Hepatitis C Virus
Stuart C. Ray
Justin R. Bailey
David L. Thomas
History
By the mid-1970s, it was apparent that at least one viral hepatitis agent other than hepatitis A virus (HAV) or hepatitis B virus (HBV) was the primary agent of posttransfusion hepatitis, a syndrome termed “non-A, non-B” hepatitis (NANBH).197,335 Studies of transfusion recipients revealed that NANBH tended to be milder in its acute form than HBV but could cause severe complications including cirrhosis and liver failure.16 Inoculation of chimpanzees with blood components from humans having both acute and chronic NANBH resulted in characteristic elevations of hepatic transaminases, providing a valuable animal model for NANBH and establishing the chronic nature of NANBH.17 By the mid-1980s, physicochemical studies of infectious inocula had revealed that the NANBH agent was a small (less than 80 nm), enveloped virus; however, the agent defied efforts directed at conventional viral cultivation and immunological detection.76,159
Serial passage of NANBH in chimpanzees provided key pathologic, physiologic, and biochemical insights, as well as a well-characterized pool of specimens in which the agent was known to be present. A team led by Michael Houghton assembled a lambda phage library of complementary DNA (cDNA) derived from one such high-titer chimpanzee plasma specimen and then screened more than 1 million expression clones using serum from a chronic NANBH patient to find a single positive cDNA clone called 5-1-1.116 This discovery led to initial assays for detection of antibodies to the newly named hepatitis C virus (HCV),18,116,352 and the 5-1-1 antigen continues to be a component of anti-HCV serologic tests.
The first cDNA clone enabled further characterization of the genome as a positive-strand RNA molecule of almost 10,000 nucleotides containing a single open reading frame with an organization consistent with the Flaviviridae.117 Discovery of the authentic 5′ and 3′ untranslated regions (UTRs) led to a full-length cDNA clone of the HCV genome that, when transcribed, was infectious by direct intrahepatic injection in chimpanzees.337 The development of in vitro model systems was relatively intractable until the development of subgenomic RNA replicons387 and then successful passage in cell culture of a clone from one strain.670
HCV continues to present unresolved scientific and clinical challenges. Questions persist regarding fundamental aspects of the HCV life cycle, replication dynamics in vivo, mechanisms of persistence, and pathogenesis. Screening of blood products using antibody- and then nucleic acid–based testing, combined with other blood banking practices, provides a sound basis for the virtual elimination of transfusion-transmitted HCV infection; nonetheless, new infections continue to occur via other routes. Nearly 3% of humans remain chronically infected with
HCV, and although treatment continues to improve in efficacy and availability, HCV infection remains a major cause of death and disability worldwide.
HCV, and although treatment continues to improve in efficacy and availability, HCV infection remains a major cause of death and disability worldwide.
Pathogenesis and Pathology
Entry into the Host
As discussed in the Transmission section later (under Epidemiology), the primary route of HCV entry is percutaneous, although permucosal infection has also been described. Experimentally, HCV infection can be achieved by intravenous injection of HCV virions or intrahepatic injection of HCV genomic RNA.337,694
Cell and Tissue Tropism
As depicted in Figure 27.1, HCV replication in vivo occurs primarily or exclusively in hepatocytes, the major parenchymal cell of the liver.505 The basis for this tropism is likely to be multifactorial, including entry facilitated by proteins expressed at particularly high levels on hepatocytes (e.g., low-density lipoprotein receptor [LDL-R]446 and scavenger receptor class B type I [SR-BI]185,248,557), dependence on liver-specific miR-122 for efficient replication,299 and utilization of the liver’s lipoprotein assembly pathway for virion production.280 Tissue-specific subcellular localization and dynamic interaction between viral components and more broadly expressed proteins on which HCV replication depends (such as CD81,195,248,270,320,499 claudins [CLDNs],183,267,270,422 occludin [OCLN],500 epidermal growth factor receptor,392 and cyclophilins257) may also contribute to liver-specific tropism. HCV entry is discussed in detail in Chapter 25.
Extrahepatic Replication
Productive infection of other cell types is controversial,70,362 but there is evidence for extrahepatic detection of HCV negative-strand replicative intermediates,106,375,701 sequence variant compartmentalization,459,562 and in vitro replication in a variety of cell types.602,608 Viral dynamic modeling of data from the anhepatic phase of liver transplantation suggested that in a subset of patients with end-stage liver disease, an extrahepatic compartment exists that contributes no more than 3% to 4% of plasma viremia.142,505
Model Systems In Vitro
During the first decade after HCV was discovered, the only robust model was the chimpanzee infected with primary or chimpanzee-passaged HCV isolates. Efforts to culture HCV from human and chimpanzee serum using primary hepatocytes or hepatoma cell lines were limited by relatively insensitive tools for measuring and visualizing infection, inconsistent cell lines, and the inherent variability of HCV isolates (see Genetic Diversity, later).284,290,313,444,445,454,570,697 Without an efficient culture system, screening of candidate antivirals was hampered.
The First Infectious Clone
In 1997, two groups separately reported chimpanzee infection with infectious clones of HCV,337,694 generated from acute phase isolate H7717 by using overlapping sequences spanning the genome (Fig. 27.2) to identify low-frequency polymorphisms
that were likely to be either artifactual (generated during cDNA cloning) or biologically genuine but less infectious. By removing such minor sequence variations, they constructed consensus sequences that were infectious, paving the way for the generation of consensus sequences from other isolates in search of clones that would replicate efficiently in vitro.51
that were likely to be either artifactual (generated during cDNA cloning) or biologically genuine but less infectious. By removing such minor sequence variations, they constructed consensus sequences that were infectious, paving the way for the generation of consensus sequences from other isolates in search of clones that would replicate efficiently in vitro.51
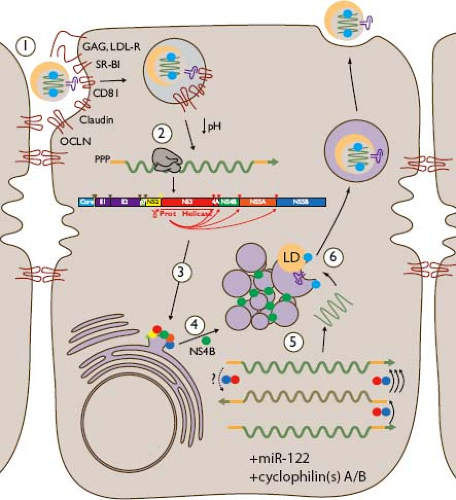 Figure 27.1. Life cycle of hepatitis C virus (HCV). Initial binding and internalization (1) probably involve glycosaminoglycans (GAGs) and low-density lipoprotein receptor (LDL-R), which may interact with viral envelope proteins or with virion-associated lipoproteins. Entry depends directly on binding of E2 with the tetraspanin CD81, as well as interactions with scavenger receptor BI (SR-BI) and tight junction proteins claudin-1 and occludin (OCLN). The viral genome is released from late endosomes (2) in a pH-dependent manner, followed by internal ribosome entry site (IRES)-dependent polyprotein synthesis (3) with initial cleavages among the structural proteins mediated by signalase and signal peptide peptidase followed by cleavage of the NS2–NS3 junction by NS2-NS3 cysteine protease; the remaining junctions are cleaved by the NS3-NS4A serine protease. NS4B recruits and rearranges endoplasmic reticulum (ER) membranes (4) to form a membranous web, the principal site of viral replication. Minus-strand and subsequent plus-strand RNA syntheses are affected by the NS5B RNA-dependent RNA polymerase (RdRp) (5) and depend on miR-122 and cyclophilin B, as well as conserved structural elements at the 5′ and 3′ ends of the genome. Core protein associates with lipid droplets (LDs) in the lipoprotein assembly pathway (6), linked to NS5A and other members of the replication complex by interaction with NS2. Viroporin p7 is necessary for production of stable viral particles coated with E1 and E2, which fold in a cooperative manner and are glycosylated in a manner consistent with ER but not Golgi processing. |
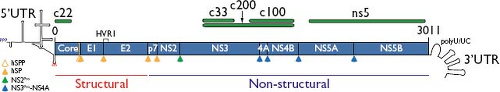 Figure 27.2. Map of the hepatitis C virus (HCV) genome, depicting the 5′ untranslated region (5′UTR), capsid core, envelope genes E1 and E2, viroporin p7, membrane-anchored cysteine protease NS2, serine protease-helicase NS3, NS3 protease co-factor NS4A, membrane remodeling protein NS4B, phosphoprotein NS5A, RNA-dependent RNA polymerase NS5B, and the 3′UTR. Depicted as green bars are protein segments used as antigens in HCV enzyme immune assay (EIA) and recombinant immunoblot assay (RIBA) (c22,268,609 c100-3352). Cleavages of the polyprotein are due to the action of signal peptidase (solid orange), signal peptide peptidase (open orange), NS2 cysteine autoprotease (green), and NS3-NS4A serine protease (blue). |
HCV Replicons
In 1999, the efficient replication of subgenomic replicons in hepatoma cell line Huh-7 was reported, representing the first culture system that depended on HCV enzymes for propagation of a selectable marker (driven in a 2-internal ribosome entry site [IRES bicistronic construct) and permitting more direct study of the viral life cycle.387 Highly replicative subtype subgenomic replicons were developed and were shown to depend on intact viral enzymatic sequences and negative-strand subgenome intermediates, inhibited by interferon-α.64,386 This latter characteristic opened the door to “curing” of cultures with interferon, resulting in Huh-7–derived cell lines like Huh-7.565 that were more permissive for HCV replication, an important tool for subsequent achievements including authentic culture of HCV in vitro.
Although cell culture adaptation of replicons permitted greater dynamic range of replication sufficient for screening inhibitors and studying viral protein interactions, the adaptive changes64,343,386 were often found to impair infectivity in vivo,85 and when they included the structural and p7 genes, these replicons did not produce structural proteins or viral RNA in the supernatant. Strains that were assembled from human isolates as consensus clones but required no adaptive changes as replicons were identified, including subtype 1b strain HCV-N, which was also infectious in chimpanzees,285 and subtype 2a strain JFH-1.315,316 When these were assembled as full-genome replicons, JFH-1 was found to produce infectious viral particles.
HCV Cell Culture
In 2005, the complete replication cycle of HCV in culture was described, using the subtype 2a strain JFH-1 in Huh-7–derived cells that had been made more permissive by eradication of HCV subgenomic replicon infection with interferon.670,706 Subsequently, the HCV cell culture (HCVcc) platform was broadened to include subtype 1a strain H77-S.696 As discussed in Chapter 25, this system and derivatives such as chimeric structural and nonstructural regions248 and reporter constructs328 provide new avenues for investigation of HCV biology and immunity.
HCV Pseudoparticles
Prior to the advent of HCVcc, HCV pseudoparticles (HCVpp) were developed to study HCV entry, which was found to be similar to other members of the Flaviviridae, and CD81 was found to be necessary but not sufficient for E1E2-mediated entry.46,279 HCVpp expressing a variety of E1E2 genes have facilitated study of cross-subtype and cross-genotype neutralization436,478 and have been used to demonstrate that neutralizing antibodies drive the rapid evolution of E2 during acute HCV infection.170,382
Virion Production without HCV Replication
There are many uses for high-titer stocks of HCV, but these can be difficult to generate and achievable titers are dependent on genomic characteristics (e.g., culture adaptive mutations) that may interfere with intended uses. A novel system for generating virions was recently developed to address these challenges, using cells conditioned by replication of a West Nile virus replicon.638 Because this system does not depend on HCV replication, it is potentially HCV sequence (i.e., genotype) independent.
Model Systems In Vivo
The initial model for HCV infection was the chimpanzee, essential to the discovery of this virus and to key experiments described herein (e.g., Transmission, Immune Response, and Genetic Diversity sections). The chimpanzee remains the only model for studying the full range of host–HCV interactions, from acute to chronic infection.84 The availability and use of chimpanzees is very limited,22 and other models are available that are relevant to specific areas.
Mice with Implanted Ectopic Human Liver Grafts
The Trimera severe combined immunodeficiency (SCID) mouse model, in which liver fragments remain viable for weeks after ectopic implantation (e.g., under the kidney capsule), can be used for studying HCV infection. The level of replication is modest (104.8 IU/mL) but high enough for testing antiviral regimens.286 The liver tissue in this model does not maintain normal architecture.
Mice with Liver Injury and Human Hepatocyte Xenografts
SCID mice transgenic for urokinase plasminogen activator (uPA) driven by the albumin promoter develop severe neonatal liver injury that is rescued by infused hepatocytes that engraft and
occupy the space of the involuting liver.431 Similarly, RAG−/−/interleukin-2 receptor-γ–deficient (IL2Rγ−/−) mice bred for fumaryl acetoacetate hydrolase deficiency (FAH−/−) develop hepatic toxicity but can be rescued pharmacologically with NTBC (2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione) or by transfer of the FAH gene. These FRG mice (FAH−/−, RAG−/−, IL2Rγ−/−) will accept infusions of human hepatocytes after infection with a uPA-expressing adenovirus (presumably to proteolytically damage the liver stroma).35 These mice can achieve physiologic levels of human albumin and lipoprotein levels, and after infection with HCV they can develop high levels of HCV RNA (106 IU/mL) and maintain these levels for months. The mice are difficult to breed and remain immunodeficient, and hepatocyte engraftment is highly variable; however, they are a useful model for HCV replication and have enabled mechanistic study of phenomena such as HCV neutralization in vivo.434
occupy the space of the involuting liver.431 Similarly, RAG−/−/interleukin-2 receptor-γ–deficient (IL2Rγ−/−) mice bred for fumaryl acetoacetate hydrolase deficiency (FAH−/−) develop hepatic toxicity but can be rescued pharmacologically with NTBC (2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione) or by transfer of the FAH gene. These FRG mice (FAH−/−, RAG−/−, IL2Rγ−/−) will accept infusions of human hepatocytes after infection with a uPA-expressing adenovirus (presumably to proteolytically damage the liver stroma).35 These mice can achieve physiologic levels of human albumin and lipoprotein levels, and after infection with HCV they can develop high levels of HCV RNA (106 IU/mL) and maintain these levels for months. The mice are difficult to breed and remain immunodeficient, and hepatocyte engraftment is highly variable; however, they are a useful model for HCV replication and have enabled mechanistic study of phenomena such as HCV neutralization in vivo.434
Humanized Mouse
A different approach from those described earlier was the recent development of a genetically humanized mouse model of HCV infection that partially addresses host restriction factors that block HCV infection of mouse hepatocytes.168 Using adenovirus gene delivery to induce expression of potential restriction factors for entry, CD81,499 SR-BI,557 CLDN-1,183 and OCLN,500 they found that human CD81 and OCLN were required for entry in the mouse. Mouse SR-BI knockout and human SR-BI complementation confirmed the necessity of SR-BI for HCV entry and that mouse SR-BI could substitute for human SR-BI in HCV entry. The remaining host restrictions for HCV infection of mice are unknown, but the use of adenovirus gene delivery may have enhanced innate antiviral responses, and the stable expression of CD81 and OCLN in the mouse will facilitate further study. At present, the genetically humanized mouse model supports entry but not replication of HCV.
Spread
The mode of spread of HCV throughout the liver is poorly understood. High-level viremia achieved by HCV provides ample opportunity for virions to interact with hepatocytes, yet it appears that only about 10% to 20% of hepatocytes are infected during chronic infection.378 Lack of uniform infection may be explained by innate responses that could render cells refractory and adaptive immunity that could interfere with entry of free virions.588 Cell-to-cell spread449 within the liver could circumvent antibody responses, and data from in vitro culture on human hepatoma cell lines support this mode of spread,630 suggesting that virus spread in vivo is relatively resistant to neutralizing antibody compared with infection with free virions, yet is dependent on the same key entry factors (HCV envelope, CD81, SR-BI, OCLN, and CLDN family members).79 These data are supported by the observation of foci of infection during in vitro culture, suggesting that this is a potential mode of local spread; however, it is clear that humoral immune pressure drives HCV evolution during chronic infection,382 suggesting that a major component of spread during chronic HCV infection remains subject to antibody-mediated neutralization.
Immune Response
Each component of the host immune response to HCV is balanced in some way by viral components. As a result, multiple viral proteins (depicted in Figs. 27.1 and 27.2, with a detailed functional discussion in Chapter 25) have immune-evasive roles in addition to more direct functions in the viral life cycle. These include Core (capsid), E1 and E2 (envelope), NS3-NS4A (serine protease), NS5A (polyfunctional phosphoprotein), and NS5B (RNA-dependent RNA polymerase).
Innate Immune Response
The innate immune response is of great importance in control of HCV infection,214 and the virus has evolved a variety of mechanisms to evade this response (Fig. 27.3). Interferon signaling is a key component of the innate responses against HCV. Type I, II, and III interferons (IFN-α and IFN-β; IFN-γ; and IFN-λ, respectively) have all been shown to be important, early intrahepatic responses in HCV infection.60,363,374,626 Type I and II interferons are induced by overlapping signaling pathways. IFN-regulatory factor 3 (IRF3), a latent cytoplasmic transcription factor, can be activated by viral infection and translocated to the nucleus where it induces the transcription of IFN-β. In autocrine and paracrine fashion, IFN-β stimulates activation of the Janus-activated kinase and signal transducer and activators of transcription (JAK-STAT) signaling pathway and synthesis of IFN-α, as well as multiple other antiviral cytokines and chemokines, inhibiting viral replication and orchestrating the subsequent adaptive immune response.221 HCV NS3-NS4A blocks IRF3 activation by proteolytically cleaving TIR domain–containing adapter-inducing interferon-β (TRIF) and mitochondrial antiviral signaling protein (MAVS). TRIF is an adapter protein for the double-stranded RNA (dsRNA) sensing molecule Toll-like receptor 3,377 and MAVS is an adapter protein in the retinoic-acid-inducible gene I (RIG-I) signaling cascade.439 These cleavages underscore the importance of these pathways for antiviral immunity, and impairment of interferon stimulated gene (ISG) expression was reversed by treatment with small-molecule inhibitors of the NS3-NS4A protease.377,389,439
Defects in JAK-STAT signaling have also been described in HCV transgenic mice.66 In HCV transgenic mouse and human liver biopsies, impairment of JAK-STAT signaling is linked to hypomethylation of STAT1 and increased expression of protein phosphatase 2A (PP2A).175 There is also evidence that NS5A protein can stimulate IL-8, inhibit dsRNA-activated protein kinase (PKR), and interfere with 2′,5′-oligoadenylate synthetase (2,5-OAS), antagonizing type 1 interferon signaling.223,305,344,501 Overexpression of HCV core protein also interferes with IFN signaling, likely through direct interaction with STAT1.67,381 Other studies suggest that ubiquitin-specific peptidase 18 (USP18) may be up-regulated by long-term interferon stimulation, blocking activation of ISG15 and suppressing JAK-STAT signaling, and leading to refractoriness to type 1 IFN stimulation.401,553 The inhibitory effect of HCV proteins on the interferon activation cascade is incomplete, as gene expression microarray studies have shown type 1 interferon responses in the livers of acutely and chronically HCV-infected chimpanzees.60,592 Hepatic levels of ISG expression in chronically infected humans vary significantly for unclear reasons.109,552
Natural killer (NK) cells are also likely to play a very important role in control of HCV infection. NK and natural killer T-lymphocyte (NKT) cells are abundant in the liver and prime cellular immune responses through production of IFN-γ and other cytokines.138,639 Binding of the E2 protein to CD81 has been associated with inhibition of NK cell activity.138,639 HLA Cw*04 and related haplotypes, which bind inhibitory
killer immunoglobulin-like receptors (KIR) on NK cells, have been associated with viral persistence.325 The least inhibitory human leukocyte antigen (HLA)-C-KIR haplotypes are most strongly associated with recovery.
killer immunoglobulin-like receptors (KIR) on NK cells, have been associated with viral persistence.325 The least inhibitory human leukocyte antigen (HLA)-C-KIR haplotypes are most strongly associated with recovery.
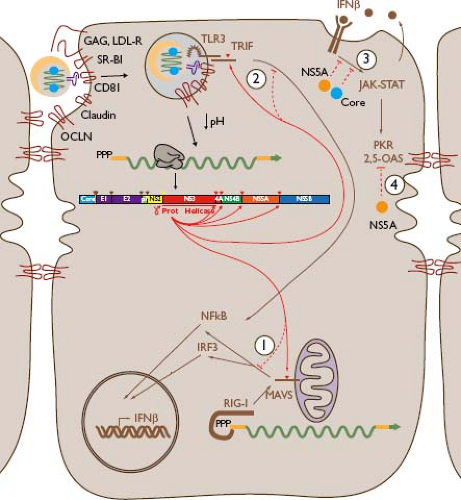 Figure 27.3. Innate responses to hepatitis C virus (HCV) and their evasion by the virus. Cytoplasmic HCV double-stranded RNA (dsRNA) can be sensed by RIG-I (1), resulting in signaling through mitochondrial antiviral signaling protein (MAVS) and subsequent nuclear translocation of nuclear factor (NF)-κB and phosphorylated IRF3 that activate an antiviral program including secretion of interferon-β (IFN-β), which has autocrine and paracrine activity; HCV NS3-NS4A protease cleaves MAVS, blocking this signaling.213,389,439 Viral dsRNA may be sensed in endosomes by Toll-like receptor 3 (TLR3) (2), which signals via the adapter molecule TIR domain–containing adapter-inducing interferon-β (TRIF), resulting in nuclear translocation of NF-κB and expression of IFN-β; NS3-NS4A protease cleaves TRIF, interfering with this response.377 IFN-β effects depend on the JAK-STAT signaling pathway, which is inhibited (3) by HCV core and NS5A proteins. Inflammatory responses including type I interferons activate host antiviral molecules including protein kinase (PKR) and 2′,5′-oligoadenylate synthetase (2,5-OAS), which are antagonized (4) by NS5A. Further details and additional innate responses and evasive mechanisms are described in the text and Table 27.1. |
Dendritic cells are critical for orchestration of both innate and adaptive immune responses. Toll-like receptor 7 (TLR7) is expressed by plasmacytoid dendritic cells (pDCs), and pDCs produce type I interferon when co-cultured with Huh-7 cells containing replicating HCV RNA606; however, data regarding the effect of HCV infection on pDCs in vivo are less clear.153,306 HCV may interfere with NK cell activation of dendritic cells,297 and there is some evidence that HCV infection may be associated with impaired peripheral dendritic cell function.40,307 This impairment may explain the collapse of the cellular immune response during the transition from acute to chronic infection.133,564
Cellular Immune Response
There is strong evidence that both CD4 and CD8 T-cell responses are critical for control of HCV infection, but there is limited understanding of failure of these responses leading to chronicity. HCV-specific T cells develop rapidly during acute infection and are then detectable for years in blood and liver in individuals after clearance of infection. During chronic infection, stronger polyclonal CD8 T-cell responses in the liver and circulation have been associated with lower circulating HCV loads,465,525 though CD8 T-cell responses to HCV epitopes are only detectable ex vivo in half of human immunodeficiency virus (HIV)-negative individuals chronically infected with HCV, whether obtained from peripheral blood or liver.327,340
Induction of T-Cell Responses
HCV-specific T cells typically become detectable in the blood 5 to 10 weeks after infection.127,371,573,615,643 In experimentally infected chimpanzees, intrahepatic T-cell responses appear another 4 to 8 weeks later.127,615 In intravenous drug users, there is significant overlap in the number of T-cell epitopes targeted during acute infection in individuals with subsequent resolving versus persistent infection outcomes.133,564 Even at the height of the response, HCV-specific CD8 T cells rarely target more than 10 epitopes, regardless of outcome, with little evidence of immunodominance.133,340 While acute phase CD4 and CD8 T-cell responses are usually detectable regardless of outcome, in individuals who progress to chronic infection, they disappear rapidly and may be less vigorous.133,160,259,308,441,564,616,644 HCV-specific T cells produce IL-2 and IFN-γ in individuals who go on to clear infection, and acquisition of full effector function may be a key factor leading to viral control in individuals with spontaneous clearance. In individuals who clear infection, functional effector CD8 T cells peak in the blood just after the initial drop in viremia, usually about 8 to 12 weeks after infection.127,251,368,371,615,616
Anti-HCV T-cell responses are not focused on one viral protein or genomic region, and there is little evidence of immunodominance in general133,340,564 when compared with responses to HIV and influenza.10,379 Comprehensive analyses of CD4 and CD8 T-cell responses in persons with acute infection, using overlapping peptides composing the HCV polyprotein, have revealed widely dispersed epitopes (Fig. 27.4).133,368,564 In persons with certain uncommon alleles, such as HLA-B*27 and HLA-B*57, immunodominant responses to functionally
constrained epitopes have been described,469,470,477,549 whereas each person with the more common allele HLA-A*02 targets a few of the dozens of epitopes restricted by that allele.673
constrained epitopes have been described,469,470,477,549 whereas each person with the more common allele HLA-A*02 targets a few of the dozens of epitopes restricted by that allele.673
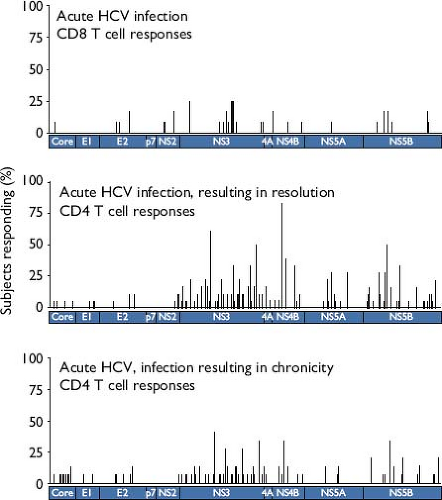 Figure 27.4. Anti–hepatitis C virus (HCV) T-cell epitopes are widely dispersed across the polyprotein during acute infection. The per-person frequency at which responses were detected using ELISpot is indicated for epitopes centered at positions indicated along the polyprotein. CD8 T-cell responses in 12 persons with acute HCV infection are indicated in the upper panel (adapted from two reports, with many outcomes unknown due to early interferon treatment133,368), while the middle and lower panels depict CD4 T-cell responses from 18 subjects with resolving acute HCV and 13 subjects who progressed to chronicity, respectively.564 (Middle and lower panels © 2012, Schulze zur Wiesch et al. Originally published in The Journal of Experimental Medicine. 209:61–75.) |
ROLE OF T CELLS IN CLEARANCE OF HCV
A central role for T cells in clearance of HCV was illustrated by studies in which T cells (CD4 or CD8) were depleted in chimpanzees in the context of acute HCV infection.251,573 Depletion of CD8 T cells, from an animal that had rapidly and spontaneously cleared HCV infection twice, led to prolonged viremia after reinfection, with control occurring when CD8 T cells returned.573 Depletion of CD4 T cells had a somewhat different effect, with widely fluctuating levels of viremia associated with progressive escape mutations at epitopes targeted by previously primed CD8 T-cell epitope responses.251,573
In humans, spontaneous clearance of HCV has been associated with expression of certain major histocompatibility complex (MHC) class I molecules, with various studies showing association with the presence of HLA-B*57, HLA-B*27, HLA-A*11, HLA-A*03, or HLA-Cw*01, and the absence of HLA-Cw*04.326,349,420,617 Some of the differences in observed associations may be due to variation in predominant circulating HCV genotypes. The mechanism by which these particular alleles favor clearance of viremia is generally unknown, though there is evidence that some protective alleles bind and present to T cells epitopes that are particularly immunogenic and/or functionally conserved,470 whereas risk alleles may be ligands for inhibitory receptors of NK cells.617 The latter mechanism is supported by evidence that polymorphisms in NK receptors may also play an important role in HCV clearance.325
HCV clearance has also been associated with MHC class II genes, particularly DQB1*0301 and HLA-DRB1*1101.269,617 In a study using peptides spanning the HCV polyprotein, individuals with resolved infections targeted an average of 10 MHC class II epitopes (range 3 to 28), whereas individuals with chronic infection targeted an average of 1 epitope (range 0 to 8). Epitopes most frequently recognized were in core and nonstructural proteins, which may reflect differences in protein processing or mismatch in other regions between the circulating virus and library peptides.150,235,489,560,565 In chimpanzees, a subset of dominant CD4 T-cell epitopes were targeted prior to clearance of infection, and subdominant populations were detected only after clearance.269
Memory Responses
Anti-HCV CD8 T-cell responses have been detected in individuals who have been exposed to HCV but have not seroconverted.81,340 Studies in chimpanzees have also shown that CD4 and CD8 memory T-cell responses are important for protection against reinfection with HCV.48,189,251,400,458,507,573,681 Control of
a second infection is associated with rapid expansion of memory CD4 and CD8 cells.363 Expansion of HCV-specific T cells was observed 2 to 3 weeks after reinfection compared to 10 to 12 weeks after initial exposure to virus. Depletion of CD8 T cells led to prolonged viremia after reinfection, and depletion of CD4 T cells also led to impaired control, despite the presence of previously primed HCV-specific CD8 T cells.251,573 Even more importantly, a recent study demonstrated that reinfected humans tend to develop broader T-cell responses and lower peak viremia and are more likely to spontaneously clear their second HCV infection.478
a second infection is associated with rapid expansion of memory CD4 and CD8 cells.363 Expansion of HCV-specific T cells was observed 2 to 3 weeks after reinfection compared to 10 to 12 weeks after initial exposure to virus. Depletion of CD8 T cells led to prolonged viremia after reinfection, and depletion of CD4 T cells also led to impaired control, despite the presence of previously primed HCV-specific CD8 T cells.251,573 Even more importantly, a recent study demonstrated that reinfected humans tend to develop broader T-cell responses and lower peak viremia and are more likely to spontaneously clear their second HCV infection.478
T-Cell Responses During Chronic Viremia
Despite T-cell responses, most HCV-infected individuals remain persistently infected. In individuals who progress to chronic infection, HCV-specific CD8 T cells become dysfunctional, possibly due to CD4 T-cell dysfunction.29,258,390,564,675 HCV-specific CD8 T cells obtained from peripheral blood during chronic infection show poor ex vivo proliferation and IFN-gamma production, low intracellular stores of perforin, and decreased ability to lyse target cells.29,258,390,675 These functions are not consistently restored after successful treatment.442
Anti-HCV T-cell responses have been studied primarily in peripheral blood; due to compartmentalization of the T-cell response, such studies are likely to underestimate the breadth and magnitude of intrahepatic responses. Approximately a third of chronically infected individuals have intrahepatic anti-HCV T-cell responses that can be expanded ex vivo.339,687 Intrahepatic anti-HCV T-cell responses may be associated with lower serum HCV RNA levels, higher degrees of hepatic inflammation, and higher rates of response to interferon-based treatment465,466; thus, responses that contribute to clearance may, if unsuccessful, contribute to injury.102 In spite of quantitative differences in number and breadth and some differences in phenotype53 of T-cell responses, there are many similarities.585 Such inferences from PBMCs have been supported by indirect correlations such as viral escape substitutions in epitopes that were detected in assays of PBMCs.134,629
Many HCV-specific CD8 T cells express the counterregulatory molecule PD-1.104,258,541,675 Studies in murine models have shown that PD-1 binds to programmed death ligands 1 and 2 (PD-L1 and PD-L2), and ligation leads to dephosphorylation of signaling molecules downstream of the T-cell receptor (TCR), decreasing T-cell sensitivity to stimulation.108,366,476 Blockade by anti–PD-L1 antibodies leads to increased proliferation of both CD8 and CD4 T cells directed against HCV.312,455,495,511,524,637 Interestingly, levels of PD-1 expression in acute infection do not appear to correlate with outcome of infection. In model systems, direct activation of CD8 T cells by hepatic parenchymal cells, without help from CD4 T cells, may result in impaired CD8 T cells that express high levels of PD-1.692 Some recent studies have suggested that high levels of PD-1 may indicate a high level of immune activation, but not necessarily T-cell exhaustion.176
T-cell immunoglobulin and mucin domain–containing molecule-3 (TIM-3) may also play a role in modulation of HCV-specific T-cell responses. Expression of both TIM-3 and PD-1 on CD8 cells during acute infection was associated with persistence, and like PD-1, blockade of TIM-3 increased proliferation of HCV-specific CD8 T cells.421 Recent studies have suggested that 2B4 (CD244), another inhibitory molecule on exhausted T cells in the lymphocytic choriomeningitis virus model of chronic infection, may also play a role in modulating HCV-specific CD8 T cells. HCV-specific CD8 T cells show increased expression of 2B4, and 2B4 stimulation reduced the increase in proliferation of HCV-specific T cells usually seen after PD-1 blockade.561
Interaction with other immune cells likely also modulates antiviral T-cell activity. Liver-infiltrating CD8 T cells may have decreased expression of the co-stimulatory molecule CD86.301,355 Regulatory T cells (Treg)298,376 may modulate HCV-specific T-cell activity, and increased early IL-10 production during chronic HCV infection may drive CD4 T cells to become Treg.209 These CD4CD25high T cells are enriched in peripheral blood during chronic HCV infection and may infiltrate the chronically infected liver, potentially protecting it from injury.69,71,92,590,678
HCV Evasion of T-Cell Response
In addition to innate and adaptive host responses that are functionally inadequate for clearing infection, there appears to be selection for HCV mutations that enable response evasion while maintaining adequate replicative fitness to sustain infection. T-cell recognition of HCV is reduced by amino acid replacements that occur in vivo,103,134,628 and such changes have been correlated with persistence.134,181 Multiple cross-sectional studies have shown enrichment of amino acid changes in predicted or confirmed cytotoxic T-lymphocyte (CTL) epitopes among chronically infected individuals with corresponding HLA types.207,232,365,521,540,629 Observed mechanisms of reduced recognition during HCV infection (Fig. 27.5) include changes adjacent to epitopes that result in impaired processing for MHC class I presentation,329,566 changes in anchor residues that reduce binding affinity for MHC class I,134,628 and mutations that affect TCR contact residues.686
Amino acid replacements have potential fitness costs that may balance the fitness gain associated with escaping a T-cell response. These substitutions could disrupt functions of HCV proteins or RNA genomic elements299,642 or create neoantigen. Loss of protein function in this context has been observed, as have compensatory changes that appear to restore function470,477,539,549; such compensatory changes must be considered when analyzing HCV evolution in the context of an immune response and may be detected in searches for long-range interactions across the genome.165 Neoantigen could be recognized by other T cells, as has been observed for HIV15; however, this does not appear to be common in HCV. Lack of recognition of the neoantigen produced by escape substitutions could be due to repertoire fixation,686 analogous to the phenomenon of original antigenic sin observed in repeated infections.334 A novel additional mechanism, the exploitation by HCV of a hole in the human T-cell repertoire686 such that the mutant form is not recognized at all, may not be surprising for a virus that has been adapting to humans for a very long time.576 Consistent with that mechanism, CD8 T cells specific for epitopes that have escape substitutions, though low in frequency, may express high levels of the memory marker CD127 similar to those found in persons with spontaneous clearance.53,311 Escape mutations are not generally observed in epitopes targeted by CD4 T cells220,564; this is not surprising given the indirect role of CD4 T cells in antiviral responses, because a viral variant with an escape substitution in a CD4
T-cell epitope might not have a survival advantage relative to nearby variants lacking such a substitution.
T-cell epitope might not have a survival advantage relative to nearby variants lacking such a substitution.
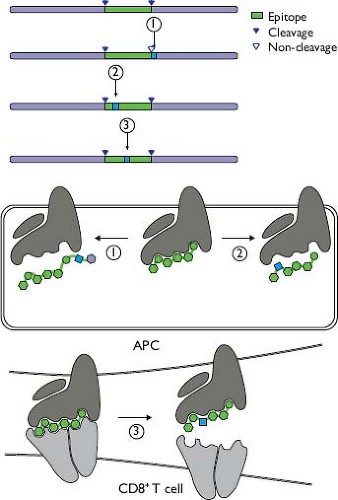 Figure 27.5. Evolutionary escape from CD8 T-cell response. Mechanisms for escape from recognition from CD8 T cells that have been demonstrated for hepatitis C virus (HCV) include (1) change in a residue affecting proteasomal processing, resulting in C-terminal extensions that cannot be trimmed in the endoplasmic reticulum (ER); (2) change in an anchor residue, resulting in loss of affinity for major histocompatibility complex (MHC) class I; and (3) change in a T-cell receptor (TCR) contact residue. |
Humoral Immune Response
There is increasing evidence for the importance of the humoral immune response in control and clearance of HCV infection. Antibodies against HCV are not absolutely required for clearance of infection, as demonstrated in individuals with congenital agammaglobulinemia.4 In individuals with normal humoral immunity, however, binding antibody responses against structural and nonstructural HCV proteins are detectable within weeks to months of infection.110,467 Using autologous virus, neutralizing antibodies can sometimes be detected within this same time period.170,494
Envelope proteins E1 and E2 are type I transmembrane proteins that exist on infectious virions as a cross-linked heterodimer.660 The structure of E1 is unknown, but recent mutational and computational analysis has produced a draft structure of E2342 that is supported by functional and antibody mapping studies.11,321 HCV envelope binds directly to CD81, and mutational analysis suggests that this binding involves E2 residues in domain I (Fig. 27.6). Although anti-E1 and anti-E2 antibodies can be detected in persons with acute and chronic HCV infection,467 almost all neutralizing antibodies target E2 and inhibit entry at a postattachment step.272,544,659
There are numerous direct and indirect lines of evidence to suggest that antibodies against HCV decrease the risk of infection after exposure (see also Passive Immunization section, later). First, there were fewer HCV infections in liver transplant recipients who received immune globulin prior to 1990, the first year that immune globulin preparations were screened for HCV seroreactivity.199 Second, immune globulin in a randomized controlled study reduced the incidence of sexual HCV transmission.497 In addition, inoculum-specific neutralizing antibodies directed at the hypervariable region 1 (HVR-1) reduced infection in chimpanzees; however, HVR-1 variability resulted in breakthrough infection.192,194,680 More recently, prophylactic treatment with a broadly neutralizing monoclonal antibody protected against HCV challenge in a human liver chimeric mouse model.369
Neutralizing antibodies may also play a role in modulating ongoing HCV infection. Individuals with primary hypogammaglobulinemia had more rapid progression of disease and poorer response to interferon treatment,63 and individuals with humoral immune defects have fewer amino acid changes in E2.73,231
Evasion of the Neutralizing Antibody Response
The development of pseudotyped lentiviruses for measuring neutralizing antibodies to HCV46,385,532 has revealed that chronic infection is associated with significant titers of neutralizing antibodies,385 and a case report of a chronically infected individual showed continuous rounds of escape from neutralizing antibody.666 During acute infection, neutralizing antibodies drive sequence evolution, suggesting that they have an impact on fitness in vivo, and early appearance of HVR-1–specific and/or neutralizing antibodies is associated with an increased likelihood of spontaneous viral clearance.13,170,494,708,709 Individuals reinfected after clearance of infection have lower second peak viremia, increased likelihood of clearance of the second infection, and a more broadly neutralizing antibody response.424,478
The enormous diversity of the virus and tolerance of amino acid changes in E1E2 contribute to escape from this host response (Table 27.1). As with HIV-1, heavy glycosylation of E1 and E2 may provide a “glycan shield” that obscures conserved, functionally important domains (Fig. 27.6).272 During the transition from acute to chronic infection, acceleration of evolution in HCV envelope genes is likely to be due to the appearance of neutralizing antibodies.382 Because they are immunodominant targets of humoral immunity while also tolerating extensive nonsynonymous variation, HVR-1 and, to a lesser degree, HVR-2 and the intergenotypic variable region (igVR) contribute to neutralizing antibody escape (Fig. 27.6, marker 2).42,47,193,523,659 Antibodies targeting the HVR-1 are common in vivo, but, given the variability of the region, they tend to be strain specific. In a study of neutralizing antibody development in acute HCV infection, neutralizing antibody escape mutations were mapped to the HVR-1.170 Most broadly neutralizing antibodies bind to the E2-CD81-binding site.80,320,369,479,544,659 Some of these epitopes are linear, while others are conformational in nature. While the
CD81-binding site is highly conserved, many of these broadly neutralizing antibodies can induce escape mutations in HCV cell culture, suggesting that replication-competent escape variants may also exist in vivo.320
CD81-binding site is highly conserved, many of these broadly neutralizing antibodies can induce escape mutations in HCV cell culture, suggesting that replication-competent escape variants may also exist in vivo.320
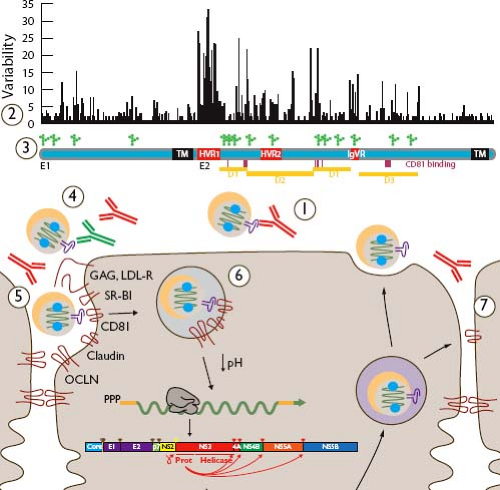 Figure 27.6. Evasion of anti–hepatitis C virus (HCV) antibody-mediated responses. Neutralization of HCV by antibodies can block infection of the cell (1). Binding of neutralizing antibodies (red) can be evaded by variability in the envelope proteins (2) illustrated here in a plot of Wu-Kabat amino acid variability691 and by dense glycosylation at approximately 15 positions (3). Nonneutralizing antibodies (4, green) and lipoproteins (5) may hinder neutralizing antibody binding to HCV envelope glycoproteins, and delayed exposure of conserved domains until late in the entry process may prevent their recognition on free virions (6). Cell-to-cell transfer of virions is resistant to neutralizing antibodies in vitro, suggesting an additional mode of escape for local spread of infection (7). Along the envelope gene map are indicated transmembrane regions (TMs), hypervariable regions (HVRs), intergenotypic variable regions (IgVRs), putative tertiary domains (D1–D3), and CD81-binding residues (vertical lines). |
Several other mechanisms in addition to antigenic variability may contribute to HCV resistance to antibody-mediated neutralization. It appears that the CD81-binding site may be partially shielded from neutralizing antibodies by the HVR-1 and by N-linked glycosylation (Fig. 27.6, marker 3).42,187,272 Lipid shielding of the virion may also play a role, because studies have suggested that some neutralization epitopes are less accessible in particles associated with very low-density lipoproteins (VLDLs) or high-density lipoproteins (HDLs) (Fig. 27.6, marker 5).90,665 Additional mechanisms of evasion of neutralizing antibodies may include nonneutralizing antibodies that bind E1E2 in a manner that interferes with binding of neutralizing antibodies, and postendocytic conformational changes in E1E2 revealing conserved determinants of entry (Figure 27.6, markers 4 and 6, respectively).545,705 In vitro demonstration of direct cell-to-cell spread of HCV, resistant to most neutralizing antibodies, suggests an additional potential mechanism for immune evasion (Fig. 27.6, marker 7)79,630,685; however, strong evidence for antibody-driven HCV evolution170,382 suggests that neutralizing antibodies apply significant selection pressure in vivo.
Release from Host and Transmission
HCV RNA has been detected in small amounts in a variety of secreted body fluids including saliva, tears, and urine,111,204,430 but transmission primarily results from percutaneous exposure
to blood or rarely from mucosal exposure to genital secretions, as discussed in the section on Transmission later.
to blood or rarely from mucosal exposure to genital secretions, as discussed in the section on Transmission later.
Table 27.1 HCV Proteins Contributing to Persistence | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Virulence
In spite of their extreme heterogeneity, genetic variants of HCV (genotypes and subtypes) have remarkably similar clinical manifestations; for example, there have been no reported outbreaks of acute fulminant hepatitis, and persons infected in common-source outbreaks have displayed a wide range of outcomes.151,322 Moreover, efforts to identify viral determinants of fibrosis progression have not revealed consistent associations. Response to treatment is strongly affected by viral genotype, with genotype 1 being relatively refractory to interferon-based therapy as discussed in the Treatment section.
HCV subverts hepatic lipoprotein metabolism (see Virus Assembly section for HCV in Chapter 25), so it is not surprising that steatosis and insulin resistance are common features in HCV infection (see Clinical Features, later).281 Multiple studies have found a significantly stronger association between genotype 3 HCV infection and steatosis151 than for other HCV genotypes; this association may be related to genotype-specific disruption of lipid biosynthesis pathways.118 Steatosis is also strongly associated with visceral obesity.5 Geographic variation in host factors as well as viral genetic types (see Genetic Diversity, later) could confound association of viral genotype with some manifestations.
During initial HCV infection, the peak of hepatic injury (illustrated by the peak in alanine aminotransferase [ALT] in Fig. 27.7) follows, rather than coinciding with, the peak of viremia.102 This consistent observation, combined with observations of liver pathology and cell culture, suggests that lysis of HCV-infected cells results primarily from the host antiviral immune response.102,115 The association of chronic infection with progressive liver disease and hepatocellular carcinoma is discussed in the Clinical Features section later.
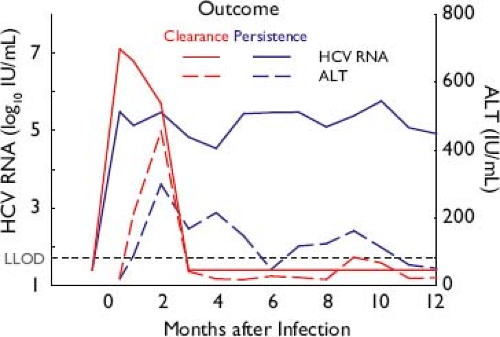 Figure 27.7. Patterns of acute hepatitis C virus (HCV) infection, resulting in spontaneous resolution or chronicity. The initial peak of viremia is followed by a peak in alanine aminotransferase (ALT) indicating cytolysis, temporally associated with the detection of cell-mediated responses to HCV that do not differ qualitatively by outcome when measured ex vivo.133 Initial level of viremia is higher in those who clear compared with those who later progress to chronic infection.383 |
Persistence
When untreated, acute infection with HCV may spontaneously resolve or persist as chronic HCV infection (Fig. 27.7, discussed further in Clinical Features, later). Spontaneous clearance of HCV occurs in approximately one-third of untreated infections. This resolution occurs in the first 2 years and is generally complete, with no residual viral RNA in serum or liver.135,424 Persistent HCV infection occurs in two-thirds of infected persons, is attributable to the evasion mechanisms discussed earlier, and is associated with persistent viremia at a level of 5 to 7 log10 IU/mL in 90% of individuals.627 Spontaneous resolution during chronicity is rare.9 Because persistence is marked by high-level viremia and constant evolution in immunocompetent hosts, HCV appears to persist dynamically and there is no evidence of a stable, latent reservoir or archive of previously dominant variants. See Clinical Features of chronic infection, later.
Epidemiology
Morbidity/Mortality
The morbidity and mortality that is most clearly caused by HCV is liver failure and/or liver cancer as a result of chronic infection. In the Unites States, the Centers for Disease Control and Prevention estimates that chronic HCV infection contributes to 15,000 deaths per year, is the leading cause of liver failure leading to transplantation, and in 2007 superseded HIV as a cause of death (Fig. 27.8).184,394,684 HCV-related liver morbidity and mortality increase with older age and greater duration of HCV infection and are expected to rise in the coming decades. Using multistate disease models, one group recently estimated that HCV-related liver failure and cancer will continue to increase until 2020–2023 without widespread treatment.147 Liver-related mortality is predicted to rise from 146,667 cases in 2000–2009 to 254,550 cases in 2010–2019 and 283,378 in 2020–2029. Reliable worldwide estimates of HCV-related mortality are not available.
HCV-infected persons are at increased risk of more than liver failure. In one study, 10,259 HCV antibody–positive blood donors were compared to donors matched by year of donation, age, gender, and zip code and followed for a mean of 7.7 years.261 Compared to the HCV-uninfected donors, the risk of death was 3.13-fold higher in HCV-infected donors, who were more likely to die of not just liver-related but also drug/alcohol-related events, trauma/suicide, and cardiovascular causes. Persons with HCV infection are also at much higher risk of some medical conditions such as mixed cryoglobulinemic vasculitis and porphyria cutanea tarda (see Clinical Features later).6,154 The degree to which HCV infection contributes to less specific medical syndromes such as chronic fatigue/arthritis or mental illness is more difficult to establish.
Origin and Spread of Epidemics
HCV infection spread during the 20th century, strongly correlated with expanded production of syringes and their worldwide use for both conventional medicine and illicit drugs.172,215,397 Drucker and co-workers172 estimated that global syringe production rose from 100,000 per year in 1920 to 7.5 million per year by 1952. Widespread use of percutaneous injections for medicinal (and then illicit) drug use antedated appreciation of blood-borne transmission of infection
and spread HCV throughout the world. This trend explains the 5- to 20-fold increased HCV prevalence rates in certain regions where unsafe injections were widespread and among injection drug users (IDUs) (see Global Burden, Incidence, and Prevalence).215 Transfusions of blood products also contributed to HCV infection, especially when donors were paid and no measures were in place to screen blood for infection.399
and spread HCV throughout the world. This trend explains the 5- to 20-fold increased HCV prevalence rates in certain regions where unsafe injections were widespread and among injection drug users (IDUs) (see Global Burden, Incidence, and Prevalence).215 Transfusions of blood products also contributed to HCV infection, especially when donors were paid and no measures were in place to screen blood for infection.399
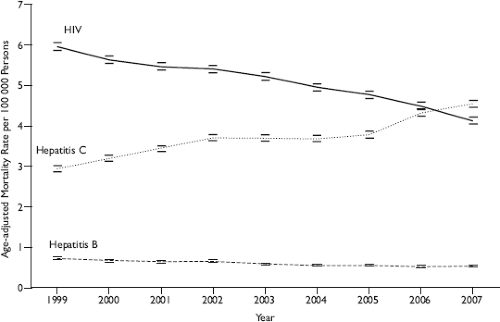 Figure 27.8. Annual age-adjusted mortality rates from hepatitis B and hepatitis C virus and human immunodeficiency virus (HIV) infections listed as causes of death in the United States between 1999 and 2007. Because a decedent can have multiple causes of death, a record listing more than one type of infection was counted for each type of infection. (From Ly KN, Xing J, Klevens RM, et al. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Intern Med 2012;156:271–278.) |
Prior to the 20th century, HCV infection was probably sustained by percutaneous practices such as scarification rituals and circumcision. This conjecture is supported by evidence of transmission by such practices where they still occur and by molecular clock estimates derived from analyses of worldwide HCV RNA sequences (see Genetic Diversity).332,509
Prevalence and Seroepidemiology
Transmission
HCV can be transmitted by percutaneous exposure to contaminated blood, from a mother to her infant, and by sexual intercourse. There is no evidence HCV can penetrate intact skin, but permucosal transmission has occurred when blood was splashed into eyes.
The likelihood that HCV transmission will occur is directly related to the inoculum and the exposure type. Blood is the usual inoculum, typically contains 5 to 7 log10 copies of HCV RNA per mL, and rarely transmits HCV when viremia is not detected.166,627 Although HCV RNA has been amplified from most other body fluids, it is not clear to what extent other body fluids harbor infectious virions.204,430,671
Percutaneous exposures such as unsafe medical procedures and injection drug use are the usual routes of HCV transmission worldwide. HCV transmission almost always occurs following very large percutaneous inocula, such as transfusion of a contaminated unit of blood.182,669 However, even very small (less than 10 μL) blood inocula may contain infectious virions to establish infection in a recipient if injected percutaneously, and nosocomial exposure may occur if strict universal precautions are not observed.14,293 Blood spiked with an HCV reporter virus was loaded into syringes and viability was recovered from 71% of tuberculin syringes kept at 22°C for 7 days.480 This finding correlates with studies of health care personnel with accidental needlestick exposures in whom transmission occurs in 1% to 2% overall and more often from hollow-bore needles, which contain a larger inoculum than a solid-bore needle.330,528 Repeated small-volume exposures to HCV explain the high rates of HCV among injection drug users (see later).
Nonmedical percutaneous exposures such as body piercing and tattooing are plausible risks and epidemiologically linked to HCV prevalence in many countries, though they are likely to be confounded by other risky behaviors in some populations.276,336,395,411,426,438,462,506,601
The frequency by which HCV is transmitted sexually is controversial. On the one hand, long-term monogamous partners of individuals with HCV infection almost never acquire HCV.30 In one study, 895 monogamous sexual partners of persons with chronic HCV infection were followed for over 8,000 person-years, and there were no instances of sexual HCV transmission, despite unprotected intercourse occurring an average of 1.8 times per week.654 On the other hand, HCV infection occurs in persons acknowledging high-risk sexual practices (and no other exposure),650 and there are multiple outbreaks among HIV-infected men who have high-risk sexual exposures with other men.145,648 One speculation is that, as with HIV, the risk of sexual HCV transmission is greater during the acute phase of infection when viremia peaks and prior to formation of neutralizing antibodies. In addition, anal intercourse may cause mucosal tears that promote HCV transmission. Permucosal spread of HCV may also explain the association of HCV infection with intranasal use of cocaine.125
Global Burden, Incidence, and Prevalence
There are an estimated 185 million HCV-infected persons in the world, or 2.2% of the human population.688,689 There are marked differences in HCV prevalence between regions (Fig. 27.9) and, even within countries, between age and risk groups. Egypt appears to have the highest HCV prevalence, which is as high as 50% in persons born before 1960.215 The history of HCV infection in Egypt is exemplary of global
transmission patterns. From the 1950s to the 1980s, the Egyptian Ministry of Health embarked on a campaign to eradicate schistosomiasis infection by intravenously administering tartar emetic to millions of citizens.589 The effort, commended at the time as a public health model, occurred before there was widespread appreciation for blood-borne transmission of infectious agents. HCV was transmitted extensively because of the widespread reuse of insufficiently cleaned injection equipment.215 Consequently, the prevalence of HCV infection can exceed 50% in persons alive during that campaign while being 1% to 2% in those born after. In addition, more than 90% of HCV infections in Egypt are genotype 4, which make up less than 10% of genotypes in most other regions of the world.520
transmission patterns. From the 1950s to the 1980s, the Egyptian Ministry of Health embarked on a campaign to eradicate schistosomiasis infection by intravenously administering tartar emetic to millions of citizens.589 The effort, commended at the time as a public health model, occurred before there was widespread appreciation for blood-borne transmission of infectious agents. HCV was transmitted extensively because of the widespread reuse of insufficiently cleaned injection equipment.215 Consequently, the prevalence of HCV infection can exceed 50% in persons alive during that campaign while being 1% to 2% in those born after. In addition, more than 90% of HCV infections in Egypt are genotype 4, which make up less than 10% of genotypes in most other regions of the world.520
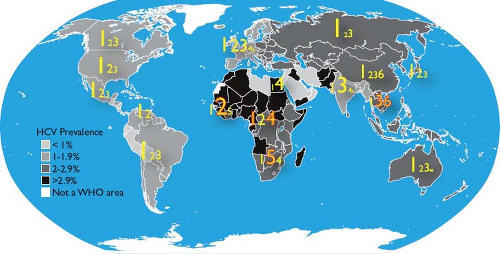 Figure 27.9. Map depicting geographic variation in the relative prevalence of hepatitis C virus (HCV) infection and genotypes. Shading of country indicates prevalence. Yellow numerals indicate prevalent HCV genotypes in different regions, with font size corresponding to relative genotype prevalence within each region; red outlining indicates genotypes with greatest intraregional diversity. HCV prevalence is highest, and genetically most diverse, in Africa. Genotype 1 is prevalent worldwide, whereas genotypes 4 and 5 are almost exclusively found in north-central and southern Africa, respectively. Genotype 7 (provisionally assigned and not depicted) has been reported very rarely in persons with epidemiologic links to central Africa. (HCV prevalence estimates adapted from World Health Organization. Global burden of disease [GBD] for hepatitis C. J Clin Pharmacol 2004;44:20–29, with permission. Relative genotype prevalence within each region based on sources cited in the text.) |
There is molecular and epidemiologic evidence of similar transmission patterns elsewhere. In studies modeling HCV sequences, Tanaka and co-workers610 estimated rapid expansion of HCV-1b in Japan in the 1920s, in Europe in the 1940s, and in the United States (HCV-1a) in the 1960s.610 Population data from southern Italy show that the HCV prevalence is 1.3% in subjects younger than 30 years and 33% in those older than 60 years of age; the odds of infection were doubled in those who recalled reusable glass syringe use.260 Thus, as mentioned earlier (in Origin and Spread), it appears HCV was widely transmitted worldwide during the 1900s due to stepped-up production of syringes and their worldwide use both for conventional and illicit drugs.172,397,509
The overall prevalence of HCV infection in Europe is 1% to 2%. However, country-specific rates vary considerably, with the lowest HCV prevalence (less than 0.5%) reported from Sweden, Germany, and the Netherlands while prevalence rates of 2% to 3% have been reported in some Mediterranean countries.271 There is less information on incident HCV infection. Although HCV surveillance is required in European countries, new infection information is restricted to symptomatic events (which are the minority of HCV infections) and thus data on trends and comparisons across regions are crude. Overall, because procedures to screen blood donations were implemented, most new infections in Europe are linked to injection drug use or recent health care exposure.408
United States Prevalence and Incidence
In the United States, an estimated 3 million persons have chronic HCV infection. Several key epidemiologic trends explain the incidence and prevalence of HCV infection. As in other parts of the world, unsafe medical injections probably contributed to an early expansion of HCV prevalence following World War II. Transfusion of blood and blood products caused new HCV infections until 1992, when the most effective screening measures were adopted. However, it was the epidemic of injection drug use from the 1950s to the 1980s that caused most HCV infections in the United States. Whereas there were probably fewer than 500,000 persons with chronic HCV infection in the early 1950s, by the mid-1990s there were an estimated 3.5 million persons with chronic HCV infection and another 1 million to 1.5 million who had recovered.31 Much of that epidemic spread was due to injection drug use.
Not only does injection drug use cause most HCV infections in the United States, but also most injection drug users have been HCV infected. HCV infection generally occurs within the first years of initiating the illicit use of injected drugs with annual incidence rates of 10% to 30%.135,228,266 In one cohort, 80% of subjects acknowledging 2 or more years of injection use were infected with the virus, a prevalence that was higher than that of HIV or HBV infection.229,625 Early acquisition of HCV is probably related to the practice of older (infected) IDUs teaching new (uninfected) initiates by demonstrating first on themselves and then on the new initiate.228 Although sharing of needles and syringes causes some HCV transmission, Hagan and co-workers265 estimated that 37% of new cases were due to sharing of other equipment.
After peaking in the 1980s, HCV incidence has dropped markedly in the United States.98 Elimination of transfusion-related transmission contributed to the reduction in incidence. However, most of the decline is attributed to a reduction of HCV due to injection drug use that is not fully explained.19 However, because HCV serology remains positive in most instances even when viremia is cleared, the 20-year surge in HCV incidence among persons born between 1945 and 1964 remains serologically evident.
The best data on HCV prevalence in the Unites States come from the serial National Health and Nutrition Examination Surveys (NHANES).32 By testing blood collected from a subset of persons representing households in the United States around 1990, it was estimated that 4.1 million individuals had been infected with HCV, or 1.6% of the general population. Approximately two-thirds of those infected were born between 1945 and 1965. The survey was repeated 10 years later and showed the same number of HCV-infected individuals in the same age cohort that was 10 years older. Omitted from this survey were nearly 2 million incarcerated persons in the United States, who probably represent another 250,000 to 500,000 HCV-infected persons.39 The prevalence of HCV infection in the United States is also higher among racial minorities than in Caucasian Americans, and greater in African Americans than in Mexican Americans. In non-Hispanic Blacks 40 to 49 years of age, the HCV prevalence was 14% compared to a general population prevalence of 1.6%. HCV infection was detected in only 1% of those 20 to 29 years of age.
Genetic Diversity
Genetic variability is one of the most remarkable features of HCV, contributing to evasion of host immune responses and complicating development of diagnostics, therapeutics, and effective vaccines. HCV genomic sequences can be clustered phylogenetically into related groups (genotypes and subtypes), are distinct between individuals, and are highly variable within each infected individual at any given point in time (i.e., quasispecies diversity314,406) and over time (i.e., quasispecies divergence).
Global Diversity of HCV
Soon after HCV was discovered, it was apparent that genetically distinct strains were prevalent in different geographic areas. International standards for nomenclature established six major genotypes that are phylogenetically distinct, and subsequent reports have resulted in the proposal of a seventh genotype (Fig. 27.10).248,346,451,530,577 Within genotypes, phylogenetically
distinct clusters may be found that are called subtypes. Clinically, HCV genotypes and subtypes are very similar (see Virulence), though they vary in responsiveness to interferons (genotypes 1 and 4 are less responsive) and, in more complex ways, in susceptibility to direct-acting antiviral agents (see Treatment).
distinct clusters may be found that are called subtypes. Clinically, HCV genotypes and subtypes are very similar (see Virulence), though they vary in responsiveness to interferons (genotypes 1 and 4 are less responsive) and, in more complex ways, in susceptibility to direct-acting antiviral agents (see Treatment).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


