Diagnostic Virology
Gregory A. Storch
David Wang
Diagnostic virology continues to evolve rapidly. Viral testing is now essential for the care of a number of patient groups, including hospitalized patients with acute respiratory infections; transplant recipients and other immunocompromised patients; patients infected with human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV); and infants with possible congenital infection. Multiple test methods continue to be used, but molecular tests are emerging as the dominant technology. A variety of commercial molecular assays have been or are in the process of being approved or cleared as in vitro diagnostic tests by the Food and Drug Administration (FDA). This is an important development because it makes viral diagnostic testing available to more laboratories and it improves the standardization of diagnostic testing. The scope of diagnostic virology has broadened. General categories of viral diagnostic testing and the viruses included in those categories are shown in Table 15.1.
History of Diagnostic Virology
Modern diagnostic virology dates to the first growth of human viruses in tissue culture reported by Weller and Enders in 1948.149 This and other landmarks in the history of diagnostic virology are shown in Table 15.2.
Specimens for Viral Diagnosis
The likelihood of making a specific viral diagnosis depends largely on the quality of the specimen that is received in the laboratory. Important variables include the timing of the specimen in relation to the patient’s illness, the type of specimen, the quality and amount of specimen material obtained, and the time and conditions of transport to the laboratory. Although this concept is so basic as to seem trivial, optimizing the variables listed requires knowledge on the part of the physician and attention to logistic considerations that can present substantial barriers to diagnosis.
For the diagnosis of acute viral infections, the best specimens are usually obtained from the site of disease. For example, in the patient with suspected viral meningitis, cerebrospinal fluid (CSF) is the best specimen. In infections involving skin or mucosal surfaces, specimens obtained from those surfaces are usually the only ones required. Viral titers are highest early in the course of an acute illness, so that specimens obtained within the first few days after onset are most likely to be positive.
Viral culture requires more attention to conditions of transport than specimens submitted for detection of viral antigens or nucleic acids, because the viability of the virus must be preserved. A number of clinically important viruses are labile and will not survive prolonged transport. General instructions for transporting specimens for viral culture are shown in Table 15.3. These conditions are usually also suitable for detection of viral antigens and nucleic acids, but consultation with the laboratory performing testing is advised.
For serologic diagnosis, an acute phase serum specimen should be obtained within the first few days of illness and a convalescent phase specimen 2 to 4 weeks later. If a virus-specific immunoglobulin M (IgM) assay is available, the acute phase specimen may be sufficient by itself. Immunoglobulins are stable in serum or plasma. Proper handling of specimens for serologic diagnosis begins with separation of serum or plasma. If testing is performed within several days, the serum or plasma can be stored at 4°C. If a longer delay is involved, the specimen should be frozen at –20°C or –70°C. For certain viral infections, serologic tests can also be performed on saliva or urine.
Table 15.1 Categories of Testing Performed in Diagnostic Virology | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Table 15.2 Landmarks in the History of Diagnostic Virology | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Significance of Viral Detection
As in other areas of microbiology, the detection of a virus in a clinical sample is not proof in and of itself that the detected virus is the cause of the patient’s illness. This problem regarding viral causality is exacerbated by polymerase chain reaction (PCR) and other very sensitive detection methods that may reveal very low-level persistent infection or even latent infection, unrelated to the patient’s current illness. A determination of whether a causal relationship exists requires consideration of several factors. Two of the most important are (a) the nature of the virus–host interaction and (b) whether the virus is known to cause the disease manifestations that the patient is experiencing. If the virus detected is one known to be associated with persistent or latent infection, its detection may require other supportive evidence before it can be accepted as causal of the patient’s current illness. For example, detection of the presence of virus-specific IgM antibodies or of seroconversion, in addition to the presence of the virus, favors acute infection, which, in turn, favors a causal relationship. An alternative approach using molecular methods may be to detect a specific viral RNA that encodes a structural or other protein that is expressed only in active infection. Obviously, detection of a virus known to cause the disease that the patient has is much more likely to be causal than detection of a virus not
known to be associated with that disease. This applies particularly to viruses such as human herpesvirus type 6 (HHV-6) and type 7 (HHV-7), human polyomaviruses such as JC virus, non–high-risk types of human papillomavirus (HPV), and even respiratory or gastrointestinal viruses such as rhinovirus or adenoviruses that may be present in patients who are not ill.
known to be associated with that disease. This applies particularly to viruses such as human herpesvirus type 6 (HHV-6) and type 7 (HHV-7), human polyomaviruses such as JC virus, non–high-risk types of human papillomavirus (HPV), and even respiratory or gastrointestinal viruses such as rhinovirus or adenoviruses that may be present in patients who are not ill.
Table 15.3 Instructions for Obtaining and Transporting Specimens for Viral Testinga | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
Methods Used in Diagnostic Virology
Viral Culture
For approximately 50 years, viral culture was the signature method of diagnostic virology laboratories, and it was the method that led to the establishment of virology laboratories as distinct entities apart from other areas of the clinical laboratory. With the advent of immunological and especially of molecular methods, which are more rapid than viral culture and potentially detect a broader range of viruses, cell culture is playing a diminishing role. For this reason, the treatment of cell culture in this chapter will be brief, and the interested reader is referred to other sources.77,78,125,144
By their very nature, viruses require living systems for propagation. In the past, inoculation of animals such as suckling mice or of embryonated eggs was used to grow viruses. These methods were largely supplanted by cell culture in the diagnostic laboratory. To grow viruses in cell culture, a clinical sample is prepared and then inoculated onto one or more cell culture types. Typically several cell culture types are inoculated, because no one type supports the growth of all clinically relevant viruses. Cell culture types that are commonly used are shown in Table 15.4. After inoculation, viral growth is detected in one of several ways. Traditionally, the principal method was the appearance of cytopathic effect, which refers to morphologic changes observable by microscopy that occur in virally infected cells (Fig. 15.1). Alternatively, hemadsorption, which refers to the surface binding of erythrocytes by virally infected cells, was used for the detection of influenza, parainfluenza, and mumps viruses. Interference, used to detect rubella virus, refers to the phenomenon that cells infected with certain viruses such as rubella virus become resistant to infection with other viruses that would readily infect the cells if they were uninfected by the first virus. Confirmation of virus growth, suspected based on one of the indicators described previously, can be achieved by immunofluorescence using specific antiviral antibodies. When cytopathic effect is present but immunological stains are negative, electron microscopy of cell culture material can be used to examine the culture. More recently, molecular methods such as VIDISCA (virus discovery based on complementary DNA [cDNA]-amplified fragment length polymorphism) have been used in this situation to detect new viruses growing in cell culture.107,132
Presently, most viral culture done in diagnostic virology laboratories is performed using centrifugation cultures, also often referred to as shell-vial cultures (Fig. 15.2). The shell-vial culture method was originally developed for cytomegalovirus (CMV)42,46 but has subsequently been applied to many other viruses. In this method, viral entry is enhanced by a low-speed centrifugation after inoculation of the clinical sample onto the cell culture, which may be grown on a cover
slip within a 1-dram vial referred to as a shell vial, but can also be grown within other devices such as 24-well plates that can be conveniently centrifuged. Viral growth is detected by immunofluorescence, typically at 16 and 40 hours after inoculation, allowing detection of viruses much sooner than could be accomplished using conventional tube cultures. It is now possible to purchase cell cultures for centrifugation cultures in which more than one type of cell is present. For example, a commercial system called R-Mix (Diagnostic Hybrids, Inc., Athens, Ohio) combines human adenocarcinoma (A549) and mink lung cell lines for growth of respiratory viruses. After incubation, the R-Mix cells are stained with a mixture of monoclonal antibodies specific for seven respiratory viruses. Similar mixed cell culture systems also exist to detect herpes simplex virus (HSV) and varicella-zoster virus (VZV) and to detect enteroviruses.
slip within a 1-dram vial referred to as a shell vial, but can also be grown within other devices such as 24-well plates that can be conveniently centrifuged. Viral growth is detected by immunofluorescence, typically at 16 and 40 hours after inoculation, allowing detection of viruses much sooner than could be accomplished using conventional tube cultures. It is now possible to purchase cell cultures for centrifugation cultures in which more than one type of cell is present. For example, a commercial system called R-Mix (Diagnostic Hybrids, Inc., Athens, Ohio) combines human adenocarcinoma (A549) and mink lung cell lines for growth of respiratory viruses. After incubation, the R-Mix cells are stained with a mixture of monoclonal antibodies specific for seven respiratory viruses. Similar mixed cell culture systems also exist to detect herpes simplex virus (HSV) and varicella-zoster virus (VZV) and to detect enteroviruses.
Table 15.4 Cell Culture Types Used to Detect Medically Important Viruses | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
Genetically Engineered Cell Lines
Techniques of genetic engineering have been used to modify cell lines either to make them susceptible to viruses to which they are not otherwise susceptible or to create novel means of detecting virus growth. An example is a cell line developed by Stabell and Olivo126 for the detection of HSV. This cell line, shown in Figure 15.3, consists of baby hamster kidney (BHK) cells that have been transfected with the β-galactosidase gene from Escherichia coli under the control of a promoter from the herpes simplex gene UL39. The promoter is activated by exposure to HSV proteins ICP0 and VP16. Exposure of the cells to
a specimen containing HSV specifically activates the promoter, causing production of β-galactosidase, which can be detected by a simple histochemical stain of the culture, performed 16 to 24 hours after inoculation. A commercial version of this system, called ELVIS HSV (Diagnostic Hybrids, San Diego, CA), has been shown to have sensitivity comparable to conventional viral culture for detection of HSV in clinical specimens.127 The system has also been adapted to the performance of HSV antiviral drug susceptibility assays.133 Another example is the creation of a line of buffalo green monkey (BGM) cells that have been modified to express human decay-accelerating factor, which is a receptor for some enterovirus serotypes.56 Addition of this molecule increases the ability of unaltered BGM cells to grow enteroviruses. These cells are combined with A549 cells in a commercial product called Super E-Mix (Diagnostic Hybrids).
a specimen containing HSV specifically activates the promoter, causing production of β-galactosidase, which can be detected by a simple histochemical stain of the culture, performed 16 to 24 hours after inoculation. A commercial version of this system, called ELVIS HSV (Diagnostic Hybrids, San Diego, CA), has been shown to have sensitivity comparable to conventional viral culture for detection of HSV in clinical specimens.127 The system has also been adapted to the performance of HSV antiviral drug susceptibility assays.133 Another example is the creation of a line of buffalo green monkey (BGM) cells that have been modified to express human decay-accelerating factor, which is a receptor for some enterovirus serotypes.56 Addition of this molecule increases the ability of unaltered BGM cells to grow enteroviruses. These cells are combined with A549 cells in a commercial product called Super E-Mix (Diagnostic Hybrids).
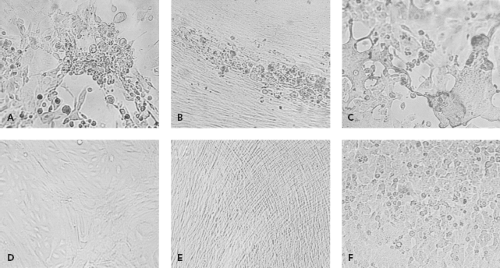 Figure 15.1. Cytopathic effect caused by viruses growing in cell culture. Herpes simplex virus growing in primary rabbit kidney cells (A); uninfected primary rabbit kidney cells (D). Cytomegalovirus growing in human embryonic lung fibroblast cells (B); uninfected human embryonic lung fibroblast cells (E). Respiratory syncytial virus growing in HEp-2 cells (C); uninfected HEp-2 cells (F). |
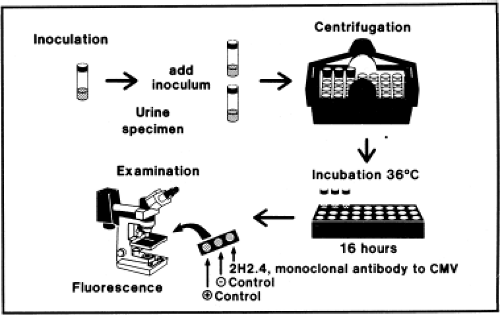 Figure 15.2. Shell-vial assay for cytomegalovirus. (From Shuster EA, Beneke JS, Tegtmeier GE, et al. Monoclonal antibody for rapid laboratory detection of cytomegalovirus infections: characterization and diagnostic application. Mayo Clin Proc 1985;60:577–585, with permission.) |
Electron Microscopy
Viral infections are rarely diagnosed using electron microscopy for the direct visualization of viral particles in specimens. Advantages include speed, lack of requirement for viral viability, and the potential to visualize many different kinds of viral particles. Disadvantages include the cost and complexity of maintaining an electron microscopy, the need for a skilled operator, and limited sensitivity related to the relatively high concentration of viral particles (105 to 106/L) that is required for visualization.89 Historically, electron microscopy was used for the evaluation of stool specimens from patients with suspected viral gastroenteritis. Viruses such as rotavirus, astrovirus, and adenovirus could be identified based on their characteristic morphology, but this has been replaced by rapid immunoassays and PCR assays. Electron microscopy can also be used to detect and classify viral particles in fixed tissue obtained by biopsy or at autopsy. Screening of tissue samples by electron microscopy is not useful because of the small area of tissue that can be examined. Thus, electron microscopy is best employed when it is directed by evidence of viral infection detected by routine histology.
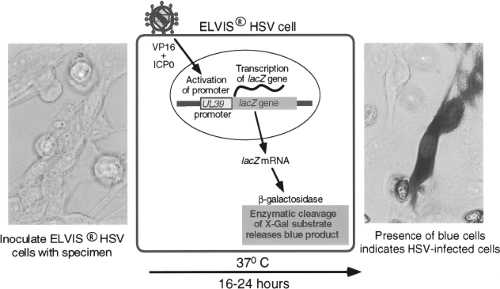 Figure 15.3. ELVIS herpes simplex virus (HSV), a genetically engineered cell line for the detection of HSV clinical samples. ELVIS cells are baby hamster kidney (BHK) cells that contain a β-galactosidase gene under the control of the HSV promoter UL39. If HSV is present in the specimen, viral particles enter the cells and the HSV proteins ICP0 and VP16 activate the UL39 promoter, leading to synthesis of β-galactosidase. The presence of β-galactosidase is detected by adding X-gal, a substrate for the enzyme. The activity of β-galactosidase on X-gal results in blue staining of ELVIS cells, visible microscopically. |
Table 15.5 Viral Inclusions | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Light Microscopy
Although viral particles, with the exception of poxviruses, cannot be directly visualized by light microscopy, indirect evidence of viral infection can be detected. The most characteristic signs of viral infection are inclusion bodies (composed of masses of virions), multinucleated cells, and syncytial cells. These findings may be present in cytologic specimens or in tissue examined after histologic staining. Viral inclusions can be found in the nucleus or the cytoplasm of infected cells. The location is characteristic of the responsible virus. The location of viral inclusions is summarized in Table 15.5. Microscopy for detection of the effects of viral infections in clinical specimens is generally carried out in surgical pathology rather than diagnostic virology laboratories.
Cytology and Histology
Cytologic examination for evidence of viral infection can be performed on smears prepared by applying a specimen directly to a microscope slide, on slides prepared by cytocentrifugation of fluids (e.g., bronchoalveolar lavage fluid), and on touch preparations prepared from pieces of unfixed tissue. Viruses that can produce cytologic evidence of infection in respiratory specimens include HSV, CMV, adenoviruses, polyomaviruses, and measles virus (Warthin-Finkeldey cells). Cytologic examination is usually not a sensitive method for detection but provides strong evidence for tissue involvement. Examples of cytologic findings suggestive of viral infection are shown in Figure 15.4. The Tzanck smear is a method sometimes used to detect cytologic evidence of HSV or VZV infection. It is rarely used in diagnostic virology laboratories because techniques such as fluorescent antibody staining and PCR are more sensitive and specific. The Papanicolaou stain (Pap smear) is used to detect evidence of HPV infection in cells obtained from the uterine cervix. HPV infection produces characteristic changes in keratinocytes, including a condensed nucleus with a prominent perinuclear clear zone referred to as koilocytosis. More specific evidence of the presence of HPV can be provided by histochemical stains or molecular techniques. Urine cytology may reveal intranuclear inclusions indicative of infection with either CMV or the polyomaviruses, JC and BK. Culture or PCR for CMV and PCR for polyomaviruses, however, are more sensitive and specific techniques. Cytology does not distinguish between JC and BK viruses.
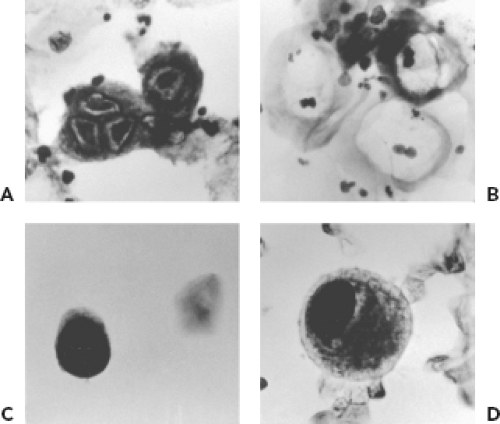 Figure 15.4. Cytologic findings suggestive of viral infection. A: Cervical smear showing multinucleated cells and the Cowdry type A intranuclear inclusions of herpes simplex infection. B: Papanicolaou smear showing binucleate squamous epithelial cells with distinct perinuclear halos. These characteristics, described as koilocytosis, are the cellular features associated with human papillomavirus infection. C: Urinary epithelial cell containing an enlarged nucleus with smudgy chromatin and a small pale glassy intranuclear inclusion indicative of polyomavirus infection. D: Cell from bronchoalveolar lavage with a large intranuclear inclusion with a perinuclear clear space (owl’s eye cell) indicative of cytomegalovirus infection. (Photographs provided by Dr. Leslie Boucher, Department of Pathology, Washington University, St. Louis, Missouri.) |
Histologic examination of stained tissue can provide unique information about the role of viral infection in producing tissue inflammation and injury and can be very useful in distinguishing between asymptomatic viral shedding and clinically significant infection. This is particularly useful for CMV infections in which prolonged viral shedding can occur without producing disease. For example, the presence of cytomegalic inclusion cells in tissue obtained by biopsy or at autopsy is often considered to be the gold standard for diagnosing clinically significant CMV infection localized in a specific organ.
Antigen Detection
The detection of viral antigens directly in clinical specimens is widely used to obtain rapid evidence of viral infection. The lack of requirement for virus viability is an important advantage. For some viruses, especially respiratory syncytial virus (RSV) and CMV, the sensitivity of antigen detection may exceed that of viral culture.111,135 Antigen detection methods can be applied when the following conditions are met: (a) viral antigen is expressed and is present in an accessible specimen, (b) an appropriate antibody (usually but not necessarily monoclonal) is available, (c) antigenic variability does not preclude recognition by immunologic reagents of different strains of the target virus, and (d) the antigen being detected is sufficiently stable that it does not degrade during transport and processing of the specimen. Methods used for viral antigen detection include fluorescent antibody (FA) staining, immunoperoxidase staining, and enzyme immunoassay. The latter can be subdivided
into solid phase and lateral flow immunochromatographic assays. The viruses for which antigen detection assays are in widespread use are shown in Table 15.6.
into solid phase and lateral flow immunochromatographic assays. The viruses for which antigen detection assays are in widespread use are shown in Table 15.6.
Table 15.6 Viral Antigen Detection: Specimens Used and Viruses Detected | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Antigen detection can also be used to detect viral antigens in fixed tissue (immunohistochemistry [IHC]). This method can document the specific viral etiology of a nonspecific finding such as an inclusion body that could be produced by several different viruses. In some cases, IHC also enhances the sensitivity of detection of viruses in tissue compared to standard light microscopy without immunostaining. Immunoperoxidase staining is the most common technique used for viral antigen detection in fixed tissue. IHC can often be performed on formalin-fixed tissue, but for some antigen–antibody combinations, sensitivity is better on fresh frozen tissue.
Fluorescent Antibody Staining
Fluorescent antibody staining is widely used to detect cell-associated viral antigens. In the direct format, a fluorescent label, usually fluorescein isothiocyanate (FITC), is conjugated directly to the antibody that recognizes the viral antigen. In the indirect format, the antiviral antibody is unlabeled and is detected by a second antibody that recognizes immunoglobulins from the animal species of origin of the antiviral antibody. The second antibody carries the fluorescent label. After staining, the specimen is viewed with epi-illumination using ultraviolet (UV) light of the wavelength needed to excite the fluorescent label. The direct method is simpler to use but requires conjugation of each antiviral antibody with the fluorescent label. The indirect method is slightly more sensitive and more versatile, because only the anti-immunoglobulin antibody has to be conjugated with the fluorescent label.
The main applications of FA staining have been to detect respiratory, ocular, cutaneous, and bloodstream pathogens. Many different respiratory specimens can be used, including nasopharyngeal swabs or aspirates, nasal washes, tracheal aspirates, and bronchoalveolar lavage fluid. In each case, the specimen is processed in the laboratory to prepare a pellet of cells, which is spotted onto one or more microscope slides. The cells are air dried, fixed in acetone, and stained with monoclonal antibodies to one or more viruses. For ocular infections, material is scraped from the conjunctiva or cornea, placed on microscope slides, and processed as described for respiratory specimens. Staining is usually directed at detection of HSV and adenovirus antigens. For cutaneous infections, the lesion is scraped with a scalpel blade or swab, and cellular material obtained is placed directly on one or more microscope slides. Alternatively, the base of the lesion can be rubbed vigorously with a swab and the swab submitted to the laboratory, where cellular material is washed from the swab and spotted onto a microscope slide. The use of cytocentrifugation to prepare the slides has been shown to improve the results.75 Staining is directed at detection of HSV and VZV. In the CMV pp65 antigenemia assay, peripheral blood leukocytes are separated from anticoagulated blood; spotted onto a microscope slide, usually by cytocentrifugation; and stained using a monoclonal antibody specific for the CMV pp65 antigen. The major limitation of FA staining is having an adequate number of cells in the specimen. Immunoperoxidase (IP) staining is similar to FA staining, except that horseradish peroxidase is used in place of a fluorescent label, making it possible to view the stain using light microscopy rather than requiring fluorescence microscopy. It is more often used to detect viral antigens in fixed tissue rather than directly in patient samples.
Enzyme Immunoassay
Enzyme immunoassay (EIA) is a versatile and widely used method that can be applied to the detection of antigens, regardless of whether they are cell associated or in fluid phase. Because intact cells in the specimen are not required, specimen integrity is less important than for FA or IP staining. This may be advantageous when specimen transport time is prolonged. A common assay format for antigen detection is the double antibody sandwich technique, shown in Figure 15.5. In this assay, a capture antibody specific for the viral antigen being sought is bound to a reaction surface, for example, the wells of a plastic microtiter tray or the surface of a plastic bead. When the specimen is added, viral antigen present in the specimen binds to the capture antibody. Bound antigen is detected using a different antiviral antibody, the detector antibody. The detector antibody can be labeled with an enzyme or can be detected by
the addition of an enzyme-labeled third antibody with specificity for immunoglobulin of the species from which the second antibody was derived. The addition of an enzyme substrate produces a color change or light emission if the enzyme is present. Thus, color change or light emission indicates that viral antigen was present in the specimen being tested. Advantages of EIA include applicability to diverse specimens and potential for automation. Laboratory instruments are now available that can perform EIA to detect either antigens or antibodies. Viruses for which antigen EIA have been widely used are RSV, influenza, rotavirus, enteric adenoviruses, HSV, HBV, and HIV.
the addition of an enzyme-labeled third antibody with specificity for immunoglobulin of the species from which the second antibody was derived. The addition of an enzyme substrate produces a color change or light emission if the enzyme is present. Thus, color change or light emission indicates that viral antigen was present in the specimen being tested. Advantages of EIA include applicability to diverse specimens and potential for automation. Laboratory instruments are now available that can perform EIA to detect either antigens or antibodies. Viruses for which antigen EIA have been widely used are RSV, influenza, rotavirus, enteric adenoviruses, HSV, HBV, and HIV.
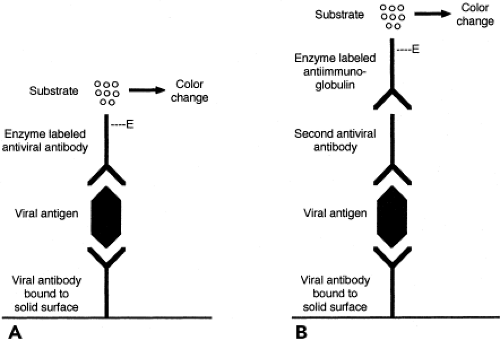 Figure 15.5. Enzyme immunoassay (EIA) for antigen detection. A: Direct. B: Indirect. |
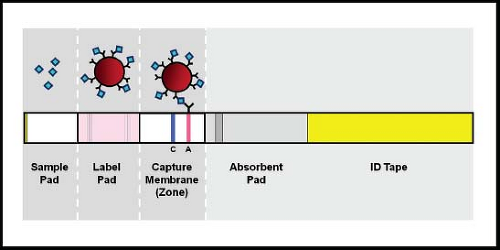 Figure 15.6. Lateral flow immunochromatographic assay. The sample is applied to the sample pad. The sample flows across the unit from left to right, drawn by the absorbent pad. Located in the label pad are one or more antibodies, each of which is conjugated to a label such as gold particles. These antibodies bind to antigen present in the sample as it moves across, forming complexes of antigen with conjugated antibody. If present, these complexes are then captured by second antibodies, located in lines in the capture membrane zone. When complexes are captured, the presence of the conjugated label causes the line to become visible. In the figure, antibodies against the target antigen are indicated by “A,” and antibodies against a control antigen are indicated by “C.” Presence of a colored line corresponding to A is a positive test. Presence of a line corresponding to C in the absence of a line corresponding to A indicates a negative test. Absence of both lines indicates that the test is not valid. (Figure provided by John Tamerius, Quidel Inc.) |
Membrane Immunoassay
The lateral flow immunochromatographic assay is a variant of EIA that was first used in home pregnancy tests. A schematic diagram of a lateral flow unit is shown in Figure 15.6. In these tests, a sample is applied directly to a membrane and is drawn across the membrane by capillary action. Antigens in the sample react first with an antibody with specificity for the antigen being detected. This antibody is conjugated to a detector label such as gold particles or fluorescein. If binding occurs, the antigen–antibody complexes sweep across the membrane until they are captured by a second antibody that is bound to the membrane. When the labeled antigen–antibody complexes are captured, a line becomes visible because of the concentration of the label into a limited physical space. Most assays also include a positive control. These assays can be configured as dipsticks or as self-contained cassettes. Standard lateral flow immunochromatographic assays do not require an instrument and are convenient for testing single samples, with results available in 5 to 20 minutes. Multiple commercial versions are now available, mainly for detection of influenza, RSV, and rotavirus. Some of the tests are sufficiently simple to perform that they have been assigned waived status under the Clinical Laboratory Improvement Amendments (CLIA), meaning that they can be performed by individuals without specific training who can be working within or outside of a certified clinical laboratory. Unfortunately, sensitivity of these tests has generally been less than that for fluorescent antibody staining, culture, or PCR. This problem was especially true for commercial influenza lateral flow tests in detecting the 2009 HIN1 pandemic.106 A recent test developed by 3M (3M Rapid Detection RSV or Influenza tests, 3M, St. Paul, MN) achieves increased sensitivity by using fluorescent beads conjugated to a detector antibody and reading the captured antigen–antibody complexes in a fluorescence reader.41
Nucleic Acid Detection
Diagnostic virology has been revolutionized by the application of nucleic acid detection techniques, which detect specific DNA or RNA sequences, and can be applied to the detection of virtually any virus. Depending on the target sequence, the assays can be specific for a single virus species or for a group of related viruses. The latter characteristic is particularly advantageous, because it allows nucleic acid detection techniques to be applied to groups of viruses (e.g., the enteroviruses) for which antigenic diversity has precluded successful application of antigen detection techniques. For example, enteroviruses, for which a rapid nonmolecular detection method had not been available, can be detected using reverse-transcription PCR assays that amplify a highly conserved sequence in the 5′ nontranslated region of the genome.14,115 An overview of applications of nucleic acid testing in diagnostic virology is shown in Table 15.7.
Table 15.7 Nucleic Acid Detection for Viral Diagnosis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The earliest attempts at nucleic acid–based diagnosis involved direct hybridization of nucleic acid probes to viral nucleic acids present in clinical specimens. Direct hybridization was never widely adopted because it lacked adequate sensitivity, requiring the presence of 104 to 105 copies of the target nucleic acid. The development of PCR118 and other nucleic acid amplification techniques overcame that sensitivity barrier and has led to the development of nucleic acid–based diagnostic tests for many viruses. Nucleic acid amplification tests (NAATs) were originally directed at viruses that were difficult or impossible to cultivate, viruses that grew very slowly in culture, and viruses for which antigen detection could not be applied
because of antigenic diversity or because the level of viral antigen in clinical specimens was too low to permit successful detection. NAATs were also very advantageous for specimens such as CSF or ocular fluid for which sample volume could be limiting. Still another advantage of nucleic acid detection as a diagnostic method is the stability of DNA as an analyte, so that detection of viral nucleic acids can be done even when conditions of transport lead to loss of virus viability. Currently NAATs are being applied to all viruses that are of interest in diagnostic virology.
because of antigenic diversity or because the level of viral antigen in clinical specimens was too low to permit successful detection. NAATs were also very advantageous for specimens such as CSF or ocular fluid for which sample volume could be limiting. Still another advantage of nucleic acid detection as a diagnostic method is the stability of DNA as an analyte, so that detection of viral nucleic acids can be done even when conditions of transport lead to loss of virus viability. Currently NAATs are being applied to all viruses that are of interest in diagnostic virology.
Nucleic Acid Amplification Assays
Target Amplification
Polymerase Chain Reaction
PCR, which is the prototype of target amplification assays, employs short oligonucleotide primers and a thermostable DNA polymerase such as Taq polymerase to amplify a segment of target DNA that is typically 100 to 1,000 base pairs (bp) in length. Classic PCR includes repetitive cycles, each consisting of denaturation, primer annealing, and extension steps that take place at different temperatures. Progression through the steps of the cycle is controlled by a thermal cycler that controls the temperature of the reaction. After PCR amplification, the PCR product (also known as the amplicon) is detected by gel electrophoresis or by one of several probe-hybridization techniques (e.g., Southern blotting). The analytic sensitivity of PCR can be as low as 1 to 10 copies of target DNA. Because of its simplicity and broad applicability, PCR remains the most widely used NAAT.
Currently, most PCR in diagnostic virology laboratories is carried out using real-time PCR in which reaction products are detected as they are synthesized.40,52 Numerous specialized instruments for running real-time PCR are now commercially available, including the LightCycler (Roche Diagnostics, Indianapolis, IN), the SmartCycler (Cepheid, Sunnyvale, CA), the ABI TaqMan 7000 series (Applied Biosystems, Foster City, CA), the Rotor Gene (Qiagen, San Diego CA), and many others. Compared with conventional PCR, real-time PCR has several important advantages. Because the accumulation of PCR product is monitored in the reaction tube, no separate detection method (e.g., gel electrophoresis) is required, thus shortening the effective assay time markedly and decreasing the risk of contamination of the laboratory environment by the amplified PCR product. The time from setting up the assay to completion can be less than 1 hour. The use of multiple fluorescent dyes with different emission wavelengths makes it possible to perform multiplex reactions with simultaneous amplification of more than one product. Of great importance, quantification of PCR targets is readily achieved because the generated fluorescence is proportional to the amount of PCR product.
Detection of PCR products in real-time PCR has generally used one of three methods, although other detection methods have also been introduced more recently. The simplest system uses the DNA binding dye SYBR Green, which emits fluorescence when it is bound to double-stranded DNA (dsDNA). When SYBR Green is included in a PCR reaction, the intensity of fluorescence is proportional to the amount of PCR product. Because SYBR Green binds nonspecifically to any dsDNA, signal is generated by undesired amplification products such as primer dimers, as well as by intended amplicons. Discrimination between different amplification products can be achieved through the use of dissociation curves, referred to as melting point analysis. Melting point analysis is performed after completion of the amplification reaction by recording fluorescence as the temperature of the reaction mix is gradually increased. When the dissociation temperature (melting point) of the dsDNA reaction product is reached, SYBR Green is released and fluorescence decreases. The melting point is affected by both length and sequence of the PCR product, and thus is precisely defined for a specific PCR product. Unintended amplification products will usually have different melting points, allowing easy discrimination from the intended PCR product.
The other two detection systems are based on the use of oligonucleotide probes that are labeled with fluorescent dyes that interact with one another according to principles of fluorescence resonance energy transfer (FRET). The hybridization probe assay format requires two oligonucleotide probes that are homologous to adjacent portions of one of the strands of the amplified DNA. The probes are chosen so that the 5′ end of one probe is within a few nucleotides of the 3′ end of the other probe. These adjacent ends are each labeled with a fluorescent dye. The dye on the 3′ end is termed the donor dye and the dye on the 5′ end is termed the acceptor dye. The required property of these dyes is that when they are in close proximity (within a distance of several nucleotides), excitation of the donor dye leads to emission of light by the acceptor dye. When both probes bind to their target sequences, the two dyes are within the proximity required for excitation of the acceptor dye and fluorescence occurs. The fluorescence intensity is proportional to the amount of PCR product.
The other detection system based on interacting fluorescent dyes is the fluorogenic 5′ exonuclease assay (also referred to as the Taqman assay). This assay uses a probe that is complementary to a segment of the intended PCR product located between the PCR primers. This probe is labeled with two fluorescent dyes, one called the reporter that is linked to the 5′ end of the probe, and the other called the quencher that is linked to the 3′ end. When they are in close proximity (i.e., bound to opposite ends of an oligonucleotide probe), the quencher prevents fluorescence by the reporter. During the extension step of PCR, the probe labeled with both dyes binds to the PCR product as it is being synthesized. During extension, the 5′ exonuclease activity of Taq polymerase cleaves nucleotides from the 5′ end of the bound probe, releasing the reporter dye away from the quencher, thus allowing it to emit light on excitation. As in the hybridization probe assay, the intensity of fluorescence is proportional to the amount of PCR product.
Because Taq polymerase uses only DNA as a template, the use of PCR to detect viral RNA sequences requires the inclusion of a reverse-transcription (RT) step before PCR (RT-PCR). The RT reaction can be performed using a devoted enzyme such as Moloney murine leukemia virus RT or avian myeloblastosis virus RT. Alternatively, heat-stable, multifunctional enzymes such as recombinant Thermus thermophilus DNA polymerase are now available that can carry out RT as well as DNA polymerase reactions. Special care must be used in specimen processing because of the susceptibility of RNA to digestion by ribonucleases that may be present in clinical samples. In addition to detecting virion RNA, RT-PCR can also be applied to the detection of viral messenger RNA. This may be particularly useful in the diagnosis of infection caused by viruses that have a latent phase in their life cycle. For these viruses, detection of viral DNA might not distinguish between
latent and productive infection, whereas detection of a messenger RNA (mRNA) expressed only in productive infection would be evidence of active viral infection.
latent and productive infection, whereas detection of a messenger RNA (mRNA) expressed only in productive infection would be evidence of active viral infection.
RNA Amplification Assays
Several reactions have been developed that are directed at amplification of RNA. Transcription-based amplification (Fig. 15.7)48,73 uses three enzymes, RT, ribonuclease (RNase) H, and T7 RNA polymerase, to amplify a target RNA sequence, employing a series of reactions that mimic the retrovirus replication scheme. An advantage of transcription-based amplification assays is that they are isothermal and do not require complicated instrumentation. The assays begin with synthesis of a DNA strand that is complementary to the RNA target, using a primer that contains a T7 polymerase binding site at its 5′ end. The resulting DNA–RNA hybrid is converted to dsDNA by the action of RNase H and a second primer that also contains a 5′ T7 polymerase binding site. The dsDNA product then serves as a template for transcription driven by T7 RNA polymerase. The newly synthesized RNA transcripts serve as templates for additional cycles of the reaction. Variants of transcription-based amplification include transcription-mediated amplification (TMA),39 self-sustained sequence replication (3SR),48 and nucleic acid sequence–based amplification (NASBA).21 Currently, NASBA assays for HIV, enteroviruses, and CMV pp67 have been approved by the FDA and are marketed by bioMérieux Inc. (Durham, NC) under the trade name NucliSENS. TMA is the basis for the Procleix assays for HIV, HCV, HBV, and West Nile virus marketed by Gen-Probe (San Diego, CA).
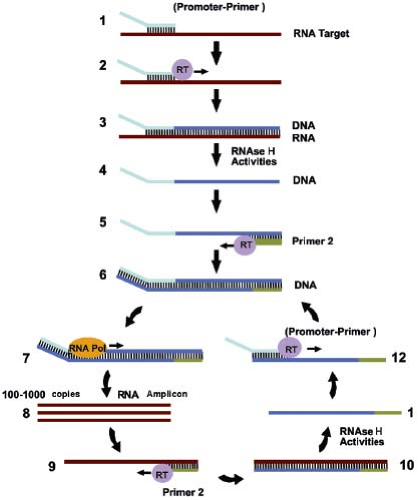 Figure 15.7. Transcription-mediated assay, also called 3SR. The 3SR reaction depends on a continuous cycle of reverse transcription and transcription reactions to replicate an RNA target by means of complementary DNA (cDNA) intermediates. Steps 1–6 depict the synthesis of a double-stranded cDNA, which is a transcription template for T7 RNA polymerase. Complete cDNA synthesis is dependent on the digestion of the RNA in the intermediate RNA–DNA hybrid (step 4) by ribonuclease (RNase) H. Transcription-competent cDNAs yield antisense RNA copies of the original target (step 7, right). These transcripts are converted to cDNA containing double-stranded promoters on both ends in an inverted repeat orientation (steps 7–12). These cDNAs can yield either sense or antisense RNA, which can re-enter the cycle. |
Signal Amplification
Examples of signal amplification assays include the branched-chain DNA (bDNA) assay,22 the hybrid capture assay,51 and the cleavase reaction.49 The bDNA assay (Fig. 15.8) uses short, branched-chain oligonucleotides to capture the target nucleic acid sequence. Other branched-chain oligonucleotides link multiple molecules of detector enzyme to the captured target. A chemiluminescent substrate allows detection of the target, and measurement of the intensity of emitted light makes it possible to quantify the input target accurately. Because the target itself is not amplified, this reaction is less susceptible than PCR to carryover contamination. Currently, FDA-approved quantitative bDNA assays for HIV and HCV are marketed by Siemens USA (Deerfield, IL) under the trade name Versant.
The hybrid capture assay (Fig. 15.9) is a signal amplification assay that involves a liquid hybridization reaction between the denatured DNA target and RNA probes specific for the viral DNA sequence of interest. If the viral DNA is present, DNA–RNA hybrid molecules are formed and are captured and detected using an antibody specific for DNA–RNA hybrids. This assay can be used as either a qualitative or quantitative assay. Currently, FDA-approved or cleared hybrid capture assays for HPV and CMV are marketed by Qiagen (Valencia, CA).83,88
The Invader assay (Fig. 15.10) is an isothermal reaction that makes use of (a) an enzyme referred to as Cleavase that functions as an FEN-1–encoded flap endonuclease,136 plus (b) two short sequence–specific oligonucleotides termed the probe oligo and the Invader oligo. To initiate the reaction, the two oligos bind to the target DNA with a short region of overlap that is designed to occur at a targeted single nucleotide polymorphism (SNP). The overlap structure occurs only when exact base pairing occurs with the targeted SNP. The resulting structure forms a substrate for the Cleavase enzyme, which cleaves the primary probe oligo at the position of overlap, releasing the 5′ portion, which is termed the flap oligonucleotide. The primary probe and the Invader oligos are present in large molar excess, resulting in generation of many cleaved flap oligos if the targeted SNP is present in the template. The flap oligo itself functions as an Invader oligo, binding to a hairpin region of a third oligo, termed a fluorescence resonance energy transfer (FRET) cassette because it contains a fluorophore and a quencher adjacent to one another at one end of the cassette. The sequence of the FRET oligo causes it to assume a hairpin configuration in which the quencher damps fluorescence from the fluorophore. Binding of the flap Invader generates another tertiary DNA structure that also functions as a substrate for the Cleavase enzyme. The resulting cleavage releases the fluorophore away from the quencher, which leads to the generation of a fluorescent signal. Reaction conditions that allow cycling of the probe oligo and the hairpin oligos cycle on and off their
respective targets result in the generation of 106– to 107-fold signal amplification per hour. Currently, FDA-approved tests using the Cleavase reaction to detect high-risk HPV types and to genotype HPV types 16 and 18 are marketed by Hologic (Bedford, MA) (formerly Third Wave Technologies) under the trade name Cervista.
respective targets result in the generation of 106– to 107-fold signal amplification per hour. Currently, FDA-approved tests using the Cleavase reaction to detect high-risk HPV types and to genotype HPV types 16 and 18 are marketed by Hologic (Bedford, MA) (formerly Third Wave Technologies) under the trade name Cervista.
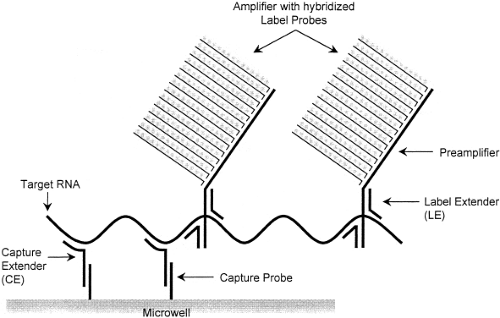 Figure 15.8. Branched-chain DNA assay as used for ultrasensitive detection of human immunodeficiency virus (HIV). After liberation of viral nucleic acid from the clinical specimen by a lysis buffer, viral nucleic acid is hybridized with two sets of bifunctional oligonucleotide probes, each of which contains sequences complementary to the target. One set of probes also contains a generic sequence complementary to a capture probe that is bound to the surface of a microtiter tray and serves to bind the target to the solid surface. The other set of probes contains a sequence complementary to preamplifier molecules. Additional specificity is achieved because two of the target-specific probes must be juxtaposed in the correct orientation to stabilize the binding of the preamplifier molecule. Each preamplifier molecule binds numerous amplifier molecules, each of which binds many subsequently added alkaline phosphatase–labeled probes. A chemiluminescent substrate is added and generated light is read by a luminometer. The cascade effect results in amplification of the signal generated from initial binding of probes to the target and allows for detection and quantification of nucleic acid present in the specimen. (Courtesy of Bayer Diagnostics, Emeryville, California.) |
The loop-mediated isothermal amplification (LAMP) (Fig. 15.11) is an amplification reaction that employs four primers and a strand-displacing DNA polymerase.96 It was developed originally by Eiken Chemical Co. (Tokyo, Japan), and additional detailed diagrams of the assay can be viewed on the Eiken web page: http://loopamp.eiken.co.jp/e/lamp/index.html. The inner primers have sense and antisense sequences. When they initiate DNA synthesis, the result is a stem-loop structure, which itself serves as a target for additional amplification. The resulting reaction can produce 109 copies of target DNA or RNA in less than 1 hour under isothermal conditions. The reaction can be monitored by measuring turbidity, which results from the accumulation of magnesium pyrophosphate, a by-product of the reaction. Alternatively, the synthesis of dsDNA can be measured using an agent such as SYBR Green, which generates fluorescence when it intercalates between the strands of dsDNA. The reaction has high specificity by virtue of the inclusion of multiple primer sets. The isothermal nature of the reaction, which obviates the requirement for a thermal cycler, makes LAMP attractive for use as a point-of-care test or for use in the developing world.
Contamination
A general concern regarding highly sensitive nucleic acid amplification assays is the occurrence of false-positive findings resulting from contamination of the reaction with exogenous nucleic acids. The source of contaminating nucleic acids can be either amplified products from previous reactions or viral nucleic acids present in a different specimen, especially one containing a high level of the intended target. Prevention of contamination requires fastidious attention to procedural detail within the laboratory. A number of specific preventive techniques are widely used to prevent carryover contamination after PCR, including the use of positive displacement pipettes or plugged pipette tips to block aerosol production, UV irradiation of reaction components (not including Taq polymerase and PCR primers that can be inactivated by UV irradiation), use of small working aliquots, and frequent cleaning of equipment and laboratory surfaces with 10% bleach. An important precaution is the physical separation of the laboratory into
sections where (a) the specimen is prepared, (b) the PCR reaction is set up, (c) the PCR amplification reaction takes place, and (d) the detection of amplification products is carried out. After PCR has taken place, the reaction tube is opened only in the section of the laboratory devoted to product detection. Techniques such as the inclusion of uracil-N-glycosylase or psoralen and isopsoralen derivatives in the reaction mixture were developed to minimize the danger of PCR contamination.104 Real-time PCR is less susceptible to contamination because product detection does not require manipulating the amplified product. For any form of nucleic acid testing, an additional concern is contamination from one specimen to another, which may occur during specimen handling in the laboratory. Inclusion of negative controls in the reaction setup is important in order to detect this form of contamination.
sections where (a) the specimen is prepared, (b) the PCR reaction is set up, (c) the PCR amplification reaction takes place, and (d) the detection of amplification products is carried out. After PCR has taken place, the reaction tube is opened only in the section of the laboratory devoted to product detection. Techniques such as the inclusion of uracil-N-glycosylase or psoralen and isopsoralen derivatives in the reaction mixture were developed to minimize the danger of PCR contamination.104 Real-time PCR is less susceptible to contamination because product detection does not require manipulating the amplified product. For any form of nucleic acid testing, an additional concern is contamination from one specimen to another, which may occur during specimen handling in the laboratory. Inclusion of negative controls in the reaction setup is important in order to detect this form of contamination.
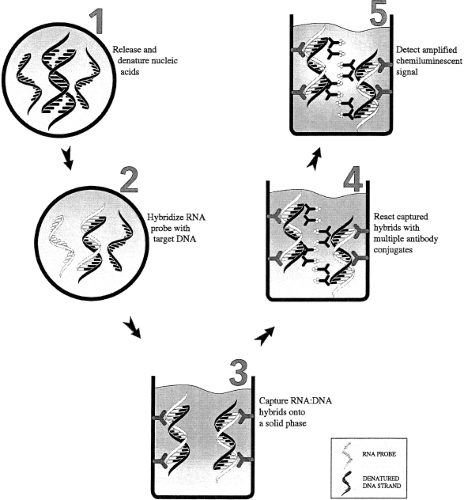 Figure 15.9. Hybrid capture assay. In step 1, target DNA is released and denatured. In step 2, RNA probe hybridizes with target DNA. In step 3, RNA–DNA hybrids are captured by an antibody to RNA–DNA hybrids that is bound to the sides of the reaction vessel. In step 4, an enzyme-conjugated antibody that recognizes RNA–DNA hybrids binds to the hybrids. In step 5, a chemiluminescent substrate is added, and light is emitted if hybrids have been formed. (Courtesy of Digene Corporation, Beltsville, Maryland.) |
Nucleotide Sequencing
Sequencing of PCR amplification products can be carried out using the cycle-sequencing reaction57 or pyrosequencing. Sequence information can be used for several purposes including precise identification of a virus, genotyping, and the presence of mutations associated with antiviral drug resistance or unusual clinical manifestations. Genotypic resistance assays are performed most commonly for HIV, CMV, HBV, and influenza A virus (see section on antiviral susceptibility testing). For HBV, nucleotide sequencing of the nucleocapsid gene
and its associated promoter is also used for detection of core promoter and precore mutations, which have been associated with unusually severe disease and progression to chronic infection.97 High-throughput (“next-generation”) sequencing will probably have important applications in diagnostic virology, including for antiviral susceptibility testing, for identification of viruses growing in cell culture that are not identifiable by conventional means,130 and for discovery of known15,154 and novel viruses.20,99
and its associated promoter is also used for detection of core promoter and precore mutations, which have been associated with unusually severe disease and progression to chronic infection.97 High-throughput (“next-generation”) sequencing will probably have important applications in diagnostic virology, including for antiviral susceptibility testing, for identification of viruses growing in cell culture that are not identifiable by conventional means,130 and for discovery of known15,154 and novel viruses.20,99
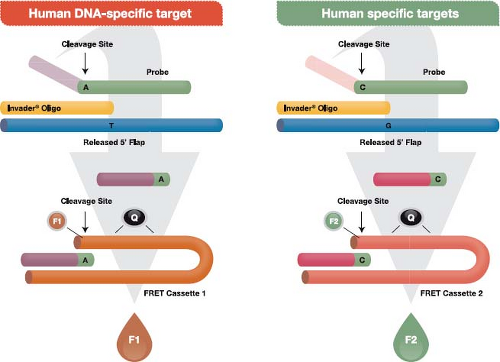 Figure 15.10. Invader assay. The left side of the figure shows a positive control reaction for human histone 2DNA and the right side shows the reaction to detect human papillomavirus (HPV). For each reaction, the probe oligo and the Invader oligo bind to the target, with a short region of overlap occurring at a targeted single nucleotide polymorphism (SNP). This results in a structure that is cleaved by the activity of an enzyme called “Cleavase,” releasing the 5′ end of the probe oligo at the position of overlap. The sequence of the released portion is homologous to the 3′ end of a third oligo, which includes a fluorophore (F1 for the human DNA reaction and F2 for the HPV reaction) and a quencher (Q) and is referred to in the figure as the FRET cassette. The hairpin configuration assumed by the FRET cassette brings the quencher sufficiently close to the fluorophore to allow its fluorescence to be quenched. Binding of the released portion of the probe oligo to the FRET cassette results in a region that is another substrate for Cleavase. The activity of Cleavase releases the fluorophore, resulting in generation of a fluorescent signal. (Courtey of Hologic.) |
Microarray Technology
High-density microarrays consist of hundreds or thousands of oligonucleotide probes bound to a solid phase, usually a small silicon chip. Amplified nucleic acid can be hybridized to the array, and binding to specific probes can be identified. Use of probes representing all possible nucleotide sequence variations within a target sequence allows very rapid determination of nucleotide sequence.45 Multiple sequence variants present within the specimen can also be detected. Microarray technology has the potential to allow simultaneous detection of multiple infectious disease pathogens, viral and nonviral. An early application in diagnostic virology has been for rapid sequencing to detect HIV mutations associated with resistance to antiretroviral drugs.70 In another application, sequences representing all sequenced viruses have been selected to create a microarray that can be used to discover unknown viruses. This microarray was successful in categorizing the agent of severe acute respiratory syndrome (SARS) as a new coronavirus.145 The major application of microarray technology in diagnostic virology to date has been the use of liquid arrays, which are used for the simultaneous detection of multiple respiratory viruses, as exemplified by the xTAG RVP described later. Future uses of liquid arrays and other microarray technology under development include assays to detect multiple viruses (as well as other classes of pathogens) associated with gastroenteritis, sexually transmitted diseases, sepsis, and bioterrorism.
Commercial Nucleic Acid Amplification Platforms and Tests
Government Regulation
In the United States, commercially marketed diagnostic tests and test platforms are regulated by the FDA. Before marketing
a test for diagnostic use, a manufacturer must receive authorization from the FDA. This authorization can take one of several forms. If there is no similar test on the market, the manufacturer must apply for premarket approval (PMA). This is a rigorous and expensive process that requires the manufacturer to provide evidence for the accuracy and utility of the proposed test. A test that is “substantially equivalent” to a test that has been previously approved or cleared by the FDA is cleared for use through a mechanism referred to as 510K, after the section of the Federal Food, Drug, and Cosmetic Act that describes the process. Some diagnostic test materials are marketed under the classification “analyte-specific reagent (ASR).” This term is used by the FDA to refer to reagents or materials, including nucleic acid primers and probes, that must meet certain specifications set by the FDA, including being produced utilizing current “good manufacturing practices.” In addition, clinical laboratory tests carried out using ASRs must have appropriate labeling appended to the reported results stating that the test was developed and validated by the laboratory and has not undergone FDA clearance or approval. ASRs are intended to be used as components of laboratory-developed tests and can be sold only to laboratories that are qualified under CLIA to perform highly complex testing. ASRs are excluded from the need for premarket approval by the FDA. They cannot be marketed as complete test kits and must be sold without instructions for use or claims regarding performance characteristics.
a test for diagnostic use, a manufacturer must receive authorization from the FDA. This authorization can take one of several forms. If there is no similar test on the market, the manufacturer must apply for premarket approval (PMA). This is a rigorous and expensive process that requires the manufacturer to provide evidence for the accuracy and utility of the proposed test. A test that is “substantially equivalent” to a test that has been previously approved or cleared by the FDA is cleared for use through a mechanism referred to as 510K, after the section of the Federal Food, Drug, and Cosmetic Act that describes the process. Some diagnostic test materials are marketed under the classification “analyte-specific reagent (ASR).” This term is used by the FDA to refer to reagents or materials, including nucleic acid primers and probes, that must meet certain specifications set by the FDA, including being produced utilizing current “good manufacturing practices.” In addition, clinical laboratory tests carried out using ASRs must have appropriate labeling appended to the reported results stating that the test was developed and validated by the laboratory and has not undergone FDA clearance or approval. ASRs are intended to be used as components of laboratory-developed tests and can be sold only to laboratories that are qualified under CLIA to perform highly complex testing. ASRs are excluded from the need for premarket approval by the FDA. They cannot be marketed as complete test kits and must be sold without instructions for use or claims regarding performance characteristics.
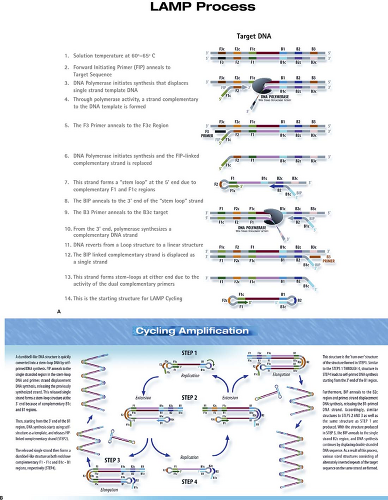 Figure 15.11. Loop-mediated amplification reaction (LAMP assay). A: Steps leading up to the formation of the “dumbbell” structure. B: The cycling steps of the reaction, which lead to extremely rapid amplification. (Courtesy of Meridian Biosciences.) |
Individual laboratories may also develop their own assays that are intended for use only in that laboratory. These tests, referred to as “laboratory-developed tests” (sometimes also referred to as “home-brew assays”), are not actively regulated by the FDA, but instead are under the jurisdiction of CLIA passed by the U.S. Congress in 1988. Laboratory-developed tests may use ASRs as components of the test and may also use reagents obtained from diverse sources not specifically labeled as ASRs. The performance of laboratory-developed tests must be verified according to specific criteria established by CLIA.
Overview of Commercial Assays and Platforms
Since the last edition of this book, there has been a dramatic increase in molecular diagnostic tests and test platforms that have been approved or cleared by the FDA for the detection of pathogenic viruses. Tests and platforms approved or cleared by the FDA at the time this chapter was written are shown in Tables 15.8 and 15.9. These tables are adapted from tables that are maintained by the Association for Molecular Pathology (AMP) and are accessible on the AMP website (http://www.amp.org). No single platform permits the detection of all medically important viruses, so laboratories whose mission is broad-based viral diagnosis will need to employ multiple platforms. Currently, laboratories vary widely in the relative proportion of testing that is carried out using commercial platforms versus laboratory-developed tests. Likewise, laboratories that provide different types of service will employ different platforms. For example, hospital-based laboratories may emphasize platforms that provide rapid turnaround time, commercial laboratories may employ platforms that allow high specimen throughput, and government laboratories may employ platforms that allow for testing of viruses of public health significance. The following paragraphs describe several categories of commercial molecular diagnostic tests.
Multiplex Assays
Multiplex PCR refers to PCR reactions in which more than one primer set is incorporated into the reaction mix, allowing the detection of multiple targets. Multiplex assays may be developed either as commercial tests or as laboratory-developed tests. Currently, multiplex assays are being developed for important specimen types to amplify various viral and other microbial agents that are pathogenic at the specific body site. This process is most highly developed for respiratory samples, for which five different multiplex assays are currently cleared by the FDA.
The Proflu+ assay marketed by Prodesse (now Gen-Probe, San Diego, CA), the Simplexa Flu A/B & RSV Test assay marketed by Focus (Cypress, CA), and the Verigene RV+ (Nanosphere, Northbrook, IL) are multiplex real-time assays that use separate primer pairs to simultaneously amplify influenza A and B viruses and RSV plus an internal control. In the Proflu+ and Simplexa assays, unique probes for each virus and the internal control are labeled with separate fluorophores, allowing determination of which virus or viruses have been amplified. The Simplexa assay is run on the 3M Integrated Cycler, an innovative instrument produced by 3M (St. Paul, MN) that has the capability to run 96 assays in 1 hour. Multiple different specimens can be loaded on the instrument and multiple assays can be run simultaneously. The Verigene RV assay uses a very innovative system to detect amplified products in which the amplicons are captured by capture probes that are bound to specific sites on a microarray. Captured amplicons are then detected by binding of silver-coated gold nanospheres that have been tagged with oligonucleotides that are specific for the amplicons and are visualized by light scatter.
The xTAG Respiratory Virus Panel (RVP) marketed by Luminex (Toronto, Ontario, Canada) was the first large multiplex assay to be cleared by the FDA. This assay detects the following viruses in nasopharyngeal swabs: influenza A and B, influenza A subtypes H1 and H3, RSV A and B, parainfluenza 1 through 3, human metapneumovirus (hMPV), enterovirus/rhinovirus, and adenovirus.85,98 Primers for influenza A hemagglutinin subtype H5, parainfluenza 4, and coronaviruses OC43, 229E, NL63, HKU1, and SARS are also included in the reaction but are not currently cleared for use in the United States. The first step in the reaction is reverse transcription followed by a multiplex PCR reaction that includes primer sets for each target. The resulting amplicons are prepared for detection through a reaction called target-specific primer extension (TSPE). The 3′ end of each of the primers used in TSPE is homologous to one of the amplicons from the multiplex PCR reaction. The 5′ end of the primer consists of a short “tag” sequence that will be used to capture the TSPE product. During the extension reaction that follows annealing of the TSPE primers, biotin-labeled deoxycytidine triphosphate (dCTP) is incorporated into the primer extension product. Detection of the TSPE products uses a set of polystyrene microspheres composed of 100 members. Each member microsphere incorporates two fluorescent dyes in a unique ratio that allows identification by flow cytometry using two lasers, each of which emits light of the appropriate wavelength to excite one of the two fluorescent dyes. Bound to each member of the microsphere set is a short oligonucleotide (“antitag”) with sequence homology to one of the tags introduced in the TSPE reaction. These tags are specific for each amplicon amplified by the
multiplex reaction. Following the TSPE reaction, the reaction mix is incubated with the microspheres, allowing the microspheres to specifically capture any TSPE products that have been produced. A fluorescent detection signal is generated through the inclusion of streptavidin-phycoerythrin, which binds to the biotin molecules incorporated into the amplicons. As the microspheres pass through the flow cell, they are interrogated by the lasers, allowing identification of the specific microsphere set member and determination of whether or not a TSPE product has been bound. Computer software analyzes the fluorescent events and determines for each virus and internal control whether a threshold of detection has been surpassed. Recently a version that requires less time to complete, called xTAG RVP Fast, was cleared by the FDA for detection of influenza A, influenza A subtype H1, influenza A subtype H3, influenza B, RSV, hMPV, rhinovirus, and adenovirus.
multiplex reaction. Following the TSPE reaction, the reaction mix is incubated with the microspheres, allowing the microspheres to specifically capture any TSPE products that have been produced. A fluorescent detection signal is generated through the inclusion of streptavidin-phycoerythrin, which binds to the biotin molecules incorporated into the amplicons. As the microspheres pass through the flow cell, they are interrogated by the lasers, allowing identification of the specific microsphere set member and determination of whether or not a TSPE product has been bound. Computer software analyzes the fluorescent events and determines for each virus and internal control whether a threshold of detection has been surpassed. Recently a version that requires less time to complete, called xTAG RVP Fast, was cleared by the FDA for detection of influenza A, influenza A subtype H1, influenza A subtype H3, influenza B, RSV, hMPV, rhinovirus, and adenovirus.
Table 15.8 Viral Molecular Tests Approved or Cleared by the Food and Drug Administrationa
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|