Coronaviridae
Paul S. Masters
Stanley Perlman
History
Coronaviruses are enveloped RNA viruses that are broadly distributed among humans, other mammals, and birds, causing acute and persistent infections. Members of this family were isolated as early as the 1930s as the causative agents of infectious bronchitis in chickens,25 transmissible gastroenteritis in pigs,142 and severe hepatitis and neurologic diseases in mice.75,186 It was not until the 1960s, however, that these viruses,27,32 as well as certain human respiratory viruses,8,391 were recognized to share characteristics that merited their being grouped together. Their most notable common feature, revealed by electron microscopy, was a fringe of widely spaced, club-shaped spikes that projected from the virion surface; these spikes were morphologically distinct from the surface projections of ortho- and paramyxoviruses. The halo of spikes was described as giving the viral particle the appearance of the solar corona, which prompted the name that was adopted for this new virus group.7
Over the next 40 years, coronaviruses were studied mainly because they cause economically significant respiratory and gastrointestinal diseases in domestic animals and because they provide unique models for viral pathogenesis. In humans, two coronaviruses were known to be responsible for a substantial fraction of common colds, particularly those that circulate in winter months. This situation changed dramatically with the emergence in 2002 of a devastating new human disease, severe acute respiratory syndrome (SARS), which was caused by a previously unknown coronavirus.143,288,440 Research stimulated by the SARS outbreak has led to great strides in our understanding of coronaviruses; by 2005, two additional, widespread human respiratory coronaviruses had been discovered.573,615 Moreover, the search for animal virus reservoirs has nearly tripled the total number of identified coronaviruses,255,394,616 although most of the recently discovered species are known only as genomic sequences and have yet to be isolated or propagated experimentally.
Classification
The coronaviruses are the largest group within the Nidovirales (Fig. 28.1), an order that comprises the families Coronaviridae, Arteriviridae,524 and Roniviridae.102 The arteriviruses, a small group of mammalian pathogens, are discussed in Chapter 29. The roniviruses, which infect shrimp, and a very recently isolated mosquito-borne virus,416,663 which is not yet classified, are currently the only members of the order having invertebrate hosts. Nidoviruses are membrane-enveloped, nonsegmented positive-strand RNA viruses that are set apart from other RNA viruses by certain distinctive characteristics.194 Their most significant common features are (a) an invariant general genomic organization, with a very large replicase gene upstream of the structural protein genes; (b) the expression of the replicase-transcriptase polyprotein by means of ribosomal frame shifting; (c) a collection of unique enzymatic activities contained within the replicase-transcriptase protein products; and (d) the expression of downstream genes via transcription of multiple
3′-nested subgenomic messenger RNAs (mRNAs). This last property has provided the name for the order, which comes from the Latin nido, for “nest”.157 It should be noted that the replicative similarities among the three nidovirus families are offset by marked differences in the numbers, types, and sizes of their structural proteins and great variation among the morphologies of their virions and nucleocapsids.
3′-nested subgenomic messenger RNAs (mRNAs). This last property has provided the name for the order, which comes from the Latin nido, for “nest”.157 It should be noted that the replicative similarities among the three nidovirus families are offset by marked differences in the numbers, types, and sizes of their structural proteins and great variation among the morphologies of their virions and nucleocapsids.
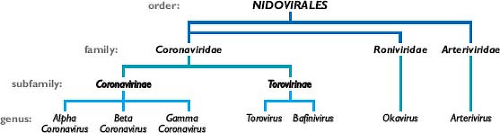 Figure 28.1. Taxonomy of the order Nidovirales. |
Coronaviruses are now classified as one of two subfamilies (Coronavirinae) in the family Coronaviridae (see Fig. 28.1). The other subfamily, Torovirinae, includes the toroviruses, which are pathogens of cattle, horses, and swine,523 and the bafiniviruses, whose sole member is the only nidovirus currently known to infect fish.505 This chapter will concentrate almost exclusively on the Coronavirinae.
Coronaviruses have long been sorted into three groups, originally on the basis of serologic relationships and, subsequently, on the basis of phylogenetic clustering.193,195 Following proposals that were recently ratified by the International Committee on Taxonomy of Viruses (ICTV),57 these groups—the alpha-, beta-, and gammacoronaviruses—have now been accorded the taxonomic status of genera (see Fig. 28.1). The ICTV classifications have also established rigorous criteria for coronavirus species definitions, in a manner consistent with those used for other viral families. As a consequence, some viruses previously considered to be separate species are currently recognized as a single species—for example, the viruses now grouped within alphacoronavirus 1 or betacoronavirus 1 (Table 28.1). Additionally, the new classification criteria resolve any previous uncertainty about the taxonomic assignment of the virus that caused SARS (severe acute respiratory syndrome coronavirus [SARS-CoV]) as a betacoronavirus.153,197,374,473,483,521,534,535
Almost all alpha- and betacoronaviruses have mammalian hosts. In contrast, the gammacoronaviruses, with a single exception, have been isolated from avian hosts. Several of the viruses listed in Table 28.1 have been studied for decades, specifically those included in the species alphacoronavirus 1, betacoronavirus 1, murine coronavirus, and avian coronavirus. The focus on these viruses came about largely because they were amenable to isolation and growth in tissue culture. However, since 2004, molecular surveillance and genomics efforts initiated in the wake of the SARS epidemic have led to the discovery of a multitude of previously unknown coronaviruses that now constitute most members of this subfamily.616 Notably, most of the newly recognized species were identified in bats, which constitute one of the largest orders within the mammals. Diverse coronaviruses have been described from bats, principally in Asia but also in Africa, Europe, and North and South America. These viruses include likely predecessors of SARS-CoV308,332 but also four unique species of alphacoronaviruses and three species of betacoronaviruses. Birds have also proven to be a rich source of new viruses. Novel avian coronaviruses have been found to infect geese, pigeons, and ducks,255 and highly divergent coronaviruses recently identified in bulbuls, thrushes, and munias617 have the potential to define a fourth genus in the Coronavirinae. It has been proposed that bats and birds are ideally suited as reservoirs for the incubation and evolution of coronaviruses, owing to their common ability to fly and their propensity to roost and flock.616
Five of the viruses in Table 28.1 are associated with human disease. The most categorically harmful of these, SARS-CoV, which is discussed at length later in this chapter, does not currently infect the human population. The remaining four human coronaviruses (HCoVs), the alphacoronaviruses HCoV-229E and HCoV-NL63, and the betacoronaviruses HCoV-OC43 and HCoV-HKU1, typically cause common colds. Remarkably, HCoV-NL63 and HCoV-HKU1 were only discovered recently, in the post-SARS era,573,615 despite the fact that each has a worldwide prevalence and has been in circulation for a long time.461,618 Although generally associated with upper respiratory tract infections, the extant HCoVs can also cause lower respiratory tract infections and have more serious consequences in the young, the elderly, and immunocompromised individuals. In particular, HCoV-NL63 is strongly associated with childhood croup,574 and the most severe HCoV-HKU1, −OC43, and −229E infections are manifest in patients with other underlying illnesses.460
Virion Structure
Virus and Nucleocapsid
Virions of coronaviruses are roughly spherical and exhibit a moderate degree of pleomorphism. In the earlier literature, viral particles were reported to have average diameters of 80 to 120 nm but were far from uniform, with extreme sizes from 50 to 200 nm.389 The spikes of coronaviruses, typically described as club-like or petal-shaped, emerge from the virion surface as stalks with bulb-like distal termini. Some of the variation in particle size and shape was likely attributable to stresses exerted by virion purification or distortions introduced by negative staining of samples for electron microscopy. More recent studies, employing cryo-electron microscopy and cryo-electron tomography,21,30,413,415 have produced images (e.g., Fig. 28.2A) in which virion size and shape are far more regular, although still
pleomorphic. These studies, which examined a number of alpha- and betacoronaviruses, converge on mean particle diameters of 118 to 136 nm, including the contributions of the spikes, which project some 16 to 21 nm from the virion envelope.
pleomorphic. These studies, which examined a number of alpha- and betacoronaviruses, converge on mean particle diameters of 118 to 136 nm, including the contributions of the spikes, which project some 16 to 21 nm from the virion envelope.
Table 28.1 Classification of Coronaviruses | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Enclosed within the virion envelope is the nucleocapsid—a ribonucleoprotein that contains the viral genome. The structure of this component is relatively obscure in images of whole virions; however, its makeup has been partially displayed by electron micrographs of spontaneously disrupted virions or of virions solubilized with nonionic detergents.59,109,183,269,366 Such studies revealed another distinguishing characteristic of coronaviruses: They have helically symmetric nucleocapsids.
Helical symmetry is common for negative-strand RNA virus nucleocapsids, although it is highly unusual for positive-strand RNA animal viruses, almost all of which have icosahedral capsids. The best-resolved images of the coronavirus nucleocapsid, which were obtained with HCoV-229E, showed filamentous structures 9 to 13 nm in diameter, with 3- to 4-nm-wide central canals59; these filaments were thinner and less sharply segmented than paramyxovirus nucleocapsids. However, widely ranging and sometimes discrepant parameters have been reported for the nucleocapsids of other coronaviruses,378 varying with both the viral species and the method of preparation.109,183,269,366,476 Thus, further work is needed to clearly define the diameter, symmetry, length, and protein: RNA stoichiometry of this virion component in isolation. More recent coronavirus ultrastructural studies suggest that when packaged within the virion envelope, the helical nucleocapsid is quite flexible, forming coils and other structures that fold back on themselves.21,413
Helical symmetry is common for negative-strand RNA virus nucleocapsids, although it is highly unusual for positive-strand RNA animal viruses, almost all of which have icosahedral capsids. The best-resolved images of the coronavirus nucleocapsid, which were obtained with HCoV-229E, showed filamentous structures 9 to 13 nm in diameter, with 3- to 4-nm-wide central canals59; these filaments were thinner and less sharply segmented than paramyxovirus nucleocapsids. However, widely ranging and sometimes discrepant parameters have been reported for the nucleocapsids of other coronaviruses,378 varying with both the viral species and the method of preparation.109,183,269,366,476 Thus, further work is needed to clearly define the diameter, symmetry, length, and protein: RNA stoichiometry of this virion component in isolation. More recent coronavirus ultrastructural studies suggest that when packaged within the virion envelope, the helical nucleocapsid is quite flexible, forming coils and other structures that fold back on themselves.21,413
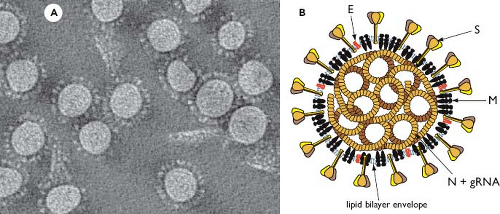 Figure 28.2. Coronavirus structure. A: Cryo-electron tomographic image of purified virions of mouse hepatitis virus (MHV), reconstructed as described in reference 415. (Courtesy of Benjamin Neuman, David Bhella, and Stanley Sawicki.) B: Schematic showing the major structural proteins of the coronavirus virion: S, spike protein; M, membrane protein; E, envelope protein; and N, nucleocapsid protein. |
Virion Structural Proteins
Coronaviruses contain a canonical set of four major structural proteins: the spike (S), membrane (M), and envelope (E) proteins, all of which are located in the membrane envelope, and the nucleocapsid (N) protein, which is found in the ribonucleoprotein core (see Fig. 28.2B).
The distinctive surface spikes of coronaviruses are composed of trimers of S molecules.30,129,529 S is a class I viral fusion protein41 that binds to host cell receptors and mediates the earliest steps of infection.95 In some cases, S protein can also induce cell–cell fusion late in infection. The S monomer is a transmembrane protein of 128 to 160 kDa, composed of a very large N-terminal ectodomain and a tiny C-terminal endodomain (Fig. 28.3). This protein is inserted, via a cleaved signal peptide,62 into the endoplasmic reticulum (ER), where it obtains N-linked glycosylation increasing its mass by some 40 kDa.224,487 Comprehensive mapping of glycosylation sites has not been carried out for any S protein; however, an analysis of the SARS-CoV S protein showed that at least half of its 23 candidate sites are glycosylated.287 The early steps of glycosylation occur co-translationally, and this modification assists monomer folding and proper oligomerization; terminal glycosylation is then completed subsequent to trimerization.129 S protein monomer folding is also accompanied by the formation of intramolecular disulfide bonds among a subset of the numerous cysteine residues of the ectodomain.425 The positions of S protein cysteines are well conserved in each coronavirus genus2,153; disulfide linkages have yet to be mapped.
In many beta- and gammacoronaviruses (e.g., mouse hepatitis virus [MHV], bovine coronavirus [BCoV], and infectious bronchitis virus [IBV]), the S protein is partially or completely cleaved by a furin-like host cell protease into two polypeptides, denoted S1 and S2, which are roughly equal in size. Correspondingly, in coronaviruses that do not have detectably cleaved mature S proteins, the N-terminal and C-terminal halves of the molecule are also designated S1 and S2, respectively. S protein cleavage occurs immediately downstream of a highly basic pentapeptide motif,2,62,361 and the extent of proteolysis correlates with the number of positively charged residues in the motif.36 The S1 domain is extremely variable, exhibiting very low homology across the three genera and often diverging extensively among different isolates of a single coronavirus.181,430,597 By contrast, the S2 domain is highly conserved.111 For those coronaviruses in which it occurs, S1-S2 cleavage is a late event in virion assembly and release from infected cells. For many other coronaviruses, an alternative type of S protein cleavage (S2′) takes place during the initiation of infection, activating the molecule for fusion.28 The differing functions of S1 and S2 and the role of proteolysis are discussed later (see the Viral Entry and Uncoating section).
A complete high-resolution structure has not yet been determined for any coronavirus S protein, although a cryo-electron microscopic reconstruction of the SARS-CoV S protein is available,30 and partial crystal structures have been solved for particular S protein domains.144,208,323,325,624,630,655
Nevertheless, all currently available structural and biochemical evidence accords well with an early proposal that S is functionally analogous to the influenza HA protein.111 In this model, the S1 domains of the S protein oligomer make up the bulbous, receptor-binding portion of the spike. The narrow stalk of the spike, distancing the bulb from the membrane, is a coiled-coil structure formed by association of heptad repeat regions (HR1 and HR2) of the S2 domains of monomers (see Fig. 28.3).
Nevertheless, all currently available structural and biochemical evidence accords well with an early proposal that S is functionally analogous to the influenza HA protein.111 In this model, the S1 domains of the S protein oligomer make up the bulbous, receptor-binding portion of the spike. The narrow stalk of the spike, distancing the bulb from the membrane, is a coiled-coil structure formed by association of heptad repeat regions (HR1 and HR2) of the S2 domains of monomers (see Fig. 28.3).
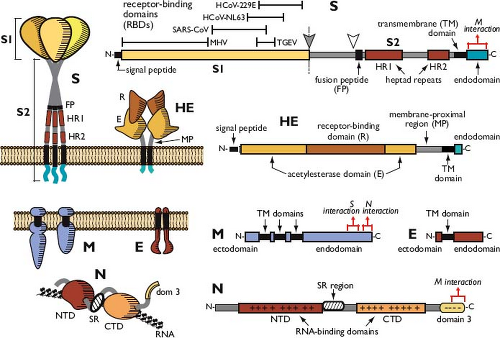 Figure 28.3. Virion structural proteins. Folded and linear representations of the spike (S), hemagglutinin-esterase (HE), membrane (M), envelope (E), and nucleocapsid (N) proteins. The size scale for the linear diagram of S is half of that for the other proteins. In the linear diagram of S, solid and open arrowheads indicate the S1-S2 and alternative (S2′) cleavage sites, respectively. In the linear diagrams of S, M, and N, red brackets indicate mapped regions involved in assembly interactions (see the Assembly and Release of Virions section). |
The most abundant structural protein in coronaviruses—the M protein544,546—gives the virion envelope its shape. The M monomer, which ranges from 25 to 30 kDa, is a polytopic membrane protein that is embedded in the envelope by three transmembrane domains.14,486 At its amino terminus is a very small ectodomain; the C-terminal endodomain of M accounts for the major part of the molecule and is situated in the interior of the virion or on the cytoplasmic face of intracellular membranes (see Fig. 28.3). Although it is inserted co-translationally into the ER membrane, the M protein generally does not bear an amino-terminal signal peptide.62,486 For IBV and MHV, either the first or the third transmembrane domain of M alone suffices as a signal for insertion and anchoring of the protein in its native membrane orientation.350,363,384 Anomalously, M proteins of the alphacoronavirus 1 species do contain cleavable N-terminal signal peptides, although it is not clear whether these are necessary for membrane insertion.263,584 The ectodomain of M is modified by glycosylation, which is usually N linked.60,251,402,536,632 However, a subset of betacoronavirus M proteins exhibit O-linked glycosylation, and the MHV M protein has served as a model for study of this type of posttranslational modification.116,349,419 Glycosylation of M influences both organ tropism and the interferon (IFN)-inducing capacity of some coronaviruses.72,113,311
M proteins are moderately well conserved within each coronavirus genus but diverge considerably across genera. The most variable part of the molecule is the ectodomain. By contrast, a short segment, overlapping the third transmembrane domain and the start of the endodomain, exhibits a high degree of sequence conservation that is seen even in torovirus M proteins.132 Like most multispanning membrane proteins, the M protein has been refractory to crystallization; however, recent cryo-electron microscopic and tomographic reconstructions have provided a glimpse of the structure of this protein within the virion envelope.21,413,415 These studies reveal that the large carboxy terminus of M extends some 6 to 8 nm into the viral particle and is compressed into a globular domain, consistent with early work showing that the endodomain is very resistant to proteases.61,384,486,490 The observed M structures are likely to be dimers, the monomers of which are associated through multiple interacting regions. M dimers appear to adopt two different conformations: a compact form that promotes greater membrane curvature and a more elongated form that contacts the nucleocapsid.415
The E protein is a small polypeptide of 8 to 12 kDa that is found in limited amounts in the virion envelope.189,344,647 Despite its minor presence, no wild-type coronavirus has been discovered to lack this protein. Engineered knockout or deletion of the E gene has effects ranging from moderate124 to severe293,296 to lethal.105,428 Thus, although E is not always essential, it is critical for coronavirus infectivity (see the Assembly and Release of Virions section). E protein sequences are widely divergent, even among closely related coronaviruses.293 However, all E proteins share a common architecture: a short hydrophilic amino terminus, followed by a large hydrophobic region, and, lastly, a large hydrophilic C-terminal tail (see Fig. 28.3). E is an integral membrane protein,100,335,582 but it does not have a cleavable signal peptide465 and is not glycosylated. Beta- and gammacoronavirus E proteins are palmitoylated on cysteine residues downstream and adjacent to the hydrophobic region38,101,335,354,647; this modification remains to be found in an alphacoronavirus E protein.189 The membrane topology of E is not completely resolved. Most evidence indicates that this polypeptide transits the membrane once, with an N-terminal exodomain and a C-terminal endodomain.101,420,465,564,582 Contrary to this are reports that E has a
hairpin conformation, placing both of its termini on the cytoplasmic face of membranes,12,368 or that E can have multiple membrane topologies.648 Also unresolved is the oligomeric state of E protein. The hydrophobic region of the SARS-CoV E protein forms multimers, from dimers through pentamers.564,610 A pentameric alpha-helical bundle structure has been solved for this domain,449 although it is not yet clear whether this reflects the organization of the native protein.
hairpin conformation, placing both of its termini on the cytoplasmic face of membranes,12,368 or that E can have multiple membrane topologies.648 Also unresolved is the oligomeric state of E protein. The hydrophobic region of the SARS-CoV E protein forms multimers, from dimers through pentamers.564,610 A pentameric alpha-helical bundle structure has been solved for this domain,449 although it is not yet clear whether this reflects the organization of the native protein.
Residing in the interior of the virion, the N protein is the sole protein constituent of the helical nucleocapsid.222 Monomers of this 43- to 50-kDa protein bind along the RNA genome in a beads-on-a-string configuration common to other helical viral nucleocapsids (see Fig. 28.2B). However, unlike the nucleoproteins of rhabdo- and paramyxoviruses, the coronavirus N protein provides little or no protection for its genome against the action of ribonucleases.366,408 The bulk of the N protein monomer is made up of two independently folding domains—designated the N-terminal domain (NTD) and the C-terminal domain (CTD)—although neither includes its respective terminus of the N molecule (see Fig. 28.3). Crystal or solution structures have been determined for NTDs and CTDs of SARS-CoV, IBV, and MHV.76,164,200,234,253,493,555,646 Flanking the NTD and CTD are three spacer segments, the central one of which contains a serine- and arginine-rich tract (the SR region), which was noted to resemble the SR domains of RNA-splicing factors.442 Another functionally distinct region of N, the carboxy-terminal domain 3, has been defined genetically.236,279,441,442 The spacer segments and domain 3 are each likely to be intrinsically disordered polypeptides.66,67 Most of the N molecule, including the NTD and CTD, is highly basic; by contrast, domain 3 is acidic. There is only a moderate degree of sequence homology among N proteins across the three genera, with the exception of a stretch of 30 amino acids within the NTD that is highly conserved among all coronaviruses.380
The N protein is a phosphoprotein,272,352,515,542 modified at a limited number of serine and threonine residues. Phosphorylation sites have been mapped for a representative coronavirus from each genus, and targeted sites, collectively, fall in every domain and spacer region of the N molecule.55,77,604,619 Thus, a general pattern for N protein phosphorylation cannot yet be discerned, nor have all responsible kinases been identified, although there is evidence linking glycogen synthase kinase-3 to phosphorylation of the SR region.619 The role of phosphorylation is also not known but is thought to have regulatory impact. Phosphorylation has been suggested to trigger a conformational change in N protein,541 and it may enhance the affinity of N for viral versus nonviral RNA.77
The most conspicuous function of the N protein is to bind to viral RNA. Nucleocapsid formation must involve both sequence-specific and nonspecific modes of RNA binding. Specific RNA substrates that have been identified for N protein include the transcription-regulating sequence (TRS)200,412,539 (see the Viral RNA Synthesis section) and the genomic RNA packaging signal96,396 (see the Assembly and Release of Virions section). The NTD and the CTD are each separately capable of binding to RNA ligands in vitro, and the structures of these domains offer some clues as to how this is accomplished. The NTD consists of a U-shaped β-platform with an extruding β-hairpin, which presents a putative RNA-binding groove rich in basic and aromatic amino acid residues.164,200,493 The CTD forms a tightly interconnected dimer, which exhibits a potential RNA-binding groove lined by basic α-helixes.253,555 Some work suggests that in the intact N protein, optimal RNA binding requires concerted contributions from both the NTD and the CTD.67,235 A significant fraction of nucleocapsid stability also results from interactions among N monomers.408 This level of association is generally attributed to the CTD67,164,253,646; however, additional regions of N–N interaction have been mapped to the NTD and to domain 3.164,235,253 Another crucial function of N protein is to bind to M protein.162,546 This capability is provided by domain 3 of N.236,295,585
A fifth prominent structural protein—the hemagglutinin-esterase (HE) protein—is found in only a subset of the betacoronaviruses, including murine coronavirus, betacoronavirus 1, and HCoV-HKU1. In virions of these species, HE forms a secondary set of short projections of 5 to 10 nm arrayed beneath the canopy of S protein spikes.204,435,550 The 48-kDa HE monomer is composed almost entirely of an N-terminal ectodomain; this is followed by a transmembrane anchor and a very short C-terminal endodomain (see Fig. 28.3). HE is inserted into the ER by means of a cleaved signal peptide and acquires an additional 17 kDa of N-linked glycosylation at multiple sites.221,271,640 The assembled protein is a homodimer, the subunits of which are connected by disulfide bonds.221 As its name indicates, the HE protein contains a pair of associated activities. First, it is a hemagglutinin—that is, it has the capability to bind to sialic acid moieties found on cell surface glycoproteins and glycolipids.54,272 Second, HE exhibits acetylesterase activity with specificity for either 9-O– or 4-O-acetylated sialic acids.274,472,520,590,591 These characteristics are thought to allow HE to act as a cofactor for S protein, assisting attachment of virus to host cells, as well as expediting the travel of virus through the extracellular mucosa.99 Consistent with this notion, the presence of HE in MHV dramatically enhances neurovirulence in the mouse host.265 Conversely, the HE protein is a burden to the virus in tissue culture, where its expression is rapidly counterselected.343 The two activities of the HE protein are strikingly similar to the receptor-binding and receptor-destroying activities found in influenza C virus,590,591 and, remarkably, the coronavirus HE gene is clearly related to the influenza C virus HEF gene.359 Moreover, toroviruses also possess a homolog of the HE gene,99,305 raising the possibility that all three of these virus groups evolved from a common ancestor.359,522 This kinship is further corroborated by the crystal structure of the BCoV HE protein, which reveals separate receptor-binding and acetylesterase domains perched atop a truncated membrane-proximal region.650 The HE protein thus resembles a squat version of its influenza virus counterpart, shortened because it lacks the fusion domain stalk of the HEF protein.
Genome Structure and Organization
Basic and Accessory Genes
The coronavirus genome, which ranges from 26 to 32 kb, is the largest among all RNA viruses, including RNA viruses that have segmented genomes. This exceptional RNA molecule acts in at least three capacities50,194: as the initial mRNA of the infectious cycle (see the Expression of the Replicase-Transcriptase Complex section), as the template for RNA replication and transcription (see the Viral RNA Synthesis section), and as the substrate for packaging into progeny viruses (see
the Assembly and Release of Virions section). Consistent with its role as an mRNA, the coronavirus genome has a standard eukaryotic 5′-terminal cap structure301 and a 3′ polyadenylate tail.302,351,503,599 The genome comprises a basic set of genes in the invariant order 5′-replicase-S-E-M-N-3′, with the huge replicase gene occupying two-thirds of the available coding capacity (Fig. 28.4). The replicase-transcriptase is the only protein translated from the genome; the products of all downstream open reading frames (ORFs) are derived from subgenomic mRNAs. The 5′-most position of the replicase gene is dictated by the requirement for expression of the replicase to set in motion all subsequent events of infection. The organization of the other basic genes, however, does not seem to reflect any underlying principle, because engineered rearrangement of the downstream gene order is completely tolerated.121
the Assembly and Release of Virions section). Consistent with its role as an mRNA, the coronavirus genome has a standard eukaryotic 5′-terminal cap structure301 and a 3′ polyadenylate tail.302,351,503,599 The genome comprises a basic set of genes in the invariant order 5′-replicase-S-E-M-N-3′, with the huge replicase gene occupying two-thirds of the available coding capacity (Fig. 28.4). The replicase-transcriptase is the only protein translated from the genome; the products of all downstream open reading frames (ORFs) are derived from subgenomic mRNAs. The 5′-most position of the replicase gene is dictated by the requirement for expression of the replicase to set in motion all subsequent events of infection. The organization of the other basic genes, however, does not seem to reflect any underlying principle, because engineered rearrangement of the downstream gene order is completely tolerated.121
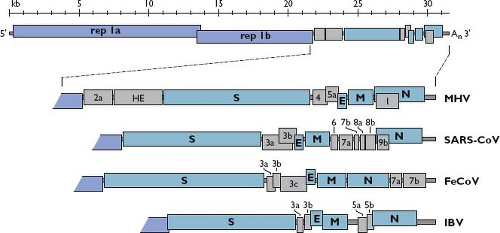 Figure 28.4. Coronavirus genome organization. A schematic of the complete genome of MHV is shown at the top. The replicase gene constitutes two ORFs, rep 1a and rep 1b, which are expressed by a ribosomal frameshifting mechanism (see the Expression of the Replicase-Transcriptase Complex section). The expanded region shows the downstream portion of the genomes of two betacoronaviruses (MHV and SARS-CoV), an alphacoronavirus (FeCoV), and a gammacoronavirus (IBV). The sizes and positions of accessory genes are indicated, relative to the basic genes S, E, M, and N. MHV, mouse hepatitis virus; ORFs, open reading frames; SARS-CoV, severe acute respiratory syndrome coronavirus; FeCoV, feline coronavirus; IBV, infectious bronchitis virus. |
Dispersed among the basic genes in the 3′-most third of the genome, there are from one to as many as eight additional ORFs, which are designated accessory genes378,407 (see Fig. 28.4). These can fall in any of the intergenic intervals downstream of the replicase gene,616 except, curiously, never between the E and M genes. In some cases, an accessory gene can be partially or entirely embedded as an alternate reading frame within another gene—for example, the internal (I) gene of MHV or the 3b gene of SARS-CoV. Accessory genes are generally numbered according to the smallest transcript in which they fall. Consequently, there is usually no relatedness among identically named accessory genes in coronaviruses of different genera, such as the 3a genes of SARS-CoV, feline coronavirus (FeCoV), and IBV (see Fig. 28.4). Some of these extra ORFs are thought to have been acquired through ancestral recombination with RNA from cellular or heterologous viral sources. The HE gene is the best-supported example of this type of horizontal genetic transfer.359 Two other such candidates are the 2a gene found in murine coronavirus and betacoronavirus 1, which encodes a putative 2′,3′-cyclic phosphodiesterase,385,485 and gene 10 of beluga whale coronavirus, which encodes a putative uridine-cytidine kinase.394 Notably, the 2a gene has a homolog embedded as a module within the replicase gene of the toroviruses,522 which is a situation also consistent with horizontal transfer. The origin of most accessory genes, however, remains an open question. It is plausible that some of them evolved through intragenomic recombination, resulting in gene duplication and subsequent divergence, as suggested for several of the accessory genes of SARS-CoV.241
Almost all accessory genes that have been examined are expressed during infection, although their functions are incompletely understood. The protein products of most accessory genes are nonstructural; however, this rule is not without exception. The HE protein, the MHV I protein,165 and the products of SARS-CoV ORFs 3a, 6, 7a, 7b, and 9b231,407,502,627 are all components of virions. Mutational knockout or deletion of accessory genes has revealed that none are essential for viral replication in tissue culture. Conversely, accessory gene ablation,103,115,206 or transfer to another virus,452,559 can have profound effects on viral pathogenesis. In some cases, the basis for this is understood to result from interactions with host innate immunity (see the Immune Response and Viral Evasion of the Immune Response section). For other accessory genes, though, potential in vivo functions have not yet been elucidated.125,165,645
Coronavirus Genetics
Classical coronavirus genetics focused principally on two types of mutants.299 The first were naturally arising viral variants, particularly deletion mutants, which offered clues to genetic changes responsible for different pathogenic traits.430,583,603 The second were temperature-sensitive (ts) mutants isolated from MHV following chemical mutagenesis.282,477,501,545 Some of these proved to be valuable in analyses of the functions of structural proteins.279,360,380,474 However, owing to the large target size of the replicase gene, most of such randomly generated mutants
had conditional-lethal, RNA-negative phenotypes. Complementation analyses of these latter mutants yielded early insights into the multiplicity of functions entailed by coronavirus RNA synthesis.22,176,177,501 There has been a recent resurgence of interest in classical replicase ts mutants, which are currently sorted into five complementation groups, because they can now be fully examined by the tools of reverse genetics.138,499,543
had conditional-lethal, RNA-negative phenotypes. Complementation analyses of these latter mutants yielded early insights into the multiplicity of functions entailed by coronavirus RNA synthesis.22,176,177,501 There has been a recent resurgence of interest in classical replicase ts mutants, which are currently sorted into five complementation groups, because they can now be fully examined by the tools of reverse genetics.138,499,543
The development of coronavirus reverse genetics proceeded in two phases.130 Initially, a method called targeted RNA recombination was devised at a time when it was uncertain whether the construction of full-length infectious complementary DNA (cDNA) clones of coronavirus genomes would ever become technically feasible. With this method, a synthetic donor RNA bearing mutations of interest is transfected into cells that have been infected with a recipient parent virus possessing some characteristic that can be selected against.279,377,380 In its current form, for manipulation of MHV, the technique uses a chimeric recipient parent virus designated fMHV (Fig. 28.5A). The fMHV chimera is a mutant of MHV that contains the S protein ectodomain from the FeCoV feline infectious peritonitis virus (FIPV) and can therefore only grow in feline cells (see the Virion Attachment to Host Cells section). The restoration of its ability to grow in murine cells, via recombination with donor RNA containing the MHV S gene, enables a strong selection for viruses bearing site-specific mutations292,381; unwanted secondary crossover events distal to the S gene are eliminated owing to the rearrangement of downstream genes in fMHV.190 Targeted RNA recombination remains a powerful method to recover structural or accessory protein or 3′ untranslated region (UTR) mutants.
To obtain access to the major part of the coronavirus genome, however, it was necessary to create full-length cDNAs, despite the barriers presented by the huge size of the replicase gene and the high instability of various regions when propagated in bacterial clones. Three innovative strategies were developed to overcome these inherent difficulties.130 In the first (see Fig. 28.5B), a full-length cDNA copy of a coronavirus genome is assembled downstream of a cytomegalovirus (CMV) promoter in a bacterial artificial chromosome (BAC) vector, which is stable by virtue of its low copy number.5,6 The infection is then launched from transfected BAC DNA through transcription of infectious coronavirus RNA by host RNA polymerase II. This method of initiating infection obviates potential limitations of in vitro capping and synthesis of genomic RNA. In the second strategy (see Fig. 28.5C), a full-length genomic cDNA is assembled by in vitro ligation of smaller cloned cDNA fragments, some of the boundaries of which have been chosen so as to interrupt regions of instability.642,643 The ligation occurs in a directed order that is dictated by the use of asymmetric restriction sites. Infectious genomic RNA is then transcribed in vitro and used to transfect susceptible host cells. An extension of this method has demonstrated the construction of a coronavirus genome entirely from synthetic cDNAs.26 In the third strategy (see Fig. 28.5D), the genome of vaccinia virus is used as the cloning vector for a full-length coronavirus cDNA that is generated by long-range reverse transcription polymerase chain reaction (RT-PCR).94,561 The cDNA is then amenable to manipulation by the repertoire of techniques available for poxvirus reverse genetics.51,94 Infections are launched from in vitro–synthesized RNA or else from transfected cDNA
transcribed in vivo by fowlpox-encoded T7 RNA polymerase.58 Collectively, these systems developed for complete reverse genetics provide an important pathway toward unraveling the complexities of the coronavirus replicase.
transcribed in vivo by fowlpox-encoded T7 RNA polymerase.58 Collectively, these systems developed for complete reverse genetics provide an important pathway toward unraveling the complexities of the coronavirus replicase.
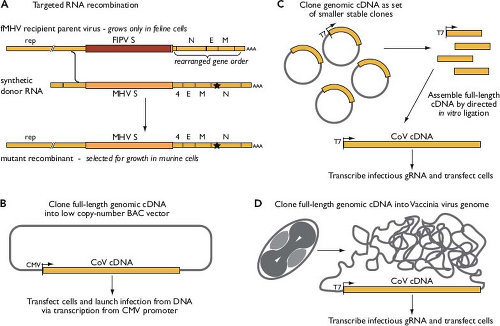 Figure 28.5. Methods for coronavirus reverse genetics. A: Targeted RNA recombination, which is applicable to the downstream third of the genome, shown here for transduction of a mutation (star) into the mouse hepatitis virus N gene. B–D: Three schemes developed for complete reverse genetics, based on stable production of full-length genomic complementary DNAs. |
Coronavirus Replication
Virion Attachment to Host Cells
Coronavirus infections are initiated by the binding of virions to cellular receptors (Fig. 28.6). There then follows a series of events culminating in the delivery of the nucleocapsid to the cytoplasm, where the viral genome becomes available for translation. Individual coronaviruses usually infect only one or a few closely related hosts. The interaction between the viral S protein and its cognate receptor constitutes the principal determinant governing coronavirus host species range and tissue tropism. This has been most convincingly shown in two ways. First, the expression of a particular receptor in nonpermissive cells of a heterologous species renders those cells permissive for the corresponding coronavirus.127,146,330,331,399,567,639 Second, the engineered replacement of the S protein ectodomain changes the host cell species specificity or tissue tropism of a coronavirus in a predictable fashion.207,292,410,453,495 The amino-terminal, more variable half of the spike protein, S1, is the part that binds to receptor. Binding leads to conformational changes that result in fusion between virion and cell membranes, mediated by the more conserved half of the spike protein, S2. The region of S1 that contacts the receptor—the receptor-binding domain (RBD)—varies among different coronaviruses (see Fig. 28.3). For MHV, the RBD maps to the N-terminal section of S1.290,554 By contrast, RBDs for SARS-CoV,614,625 HCoV-NL63,337 transmissible gastroenteritis virus (TGEV),188 and HCoV-229E34 fall in the middle or C-terminal sections of S1.
The known cellular receptors for alpha- and betacoronaviruses are listed in Table 28.2; to date, no receptors have been identified for gammacoronaviruses. The MHV receptor mCEACAM1 was the first discovered coronavirus receptor (as well as one of the first receptors defined for any virus).606,607 That this molecule is the only biologically relevant receptor for MHV was made clear by the demonstration that homozygous Ceacam1−/− knockout mice are totally resistant to infection by high doses of MHV.215 CEACAM1 is a member of the carcinoembryonic antigen (CEA) family within the immunoglobulin (Ig) superfamily and, in its full-length form, contains four Ig-like domains.146 A diversity of two- and four-Ig domain isoforms is generated by multiple alleles and alternative splicing variants of Ceacam1.97,145,147,422,423,641 The wide range of pathogenicity of MHV in mice is thought to be strongly affected by the interactions of S proteins of different virus strains with the array of receptor isoforms that are expressed in mice of different genetic backgrounds. Although their S proteins are phylogenetically very close to that of MHV, the betacoronaviruses BCoV and HCoV-OC43 do not use CEACAMs to infect their
hosts; rather, the only currently known attachment factor for these viruses is N-acetyl-9-O-acetylneuraminic acid.291,504 The recently solved structure of the MHV RBD complexed with mCEACAM1 has allowed the identification of key residues at the S protein–receptor interface.443 Coupled with mutational analysis, this structure reveals why the S proteins of BCoV and HCoV-OC43 cannot bind the MHV receptor and, conversely, why MHV does not bind to bovine or human CEACAMs.
hosts; rather, the only currently known attachment factor for these viruses is N-acetyl-9-O-acetylneuraminic acid.291,504 The recently solved structure of the MHV RBD complexed with mCEACAM1 has allowed the identification of key residues at the S protein–receptor interface.443 Coupled with mutational analysis, this structure reveals why the S proteins of BCoV and HCoV-OC43 cannot bind the MHV receptor and, conversely, why MHV does not bind to bovine or human CEACAMs.
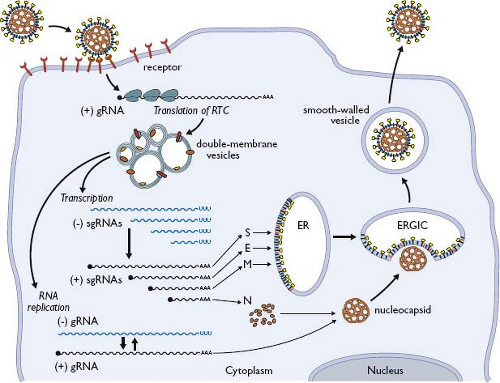 Figure 28.6. Overview of coronavirus replication (see text for details). |
Table 28.2 Coronavirus Receptors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Many alphacoronaviruses use aminopeptidase N (APN) of their respective host species as a receptor (see Table 28.2).127,567,639 APN (also called CD13) is a cell-surface, zinc-binding protease that is resident in respiratory and enteric epithelia and in neural tissue. The APN molecule is a heavily glycosylated homodimer. Mutational and inhibitor studies have shown that its enzymatic activity is not required for viral attachment and entry.126 In general, the receptor activities of APN homologs are not interchangeable among species126,281; however, feline aminopeptidase N (fAPN) can serve as a receptor not only for FIPV but also for canine coronavirus (CCoV), TGEV, and HCoV-229E.567 This circumstance has been exploited for the construction of chimeric APN molecules to map the basis for receptor recognition. Such studies have found three small, linearly discontinuous determinants in APN that govern the species specificity of this subgroup of alphacoronaviruses.29,214,280,569
The receptor for SARS-CoV—angiotensin-converting enzyme 2 (ACE2)—was discovered with notable rapidity following the isolation of the virus.331 ACE2 is a cell-surface, zinc-binding carboxypeptidase involved in regulation of cardiac function and blood pressure. It is expressed in epithelial cells of the lung and the small intestine, which are the primary targets of SARS-CoV, as well as in heart, kidney, and other tissues.209 As with APN, the receptor role of ACE2 appears to be independent of its enzymatic activity. Although the SARS-CoV S protein binds to the catalytic domain of ACE2, active-site mutation or chemical inhibition does not detectably affect the ability of ACE2 to associate with S protein or to promote syncytia formation.331,333,398 The crystal structure of the SARS-CoV S protein RBD in complex with ACE2 shows the RBD cradling one lobe of the claw-like catalytic domain of its receptor.325 Remarkably, ACE2 also serves as the receptor for the alphacoronavirus HCoV-NL63,219 and the corresponding structural complex for that virus reveals that the HCoV-NL63 RBD and the SARS-CoV RBD bind to the same motifs.624 Because the SARS-CoV and HCoV-NL63 RBDs have neither sequence nor structural homology, this finding strongly supports the notion that they have independently evolved to bind to the same hotspot on the ACE2 surface.623,624 Analyses of the SARS-CoV RBD–ACE2 interface have additionally demonstrated the structural basis for the final jump of SARS-CoV from palm civets to human hosts (see the Epidemiology section). These studies found that merely four critical residues constitute the major species barrier between the civet and human ACE2 molecules, and that mutation of only two key RBD residues was sufficient for civet SARS-CoV S protein to gain the ability to productively bind human ACE2.323,333
Viral Entry and Uncoating
The entry of virions into cells results from large-scale rearrangements of the S protein that lead to the fusion of viral and cellular membranes.41 These rearrangements are triggered by some combination of receptor binding, proteolytic cleavage of S, and exposure to acidic pH. The S proteins of many coronaviruses are uncleaved in mature virions and require an encounter with a protease at the entry step of infection to separate the receptor-binding and fusion components of the spike. The details of proteolytic activation are still incompletely understood but have been best studied for SARS-CoV. In the cell types in which this virus is most commonly grown in tissue culture, viral entry depends on cathepsins, which are acid-activated endosomal proteases. The infectivity of SARS-CoV is thus suppressed by cathepsin inhibitors or by lysosomotropic agents.517 However, cell-bound SARS-CoV can alternatively be activated by treatment with extracellular proteases, such as trypsin or elastase. This route of activation greatly enhances the infectivity of SARS-CoV and allows the virus to enter from the cell surface, thereby rendering the infection insensitive to lysosomotropic agents.383 The same pattern of proteolytic activation—cathepsin-dependence and its circumvention by exogenously added protease—is observed with a particular strain of MHV (MHV-2) that is unique in having an uncleaved S protein.464
The site of cleavage of receptor-bound SARS-CoV S protein by cathepsin or by exogenous trypsin differs from that of the S1-S2 cleavage, which occurs in other coronaviruses upon exit from cells. Cleavage at entry takes place at a locus (S2′) within the S2 half of the molecule, immediately upstream of the putative fusion peptide28 (see Fig. 28.3). It is not yet clear if cleavage at analogous S2′ sites is the pattern for all coronavirus S proteins; however, the emerging pattern is that proteolytic activation of S protein is required for infectivity and that coronaviruses have evolved in different ways to ensure that this occurs.41 Recent studies provide evidence that for the SARS-CoV S protein, the most biologically relevant protease may be TMPRSS2.187,382,514 This transmembrane serine protease, which is expressed in pneumocytes, co-localizes with and binds to ACE2. In cells expressing TMPRSS2, SARS-CoV enters at the cell surface and is insensitive to cathepsin inhibitors and lysosomotropic agents.
Just as the mechanism of S protein proteolytic activation is variable, so too is its location. Some coronaviruses, such as most strains of MHV, fuse with the plasma membrane,547,601 whereas others, such as TGEV,212 HCoV-229E,421 and SARS-CoV,517 can enter cells through receptor-mediated endocytosis and then fuse with the membranes of acidified endosomes. The boundary between these two modes of entry may easily shift. For one strain of MHV (MHV-4), as few as three amino acid changes in the heptad repeat region of S2 switches the virus from plasma membrane fusion to acid pH-dependent fusion.180 It remains unresolved whether acidic pH, per se, is required for S protein conformational changes90,154,324 or whether this reflects the requirements for activation of endosomal proteases during infection of some types of cells.517
The coronavirus S protein is a class I viral fusion protein with domains functionally similar to those of the fusion proteins of phylogenetically distant RNA viruses, such as influenza virus, human immunodeficiency virus (HIV), and Ebola virus, but on a much larger scale.41,42 As in those other viral fusion proteins, the coronavirus S2 moiety contains two separated heptad repeats—HR1 and HR2—with a fusion peptide upstream of HR1 and the transmembrane domain immediately downstream of HR2 (see Fig. 28.3). The exact assignment of the fusion peptide is not agreed upon, however.41,367,450 Receptor-mediated conformational changes in S1, and the dissociation of S1 from S2, are thought to initiate major rearrangements in the remaining S2 trimer that proceed through multiple intermediate states.133,324 These rearrangements ultimately expose the fusion peptide, which interacts with the host cellular membrane, and the two heptad repeats in each monomer are brought together to form an antiparallel, six-helix bundle. The six-helix bundle is an extremely stable, rod-like complex, the biophysical properties of which have been extensively studied.40,42,242,348,568 Highly similar crystallographic structures have been solved for the six-helix complexes from both the MHV S protein629 and the SARS-CoV S protein.144,552,630 These show the three HR1 helices forming a central, coiled-coil core some two to three times larger than its counterparts in other viruses. Arrayed around this, the three shorter HR2 helices, in an antiparallel orientation, pack into the grooves between the HR1 monomers via hydrophobic interactions. The outcome of the formation of the six-helix bundle is the juxtaposition of the viral and cellular membranes in sufficient proximity to allow mixing of their lipid bilayers and the deposition of the contents of the virion into the cytoplasm.
Expression of the Replicase-Transcriptase Complex
Following delivery of the viral nucleocapsid to the cytoplasm, the next event is the translation of the replicase gene from the genomic RNA. This gene consists of two large ORFs—rep 1a and rep 1b—that share a small region of overlap (see Fig. 28.4). Translation of the entire replicase depends on a mechanism called ribosomal frameshifting, whereby, with a fixed probability, a translating ribosome shifts one nucleotide in the –1 direction, from the rep 1a reading frame into the rep 1b reading frame.378 This repositioning is programmed by two RNA elements (Fig. 28.7A), embedded near the region of overlap, that were discovered in studies of IBV.46,47 The first element is the 5′-UUUAAAC-3′ heptanucleotide slippery sequence, which is identical for all known coronaviruses and has apparently been selected as optimal for its role.48,457 The second element, located a short distance downstream of the slippery sequence, is an extensively characterized RNA pseudoknot structure.49,405 This latter component was initially thought to be a classic two-stem (H-type) pseudoknot; however, recent analyses of SARS-CoV frameshifting support a more elaborate structure that includes a third stem loop within pseudoknot loop 2.20,141,456
The two elements act together to produce the coterminal polyprotein products pp1a and pp1ab. During most rounds of translation, the elongating ribosome unwinds the pseudoknot and translation terminates at the rep 1a stop codon, yielding the smaller product, pp1a. Some fraction of the time, however, the pseudoknot blocks the mRNA entrance channel of the ribosome.213,403,528 The consequent pause required for the ribosome to melt out the mRNA structure allows the simultaneous slippage of the P and A site transfer RNAs (tRNAs) into the rep 1b reading frame. This results in the synthesis of pp1ab when elongation resumes.20,47 Studies of reporter gene expression suggest that the incidence of coronaviral ribosomal frameshifting is as high as 25% to 30%; however, the in vivo frequency in infected cells remains to be quantitated. It is thought that the role of programmed frameshifting is to provide a fixed ratio of translation products for assembly into a macromolecular complex.457 It is also possible that frameshifting forestalls expression of the enzymatic products of rep 1b until the products of rep 1a have prepared a suitable environment for RNA synthesis.
Polyproteins pp1a (440–500 kDa) and pp1ab (740–810 kDa) are autoproteolytically processed into mature products that are designated nsp1 to nsp16 (except for the gammacoronaviruses, which do not have a counterpart of nsp1). From work begun with early studies of MHV,134,135,525 complete processing schemes have now been solved for replicases of multiple coronaviruses representing all three genera659,661 (see Fig. 28.7B). Processing also generates many long-lived partial proteolytic products, which may have functional importance. There are two types of polyprotein cleavage activity.17,358 One or two papain-like proteases (PLpro), which are situated in nsp3, carry out the relatively specialized separation of nsp1, nsp2, and nsp3. The main protease (Mpro)—nsp5—performs the remaining 11 cleavage events. Mpro is often designated the 3C-like protease (3CLpro) to point out its distant relationship to the 3C proteins of picornaviruses. Several crystal structures have been determined for PLpro and Mpro of SARS-CoV and other
coronaviruses,9,469,612,631 and these enzymes present attractive targets for antiviral drug design.468,633,634
coronaviruses,9,469,612,631 and these enzymes present attractive targets for antiviral drug design.468,633,634
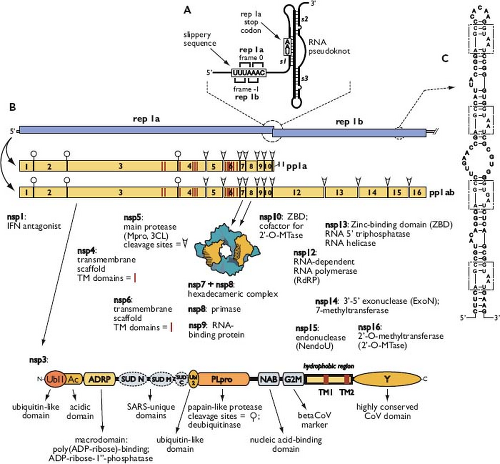 Figure 28.7. Coronavirus replicase gene and protein products. A: Ribosomal frameshifting elements of the SARS-CoV replicase gene. Pseudoknot stems are indicated as s1, s2, and s3. B: Polyprotein pp1a and pp1ab processing scheme for alpha- and betacoronaviruses. The gammacoronavirus processing scheme is identical, except for the absence of nsp1. Known functions and properties of nsp1 through nsp16 are listed; nsp11 is an oligopeptide generated when ribosomal frameshifting does not occur. Transmembrane domains in nsp3, nsp4, and nsp6 are indicated by red vertical lines. The nsp3 schematic shown is for SARS-CoV414; some modules differ in other coronaviruses. C: The RNA packaging signal located in the nsp15-encoding region of the MHV genome.81 This element is found only in a subset of the betacoronaviruses (MHV, betacoronavirus 1, and HCoV-HKU1); repeat units are boxed. SARS-CoV, severe acute respiratory syndrome coronavirus; MHV, mouse hepatitis virus; HCoV, human coronavirus. |
The processed nsps assemble to form the coronavirus replicase, which is also referred to as the replicase-transcriptase complex (RTC).660 The challenge of defining the roles of the many nsp components of the RTC was initially addressed by foundational studies in bioinformatics,196,317 which is a discipline that continues to inform the analysis of this intricate molecular machinery.414,521 Besides PLpro and Mpro, the products of rep 1a contain several activities that establish cellular conditions favorable for infection. Some of these are directly linked to RNA synthesis. Others are nonessential for viral replication in tissue culture; however, they can have major effects on virus–host interactions (see the Immune Response and Viral Evasion of the Immune Response section). The very first polyprotein product—nsp1—exhibits a broad repertoire of antagonistic activities that selectively inhibit host protein synthesis and IFN signaling.230,258,259 By contrast, nsp2 is completely expendable and, as yet, has no demonstrated function.199
Nsp3 is by far the largest of the RTC proteins. It consists of a concatenation of individual structural modules that are arranged as globular domains separated by flexibly disordered linkers414 (see Fig. 28.7B). At the amino terminus of nsp3 are ubiquitin-like (Ubl1) and acidic (Ac) domains506 that interact with the SR region of the N protein.237 It is proposed that this interaction tethers the genome to the assembling RTC to allow formation of the initiation complex for RNA synthesis. As mentioned earlier, located within nsp3 are one (in SARS-CoV and
gammacoronaviruses) or two PLpro modules (in most other coronaviruses). In addition to protease activity, PLpro domains possess deubiquitinase activity,341,469,612 which forms another part of the viral arsenal that counters host innate immunity.136,174 A highly conserved domain of nsp3 has adenosine diphosphate-ribose-1′-phosphatase (ADRP) and poly(adenosine diphosphate [ADP]-ribose)-binding activities,152,494 which, although nonessential for replication, help confer resistance to host defenses.158,297 At the C-terminus of nsp3 is a conserved region, designated the Y domain, containing three metal-binding clusters of cysteine and histidine residues.414,662 The potential functions of other domains of nsp3 (NAB, G2M, SUD),73,414,507,521 which appear only in various subsets of coronaviruses, remain to be elucidated.
gammacoronaviruses) or two PLpro modules (in most other coronaviruses). In addition to protease activity, PLpro domains possess deubiquitinase activity,341,469,612 which forms another part of the viral arsenal that counters host innate immunity.136,174 A highly conserved domain of nsp3 has adenosine diphosphate-ribose-1′-phosphatase (ADRP) and poly(adenosine diphosphate [ADP]-ribose)-binding activities,152,494 which, although nonessential for replication, help confer resistance to host defenses.158,297 At the C-terminus of nsp3 is a conserved region, designated the Y domain, containing three metal-binding clusters of cysteine and histidine residues.414,662 The potential functions of other domains of nsp3 (NAB, G2M, SUD),73,414,507,521 which appear only in various subsets of coronaviruses, remain to be elucidated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


