct nuclear localization patterns prior to the initiation of viral DNA synthesis, as the replication compartment forms around 19 hpi.467 Although IE2-p86 is believed to facilitate prereplication complex formation, it is the UL112-113 proteins that independently associate with ND10 and recruit the replisome to the viral genome, apparently via ppUL44/PPS.295 A shortage of UL112-113 gene products may underlie
the MOI-dependent replication defect in IE1-p72 mutant virus.205 The four UL112-113 gene products, ppUL84, IE2-p86, and six herpesvirus-conserved replisome proteins remain associated with the replication compartment throughout the remainder of infection.
the MOI-dependent replication defect in IE1-p72 mutant virus.205 The four UL112-113 gene products, ppUL84, IE2-p86, and six herpesvirus-conserved replisome proteins remain associated with the replication compartment throughout the remainder of infection.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


