1 Introduction
Viruses were first discovered at the end of the 19th century, although the symptoms they cause were identified much earlier. One of the earliest pieces of evidence is the symptoms of poliomyelitis in an Egyptian priest depicted on a hieroglyph. Virus discovery came about when the cause of an infectious disease (rabies), could not be explained by the presence of bacteria. Unlike bacteria, the ‘infectious materials’ were not retained by filtration and thus viruses were then referred to as ‘filterable agents ’ or ‘filterable viruses’. Until the advent of electron microscopy in the 1940s, only the chemical nature of viruses (i.e. proteins and nucleic acid) could be identifiedand their infectivity was mainly studied in animal models. The observation of a virus by electron microscopy and the development of cell tissue culture started the golden era of virology. Since then a large number of viruses have been isolated, their structure identified and their replication understood, leading to the design of potent antiviral drugs and effective vaccines. Progress in virology over the last 50 years have been considerable, leading to the eradication of smallpox following a worldwide vaccination programme, and the likely future eradication of poliovirus. For the first time in human history an infectious disease has been vanquished. Despite such an achievement, much progress is still required to combat other viruses. For example, despite large financial investment and high profile studies, a cure or vaccine for HIV is still not available. The number of commercially available antivirals is also still limited, when compared to the number of antibiotics. Within the last few years, diseases caused by viruses in human and animals have reminded us that viral infections can easily spread and cause epidemics and pandemics. Recent examples are ‘swine flu ’ and ‘avian flu’, both caused by influenza viruses, and also an outbreak of severe acute respiratory syndrome (SARS), the virus originating from Asia and spreading worldwide, and outbreaks of foot and mouth virus affecting cattle, sheep and pigs that had important economic consequences in the UK and mainland Europe.
2 General structure of viruses
Viruses are extremely diverse in size and in structure. The smallest virus is approximately 28 nm in size (poliovirus), while the largest is 750 nm (mimivirus). In simplistic terms, a virus consists of viral nucleic acid within a protein core, the capsid (also referred to as the coat), possibly surrounded by a lipidic envelope (Table 5.1). In reality, there are many differences between viruses in terms of nucleic acid, capsid structure, number of coats and envelope composition. Such differences account for the high diversity of viruses and the differences in their properties, notably their resistance to antiviral drugs and viricidal agents. Viral classification (Figure 5.1) is based on the physical and chemical properties of viruses, their structure and morphology (Figure 5.2).
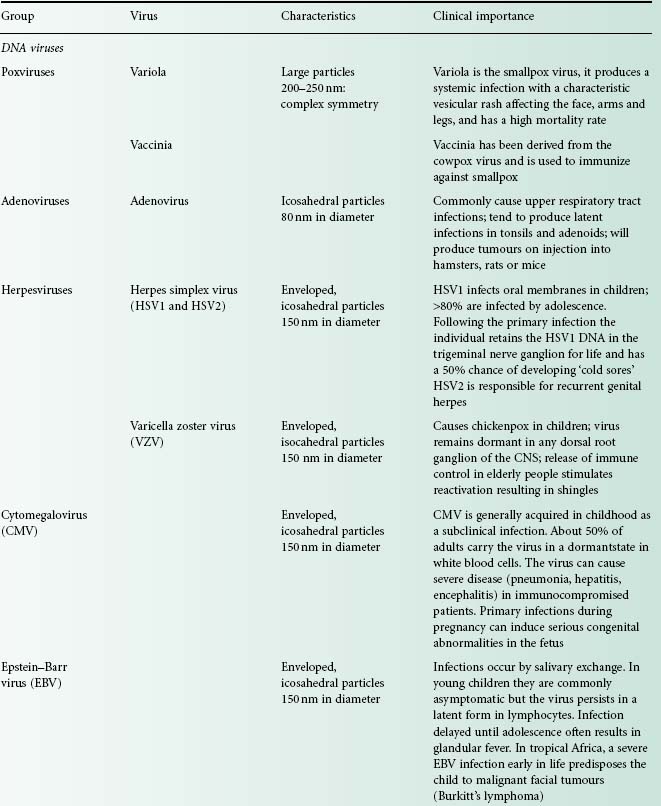
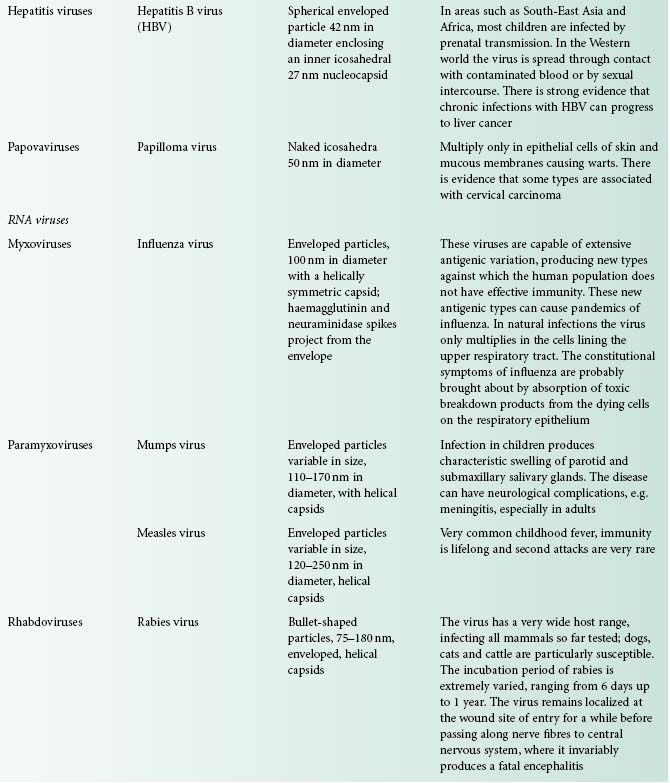


Figure 5.2 The morphology of a variety of virus particles. The large circle indicates the relative size of a Staphylococcus cell.
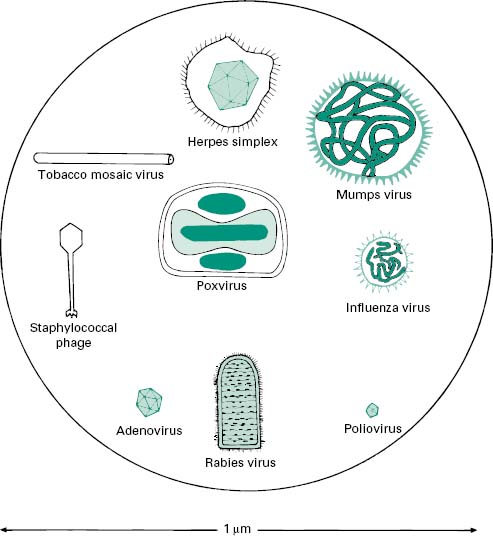
2.1 Viral nucleic acid
The viral genome is composed of either DNA or RNA. It can be double stranded (ds) or single-stranded (ss), linear (e.g. poliovirus) or circular (e.g. hepatitis B virus), containing several segments (e.g. influenza—eight segments of minus-sense ss RNA) or one molecule (e.g. poliovirus). Six groups can be distinguished depending on their nucleic acid content (Table 5.2). Viruses that contain plus-sense ss RNA (e.g. poliovirus) can have their genome translated directly by the host ribosome. The nature of the viral nucleic acid is important for the effectiveness of antiviral treatments (see below). For example, retroviridae such as HIV require a specific virus-encoded enzyme, a reverse transcriptase, to convert their ss RNA into ss DNA, to be able to replicate within the host cell. This enzyme is a primary target site of many antiviral drugs.
Table 5.2 Examples of types of nucleic acid in human and bacterial viruses
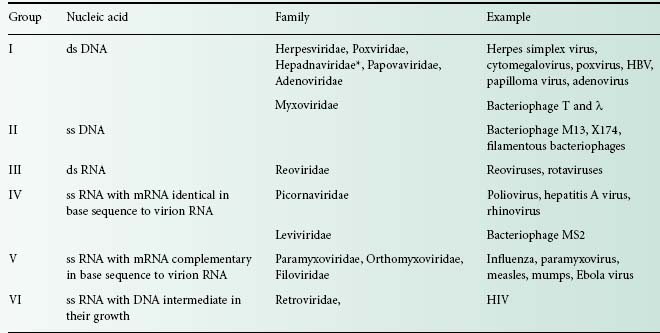
*Hepadnaviridae genome contains partially ds DNA and partially ss DNA.
On some occasions, the viral genome has been shown to be infectious, i.e. to cause an infection. Such an observation is important when one considers viricidal agents that damage the viral capsid or envelope but not the viral nucleic acid. Furthermore, in laboratory conditions, the phenomenon of multiplicity reactivation has been observed with poliovirus whereby random damage to viral capsid and nucleic acid following treatment with hypochlorite, a biocide intensively used for surface disinfection, resulted in complementary reconstruction of an infectious particle by hybridization of the gene pool of the inactivated virus. This again underlines the necessity of rendering the viral nucleic acid non-infectious following a viricidal treatment.
2.2 Viral capsid
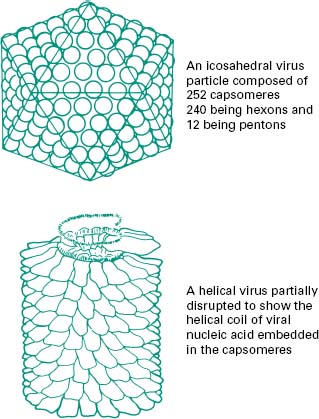
The function of the capsid is to protect the viral nucleic acid from detrimental chemical and physical conditions (e.g. disinfection). The capsid is composed of a number of subunits named capsomeres genetically encoded by the viral genome. The nature and association of the capsomeres are fundamental for the virus, as they give the shape of the capsid, but also provide the virus with resistance to chemical and physical agents. The assembly of the capsomeres results in two different architectural styles—icosahedral and helical symmetries (Figure 5.3); in mammalian viruses, a more complex structure can be found, where several proteinaceous structures envelope the viral genome core (e.g. poxviruses, rhabdoviruses). Bacterial viruses (bacteriophages) also show a complex structure consisting of a capsid head, a tail and tail fibres (see section 8.1). The nature of the capsomeres in certain viruses allows for the selfassembly of the capsid within the host cell. The capsomeres are held together by non-covalent intermolecular forces. Such assembly also allows the release of the viral genome following dissociation of the non-covalently bonded subunits. These subunits offer considerable economy of genetic information within the viral genome since only a small number of different subunits contribute to the formation of the capsid. Viruses with an icosahedral capsid usually have capsomeres in the form of pentons and hexons. The number of these subunits varies considerably between viruses: for example, adenovirus is constructed from 240 hexons and 12 pentons, whereas the poliovirus is composed of 20 hexons and 12 pentons, forming a much smaller structure. Viruses with a helical capsid (e.g. influenza and mumps viruses) have their subunits symmetrically packed in a helical array, appearing like coils of wound rope under electron microscopy. Although the core of such a virus is hollow, the viral nucleic acid is embedded into ridges on the inside of each subunit and does not fill the hollow core. Such a close association between viral nucleic acid and capsid proteins can explain the damage caused to the nucleic acid following disaggregation of the capsid after chemical or physical treatments. Table(5.1)
2.3 Viral envelope
The viral capsid can be surrounded by a lipidic envelope, which originates from the host cell. The envelope is added during the replication process or following excision of the viral progeny from the host cells. The envelope can come from the host cell nuclear membrane (e.g. herpes simplex virus) or the cytoplasmic membrane (e.g. influenza virus). One characteristic of the viral envelope is that host proteins are excluded, but proteins encoded by the viral genome are present. These viral proteins play an important serological role. Enveloped viruses are generally considered to be the most susceptible to chemical and physical conditions and do not survive well on their own outside the host cell (e.g. on surfaces), although they can persist longer in organic soil (e.g. blood, exudates, faeces). Lipids in viruses are generally phospholipids from the host envelope, although glycolipids, neutral fats, fatty acids, fatty aldehydes and cholesterol can be found.
2.4 Viral receptors
In addition to these structures, glycoproteins can be found usually protruding from the viral capsid or embedded in the envelope. These virusencoded structures are important for viral infectivity as they recognize the host cell receptor site conveying viral specificity. In bacteriophages, these structures can take the shape of tail fibres.
3 Virus-host cell interactions
Viruses need to interact with a host cell as they cannot reproduce on their own. They have no metabolism and cannot synthesize their own proteins, lipids or nucleic acids. Thus viruses can be considered as true intracellular parasites that grow within living cells and use their energy and synthetic machinery to produce viral components. The production and excision of viruses from the host cell will result in cell death, although this might not be immediate. Following the replication of one virus within the host cell, hundreds of new viruses (virus progeny or virions) can be released and infect adjacent cells (within a tissue). The propagation from one infected cell to new cells, and the subsequent destruction of tissue or cells, provides signs of the viral disease.
On the basis of host specificity, three major viral groups can be distinguished: (1) viruses of bacteria and blue-green algae, (2) plant viruses and (3) animal (including insect) viruses. Viruses are usually very specific and rarely cross species barriers, although there are some exceptions, such as rabies and influenza that can cause diseases in both animals and humans. Viruses can also be asymptomatic in certain hosts where they do not cause an infection; the host becomes a reservoir and can transmit the virus to a susceptible recipient (e.g. transmission of yellow fever to humans by mosquitoes).
Viruses can interact with the host cell in five different ways: (1) multiplication of the virus and destruction of the host cell upon release of the viral progeny, (2) multiplication of the virus and release of the virions without the immediate destruction of the host cell, (3) survival of the virus in a latent stage without noticeable changes to the infected cell, (4) survival of the infected cell in a dramatically altered or transformed state (e.g. transformation of a normal cell to one having the properties of a cancerous cell) and (5) incorporation of the viral nucleic acid in the host cell genome without noticeable changes to the infected cell. The interaction with the host cell will vary between viruses, but will generally follow one of these five routes.
There is a great diversity in viral infections and viral diseases. Many viral infections are asymptomatic or ‘silent’ whereby the virus replicates within the host but does not produce symptoms of a disease. Other common infections produce some mild symptoms, such as a low-grade fever and a ‘runny nose’. This is the case of the common cold, caused by rhinoviruses, from which patients make a full recovery within a few days. At the other end of the spectrum, some viruses kill their host very quickly following infection, as in the case of haemorrhagic viruses such as the Ebola virus. On other occasions a range of symptoms can be observed in different hosts. This has been the case recently with ‘swine flu’, which produced a range of symptoms, from a slight fever to full influenza including a high fever, vomiting, and dizziness. Other problematic viruses might not cause immediate symptoms, but following the systematic destruction of host cells will lead to an incurable disease; e.g. HIV and oncogenic (tumour) viruses.
3.1 HIV
HIV is an enveloped virus with a cone-shaped nucleocapsid containing two copies of a positive-sense ss RNA molecule and the enzyme reverse transcriptase. The viral genome encodes for the following genes: env, envelope proteins; gag, capsid proteins; pol, enzymes involved in viral multiplication; and tat, enzymes regulating host metabolism. Seventy glycoprotein spikes (gp120) project from the envelope and interact specifically with the CD4 protein receptor on the T-lymphocyte (Chapter 9). The HIV core penetrates the cell cytoplasm following membrane fusion and is uncoated releasing the two RNA molecules and the reverse transcriptase into the cytoplasm. The RNA is copied by reverse transcriptase into ss DNA, which is then duplicated to form a ds DNA copy of the original viral RNA genome. This DNA moves into the host cell nucleus where it is integrated as a provirus into a host cell chromosome.
The provirus can lie dormant in the cell or can be expressed, producing viral mRNA and proteins and resuming the multiplication cycle producing virions. The viral mRNA is polycistronic, producing polyproteins that need to be cleaved by a specific virus-encoded protease. Following the assembly of viral proteins and viral RNA, the virions bud off the infected cells.
At present there is no prospect of any drugs that can be developed to eliminate proviruses from infected cells. Current therapy has evolved around maintaining a high count of T4 helper lymphocytes by regulating the viral load produced by infected cells (up to 1010 viral particles produced continuously per day). Indeed, the ultimate decrease in T4 helper cells (below 200/ml of blood) seriously compromises the host immune system and allows infection by a range of opportunist pathogens including fungi, protozoa, bacteria and other viruses. Currently, antiviral treatments (highly active antiretroviral therapy—HAART) combining a protease inhibitor and two reverse transcriptase inhibitors, reduce the number of HIV particles and slow the progression of the disease by restoring and maintaining the number of T4 helper lymphocytes. However, this triple therapy does not eliminate the virus completely. Patients who stop using the drugs experience a rapid rebound in levels of the virus in the blood and progression of the disease. The inability to eliminate the virus completely calls for lifelong therapy that is prohibitively expensive for countries with very limited health budgets.
3.2 Tumour viruses
There is a suggestion that 20% of human cancers might have a viral origin. Virus infected cells change dramatically, acquiring the characteristics of tumour cells exhibiting uncontrolled growth. For example, the Epstein-Barr virus (EBV) has been associated with the formation of lymphomas and nasopharyngeal carcinomas, the hepatitis B and C viruses with hepatocellular carcinoma, human papilloma viruses with cervical cancer, human T-cell lymphotrophic virus type 1 with adult T-cell leukaemia/lymphoma syndrome and HIV with Kaposi’s sarcoma.
There are no identified single mechanisms by which viruses induce tumours. The acquisition of viral genes by the host must be followed by other events such as environmental or dietary exposures to chemical carcinogens for cancer to occur. For example, there might be an association between EBV and malaria to trigger Burkitt’ s lymphoma in young children in Africa. EBV and consumption of smoked fish might trigger nasopharyngeal carcinoma in adults in China. The development of liver cancer following infection with hepatitis B virus (HVB) might be triggered by high alcohol consumption, smoking and exposure to fungal toxin (aflatoxins), events that damage the liver.
It is known that the viral genome of oncogenic viruses can integrate the host DNA. Indeed, viral DNA has been recovered from infected cells. Following integration of the provirus, the regulation of cell growth and division can be affected. There is no means of eradicating proviruses. However, progress in the prevention of papillomavirus has been made by designing an effective vaccine combined to a successful vaccination programme that is responsible for an important decrease in cervical cancer cases.
4 Multiplication of h uman viruses
The objective of the replication cycle is to ensure the multiplication of the virus with the formation of identical viral progeny. Viruses differ in their replication cycle and the time to produce and release new virions. The multiplication cycle of human viruses is generally slow, from 4 to more than 40 hours. Bacterial viruses are generally faster and can take as little as 20 minutes to replicate within the bacterial host. The replication cycle can be divided into six distinct phases (Figure 5.4) that are common to all viruses, although detail within each phase varies greatly between viruses. Understanding the viral multiplication process is crucial for the development of new antiviral drugs.
Figure 5.4 Diagrammatic representation of the production and release of viral particles from an infected cell.
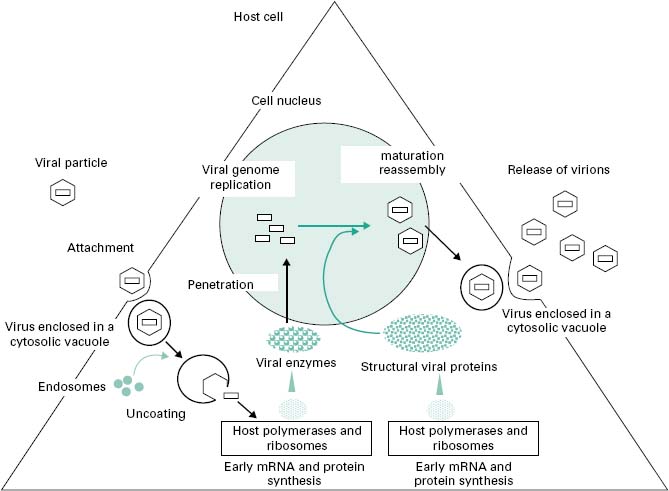
4.1 Attachment to the host cell
Viral attachment to the cell surface can be divided into three phases: (1) an initial contact mainly dependent brownian motion, (2) a reversible phase during which electrostatic repulsion is reduced and (3) irreversible changes in virus-receptor-host-receptor configuration that initiates viral penetration through the cell membrane.
All viruses possess receptors on their surface, usually in the form of glycoproteins embedded in the viral envelope or protruding as spike from the viral capsid. These structures recognize and bind receptors on the host cell and provide the virus with its high specificity although different viruses might share the same receptor. The virus-cell recognition event is similar to any protein-protein interaction in that it occurs through a stereospecific network of hydrogen bonds and lipophilic associations. For example, the haemagglutinin receptor of influenza virus binds the terminal glycoside residues of gangliosides (cell surface glycolipids) of the target cell leading directly to the virus particle adhering to the cell. Similarly, the interaction between the HIV receptor (i.e. gp120) and the T-lymphocyte receptor (i.e. CD4) has been intensively studied.
4.2 Penetration of the viral particle
Following the irreversible attachment of the virus to the host cell, penetration of the virus through the cell membrane is initiated following two energy-dependent mechanisms, endocytosis or fusion. A third mechanism has been identified in some bacteriophages that can inject their nucleic acid inside the bacterium (see section 8.1). During endocytosis, the association between virus receptor and host receptor triggers a number of mechanisms that draw the cell membrane to engulf the virus particle forming a cytosolic vacuole. This process is widespread among non-enveloped viruses, but is also used with some enveloped viruses such as influenza (orthomyxoviruses). Certain enveloped viruses (e.g. herpes simplex virus, HIV) can penetrate following fusion of their envelope with the host cell membrane, liberating the viral capsid within the cell cytoplasm.
4.3 Uncoating of the viral particle
Following penetration of the virus in a vacuole or directly into the cell cytoplasm, the viral nucleic acid then needs to be released from the capsid/coat(s) to initiate viral replication. This is the uncoating process. For viruses that penetrate by endocytoses, the acidification of the cytosolic vacuoles following endosome fusion induces a conformational change in the capsid and the release of viral nucleocapsid (some helper proteins are associated with the viral nucleic acid) into the cytoplasm. For certain viruses, such as reovirus, only a partial uncoating is necessary for the expression of the viral genome. The release of the nucleocapsid from vacuoles can occur in the cytoplasm, close to the nucleus or within the cell nucleus.
4.4 Replication of viral n ucleic a cids and t ranslation of the genome
This stage of viral replication ensures that (1) the host cell synthesis machinery is taken over by the virus, and (2) the viral genome is replicated. The structure, size and nature of the viral genome are extremely diverse and thus this stage of the viral multiplication cycle reflects this diversity. Three main mechanisms are, however, common to all viruses: the transcription of viral genes into viral mRNA, the translation of the viral genome into proteins, and the replication of the viral genome. Early transcription and translation usually occurring immediately after the release of the nucleocapsid in the cytoplasm is also common, and ensures the production of early proteins such as viral polymerases, and the hijacking of the cell synthesis machinery. In addition, some viruses can encode for genes the products of which regulate the host synthetic processes according to the needs of the virus (e.g. tat gene in HIV).
The replication of the viral genome depends on the type of nucleic acid carried by the virus. The positive strand RNA in viruses such as the poliovirus can be used directly as mRNA following the acquisition of a terminal sequence from the host cell. Negative strand RNA (e.g. in influenza virus) is transcribed into a positive RNA complementary in base sequence to the parent RNA using an RNA-dependent RNA polymerase carried by the virus. In ds DNA viruses (e.g. adenoviruses), the nucleic acid passes into the nucleus where it is usually transcribed by a host DNA-dependent RNA polymerase. In some viruses (e.g. poxvirus), this enzyme is contained within the virus and released during uncoating, allowing the viral genome to be replicated in the cell cytoplasm. In retroviruses (e.g. HIV), a singlestranded proviral DNA is produced from the viral ss RNA by a viral enzyme called reverse transcriptase. This unique enzyme acts both as an RNA-and DNA-directed DNA polymerase, and has associated RNAase activity. The proviral DNA can be transported to the cell nucleus where it can be integrated within the cell host genome by a viral integrase.
One important difference between the host cell and the virus is in the nature of their mRNA. Host cell mRNA encodes directly for functional proteins, whereas viral mRNA is polycistronic, which means several distinct proteins are encoded within a single piece of mRNA. This implies that the virus needs to use a virus-specific protease to cut at the correct place the polyprotein produced by translation to restore the functionality of viral proteins.
Late protein synthesis during the replication cycle concerns the production of structural components (e.g. capsomeres) of the new virions.
4.5 Maturation or a ssembly of virions
Towards the end of the multiplication process, large amounts of viral materials accumulate within the host. Viral capsid starts to form from individual structural proteins. In certain viruses (e.g. poliovirus) the capsid self-assembles. The replicated viral genome and some viral proteins become packaged within the capsid. Most non-enveloped viruses accumulate within the cytoplasm or nucleus and are only released when the cell lyses. Packaging of viral components can occur within the cytoplasm or in the cell nucleus. For example, with influenza virus, the capsomeres are transported to the cell nucleus where they combined with the viral RNA and assemble into helical capsids. The envelope of enveloped viruses originates from the host. With the influenza virus, viral proteins such as neuraminidase and haemagglutinin migrate to the cell membrane, displacing cell protein. The assembled nucleocapsids pass out from the nucleus to the cytoplasm and as they impinge on the altered cytoplasmic membrane they cause it to bulge and bud off completed enveloped particles from the cell. In the herpesvirus, the envelope originates from the nucleus membrane. The nucleocapsid assembles into the nucleus and it acquires its envelope as it passes through the inner nuclear membrane. The complete virus is then incorporated into a vesicle which migrates to the cell surface.
The maturation of viruses and their assembly is not well understood at present. The presence of chaperone proteins may play an important role in the interaction between the viral nucleic acid and the structural proteins.
4.6 Release of virions into the surrounding e nvironment
At the end of the multiplication process, the mature virions are released from the host cell. This can occur in a number of different ways. For most non-enveloped viruses, the virus progeny accumulates within the host cell cytoplasm and is released following cell lysis. Some viruses (e.g. bacteriophages) produce a lytic enzyme (peptide) or proteases to lyse the host enabling the release of infectious particles, although the host often self-disintegrates as it cannot maintain normal housekeeping functions during a viral infection. Enveloped viruses are usually released by a budding process over a period of hours. Ultimately the host cell will die following damage to its metabolism and housekeeping functions during viral replication.
5 Cultivation of human viruses
The study and identification of viruses (for diagnosis) depends on our ability to propagate them. In the early days of virology, viruses were propagated in a host. To some extent, this is still the case today with the use of the chick embryo (see below). However, progress in cell culture has revolutionized the cultivation of many viruses, enabling a better understanding of their replication properties, more rapid diagnosis and the easier production of vaccine (Table 5.3).
Table 5.3 Cultivation of viruses to produce vaccines
| Viruses | Cultivation |
| Hepatitis A virus | Human diploid cell |
| Infl uenza virus | Fertilized hen’s eggs-membrane bounding the amniotic cavity |
| Smallpox | Fertilized hen’s eggs—chorioallantoic membrane |
| Measles and mumps | Chick embryo cells |
| Rubella | Human diploid cells |
| Human herpesvirus 3 (shingles, herpes zoster) | Human diploid cells |
| Rabies virus | Chick embryo cells or in human diploid cells |
| Varicella zoster virus | Human diploid cells |
| Tick-borne encephalitis virus | Chick embryo cells |
| Yellow fever virus | Chick embryos |
| Poliovirus | Human diploid cell line, continuous cell line or primary, secondary or tertiary monkey kidney cells |
| Baculovirus (recombinant vaccine) | Insect vector |
5.1 Cell culture
Cells to support the growth of human viruses are usually derived from humans or other primates, or from rodents. Cell cultures may be divided into three types according to their history: (1) primary, (2) secondary and (3) continuous cell culture. Primary and secondary cells are usually diploid cell lines. Primary cell lines are derived directly from an intact tissue such as human embryo kidney or monkey kidney. Secondary cell cultures are derived from primary cultures, usually those arising from embryonic tissue. These cells are more homogenous, better characterized, but might not be as susceptible to viral infection as primary cell lines. In addition a limited number of subcultures can be performed with these cells, generally up to a maximum of about 50 before the cells degenerate. Continuous cell lines are usually derived from malignant tissue (e.g. HeLa cells derived from a cervical carcinoma) and have the capacity to multiply indefinitely in vitro.
In principle, cell culture for propagating viruses relies on the growth of cells in a semiconfluent monolayer attached to a surface (e.g. the bottom of a flask). To subculture, the cells are separated from the monolayer or relevant tissue usually with trypsin to form a suspension of single cells. These suspended cells are then used to seed a new flask. Following growth at 37 °C, cells will multiply, attach to the surface and form a new monolayer within a few days. The media used to grow the cells consists of basic nutrients and salts (the composition of the medium varies depending on the type of cell) supplemented with serum (usually bovine albumin) to provide growth factors; antibiotics and antifungals are included to prevent bacterial and fungal contamination in such a rich growth medium.
The established cell monolayer will support viral replication from which viruses can be harvested. Many types of viruses, upon inoculation of a cell culture, will produced a characteristic morphological change in the infected cells. This is called a cytopathic effect and usually indicates cell death. The cytopathic effect can take the form of cell shrinkage or ballooning, or the detachment of cells from the surface of the flask; in some instances cellular effects will be detected using various staining solutions. Cytopathic effects usually spread to adjacent cells and will result in the formation of a plaque that can easily be identified following staining. These plaques are used for the enumeration of viruses assuming one plaque results from infection by one virus.
To confirm the identity of a viral pathogen (e.g. herpes simplex virus, cytomegalovirus, influenza), viruses are grown in cell culture and following the appearance of a cytopathic effect, the identity of the virus is confirmed using an appropriate viral antisera labelled with a fluorescent dye.
Mammalian cells used for vaccine production are obtained from an approved cell bank. All such cells need to be checked for infectious agents and tumorigenicity (in the case of live vaccines). Such cells are also characterized biochemically (isoenzyme analysis), immunologically (histocompatibility antigens) and by cytogenic markers and are shown to be free from contaminating cells (nucleic acid fingerprint analysis). Depending on their origin, cells will be examined for the absence of specific infectious agents, for the absence of bacterial, fungal and mycoplasma contaminants, and for retroviruses—using product-enhanced reverse transcriptase (PERT) assay, transmission electron microscopy and, if necessary, infectivity assays. The same controls apply to insect cells.
5.2 The chick embryo
Fertile chicken eggs, 9–11 days old, are used as a convenient cell system to grow a number of human pathogenic viruses. Their usefulness derives from the many types of different tissues found in the eggs, tissues that will support the growth of different viruses (Table 5.3). The use of chicken eggs is expensive and the use of cell culture is preferred wherever possible. The use of chicken eggs for the production of vaccine is necessarily subject to many controls. The eggs have to be free from specific pathogens and originate from healthy flocks. The processing of the fertilized eggs must be conducted under aseptic conditions in an area where no other infectious agents or cells are handled at the same time.
5.3 Animal inoculation
Some animals (e.g. rodents, primates) have to be used to culture certain viruses in order to study antiviral and vaccine effectiveness, and also as a source of cell lines for cell cultures. The use of animals follows strict ethical guidelines and is extremely expensive. A number of controls ensuring that the animals are free from diseases must be observed. Where animals are used to grow viruses or to test an antiviral or vaccine, growth of the virus is indicated by signs of disease or death.
6.1 Antiviral chemotherapy
A number of antivirals are in use in the UK (British National Formulary, 2009) for a range of viral infections, including HIV, herpesvirus infections, viral hepatitis, influenza and respiratory syncytial virus. Antiviral treatments are particularly important for persons at high risk, notably immunocompromised patients. Most antivirals are prodrugs that need to be activated within the cell, usually by a kinase, and other cellular enzymes.
Antivirals can act at different stages of the viral replication cycle, with the most effective treatments targeting unique viral enzymes, such as proteases, polymerases and the reverse transcriptase (Table 5.4 and Chapter 11). A number of targets are being investigated to prevent viral attachment to the host cell: competition for CD4 receptors using a pentapeptide identical in sequence to the terminal amino acids of HIV gp120; inhibition of herpes simplex virus (HSV) ribonucleotide reductase; competition for the cell receptor using a hexapeptide fusion sequence at the N-terminus of the influenza haemagglu-tinin viral receptor. Proteases are particularly important for the uncoating process preventing the release of viral nucleocapsid and for the cleavage of viral polypeptide gene products (e.g. indinavir sulphate). The replication of viral DNA is also a well-exploited target with the use of nucleoside analogues (e.g. idoxiuridine is incorporated into viral and cellular DNA instead of thymidine), non-nucleoside analogues (e.g. nevirapine and foscarnet) and oligonucleotides (Table 5.5). These nucleic acid oligomers with base sequence complementary to conserved regions of proviral DNA have been successful use in the prevention of viral mRNA function. The inhibition of HIV reverse transcriptase has led to the synthesis of many successful antivirals (Table 5.4). The release of the mature virions after the multiplication process can also be blocked. This is the case of neuraminidase inhibitors (e.g. zanamivir and ozaltamivir) preventing the shedding of virions.
Table 5.4 Current antiviral drugs in the UK
| Type | Viral infection | Name |
| Nucleoside reverse transcriptase inhibitor | HIV | Zidovudine (AZT) |
| Abacavir | ||
| Didanosine | ||
| Emtricitabine | ||
| Lamivudine (3TC) | ||
| Stavudine (d4T) | ||
| Tenofovir | ||
| HIV, hepatitis B | Adefovir dipivoxil | |
| Hepatitis C | Entecavir | |
| Protease inhibitor | HIV | Atazanavir |
| Darunavir | ||
| (Prodrug of amprenavir) | Fosemprenavir | |
| Indinavir | ||
| Lopinavir | ||
| Nelfinavir | ||
| Ritonavir | ||
| Saquinavir | ||
| Tipranavir | ||
| Non-nucleoside reverse transcriptase inhibitor | HIV | Efavirenz |
| Etravirine | ||
| Nevirapine | ||
| Inhibition of HIV-host fusion | HIV | Enfuvirtide |
| Antagonist of CCR5 chemokine receptor | HIV | Maraviroc |
| Inhibitor of DNA polymerase | Herpes viruses | Acyclovir |
| (Prodrug of penciclovir) | Herpes zoster and genital herpes | Famciclovir |
| (Prodrug of aciclovir) | Herpes simplex, | Valaciclovir |
| Herpes zoster, cytomegalovirus | Ganciclovir | |
| (Ester of valaciclovir) | Cytomegalovirus | Valganciclovir |
| Herpes simplex, cytomegalovirus | Cidofovir | |
| Herpes simplex, Herpes zoster, Epstein-Barr, vaccinia | Vidarabine | |
| Stimulate cell mediated immune response—mode of action unknown | Herpes simplex | Inosine pranobex |
| Inactivation of virus-specific DNA polymerase and reverse transcriptase | Foscarnet | |
| Nucleoside analogue interference with viral nucleic acids synthesis | Hepatitis C, RSV, herpesvirus, adenovirus, poxvirus, Lassa fever virus, influenza, measles, mumps, HIV | Ribavirin |
| Inhibition of viral neuraminidase | Influenza | Oseltamivir |
| Zanamivir | ||
| Monoclonal antibody | RSV | Palivizumab |
Data from BNF 2009.
Table 5.5 Oligonucleotide therapies
| Methods | Principles | Effect |
| Antisense nucleotides | Production of an RNA sequence complementary to single-stranded viral RNA | Triggers the formation of double stranded duplex, inhibiting viral RNA replication |
| Antigen methods | Formation of triple helix of DNA | Inhibit transcription |
| Decoy methods | Production of synthetic decoys corresponding to a specific nucleic acid sequence which binds virally encoded regulatory proteins | Inhibit transcription |
| Ribozymes | Production of RNA molecules (oligo (ribo)nucleotides) | Cleave other RNA sequences at specific sites |
Unfortunately, antiviral chemotherapy is associated with a number of problems. Many viral diseases only become apparent after extensive viral multiplication and tissue damage have occurred, delaying treatments. Many antivirals are toxic (e.g. nucleoside analogues) since viral replication often depends on the use of host cell enzymes. There is also scope for improving the pharmacokinetic properties of antivirals, providing a better penetration and retarding drug degradation. The use of prodrugs has improved drug adsorption. Finally, antiviral monotherapy often leads to the development of virus resistance. Emerging HIV resistance has been well documented and current treatments are based on a triple therapy.
6.1.1 HIV
There is no cure for HIV infections as yet. The role of antivirals is to slow or halt disease progression. Since their discovery and use, these drugs (Table 5.4), called antiretrovirals, have considerably prolonged the life expectancy of patients, although not without some important side effects. Antiretroviral treatments aim to reduce HIV plasma levels for as much and as long as possible. Several antiretroviral drugs are usually given together to avoid emerging viral resistance. Initiation of HIV treatment (HAART) is therefore complex and involves two nucleoside reverse transcriptase inhibitors and two non-nucleoside reverse transcriptase inhibitors. Alternative regimens are possible following treatment failure and deterioration of a patient’ s condition. The use of antiretrovirals for prophylaxis after exposure is also possible, where a patient has been exposed to HIV-contaminated materials (e.g. needle injury). Such use follows guidelines available locally (e.g. hospital) or nationally (e.g. Department of Health, British Association for Sexual Health and HIV).
The immune reconstitution syndrome and the lipodystrophy syndrome have been associated with antiretro-viral treatments. The latter includes fat redistribution, insulin resistance, hyperglycaemia and dyslipidaemia. In addition, these antivirals can be damaging to liver function and have been associated with osteonecrosis following long-term combination treatments. A number of side effects are commonly associated with the use of antiret-rovirals: gastrointestinal disturbance, anorexia, pancreatitis, liver damage, dyspnoea, cough, headache, insomnia, dizziness, fatigue, blood disorders, myalgia, arthralgia, rash, urticaria and fever. Protease inhibitors are metabolized by cytochrome P-450 and therefore have a significant potential for drug interactions. Non-nucleoside reverse transcriptase inhibitors have been shown to interact with a number of drugs metabolized in the liver. They have been associated with a number of side effects such as rash, psychiatric and central nervous system disturbances, and even fatal hepatitis.
6.1.2 Herpesvirus infections
Herpesviridae is a family of viruses that include the herpes simplex virus, chickenpox (varicella), shingles (herpes zoster) and cytomegalovirus. Mild herpes simplex virus infections in healthy individuals are treated with a topical antiviral drug (e.g. treatment of cold sores). However, for primary herpetic gingivostomatitis a change of diet and analgesics are recommended. For severe infections (e.g. neonatal herpes infection, infection in immunocompromised patients) a systemic antiviral drug is used (Table 5.4). Antiviral treatments for chickenpox are recommended in patients at risk and in neonates to reduce risks of severe diseases. In healthy adults, treatment taken with 24 hours of the appearance of a rash may decrease the duration and severity of symptoms. Systemic antivirals are used to decrease the severity and duration of shingles when taken within 72 hours of the onset of rash. Antivirals for herpes are also associated with a number of side effects which vary depending on the drug, but may include nausea, vomiting, stomach pain, headache, fatigue, rash, and increase in serum and urine uric acid. Antivirals for the treatment of cytomegalovirus are usually given to immunocompromised patients and they tend to be more toxic with notable nephrotoxicity (e.g. cidofovir) and a number of documented side effects (e.g. ganciclovir, foscarnet)
6.1.3 Viral hepatitis
Hepatitis B and C are major causes of viral chronic hepatitis. The initial treatment for acute hepatitis B is with interferons (peginterferon alfa-2a) which may reduce the risk of chronic infection. However, the use of interferon is limited by a poor response rate in patients and frequent relapse. A number of antivirals are licensed for the treatment of chronic hepatitis B (Table 5.4). The choice of antivirals depends upon the initial response to peginterferon-alfa, emerging viral resistance, and co-infection with HIV. For the treatment of chronic hepatitis C a combination of ribavarin and peginterferonalfa is recommended, although the choice and duration of treatment depends upon the viral genotypes and viral load. These antivirals are also associated with a number of side effects including nausea, vomiting, abdominal pain and diarrhoea.
6.1.4 Influenza
Two antivirals are recommended for the treatment of influenza according to the National Institute of Health and Clinical Excellence (NICE) guidelines (Table 5.4). Oseltamivir was extensively used for the prevention and control of the swine flu outbreak in the UK in 2009. Following an intensive use, at least two major limitations in the usefulness of the drug have been identified. First, the drug needs to be taken within a few hours of the onset of symptoms, which proved very difficult with a number of symptoms from mild ‘cold-like ’ to severe ‘flu-like’ symptoms reported. Second, the side effects, especially in young children and adolescents, have been very severe, prompting many parents to stop the medication, decreasing the willingness to give the antivirals to children who have been possibly exposed to the virus.
6.1.5 Respiratory syncytial virus
Respiratory syncytial virus (RSV) is responsible for severe bronchiolitis notably in infants. A monoclonal antibody (palivizumab) or an antiviral drug (ribavirin) is indicated for the treatment of RSV (Table 5.4). The antiviral is associated with a number of severe side effects.
6.2 Vaccination
Vaccination is undoubtedly the most successful measure against microbial infections, and particularly viral infections. Remarkably, human protection against smallpox was achieved by Jenner in 1796 with the inoculation of cowpox, well before the ‘germ theory’ of disease was postulated. A worldwide vaccination program initiated in 1966 led to the eradication of smallpox in 1980. The poliovirus is almost completely eradicated following an intensive worldwide vaccination programme by the World Health Organization.
Vaccines are preparations containing antigens that elicit a specific and active immunity against an infecting agent; they can induce the innate and the adaptive (cellular, humoral) parts of the immune system (Chapter 9). There are currently a number of viral vaccines available against a diverse range of human viruses (Table 5.6). The success of vaccination relies on the prevention of a disease from occurring. Vaccination is particularly indicated to protect persons at risk (e.g. hepatitis B, varicella zoster vaccines for healthcare workers), prior to travelling (hep-atitis A virus, Japanese encephalitis, yellow fever, tick-borne encephalitis virus) or to prevent cancer (human papillomavirus vaccine preventing cervival cancer). A vaccine can also be given to protect from a viral outbreak (e.g. measles, mumps and rubella vaccine—MMR) or following exposure to rabies.
Table 5.6 Viral vaccines
| Vaccine | Type |
| Hepatitis A vaccine | Formaldehyde-inactivated hepatitis A virus |
| Hepatitis B vaccine | Inactivated HBV surface antigen (HBsAg) |
| HPV vaccine | Virus-like particle composed of the major capsid protein (L1) of HPV type 6, 11, 16 and 18 |
| Influenza vaccine | Formaldehyde-inactivated influenza virus |
| Japanese encephalitis vaccine | Inactivated Japanese encephalitis virus |
| MMR vaccine | Live attenuated viruses |
| Poliomyelitis vaccines | Inactivated poliomyelitis vaccine (injection) |
| Live poliomyelitis vaccine (oral) | |
| Rabies vaccine | Inactivated rabies virus |
| Rotavirus vaccine | Live attenuated rotavirus |
| Tick-borne encephalitis vaccine | Formaldehyde-inactivated tick-borne encephalitis virus |
| Varicella zoster vaccine | Varicella zoster vaccine |
| Yellow fever vaccine | Live attenuated yellow fever virus |
HBsAg, hepatitis B virus surface antigen; HPV, human papillomavirus; MMR, measles, mumps and rubella.
Viral vaccines are prepared using different methods, the most common being the use of live attenuated viruses, inactivated viruses or the use of viral components. Viruses or their components can be prepared from animals, fertilized hen ’ s eggs, in suitable cell cultures (see section 5) or in suitable tissues, or by culture of genetically engineered cells. MMR vaccine and live (oral) poliomyelitis vaccines are based on the use of live attenuated viruses, which are not as virulent as the original virus. The attenuated viruses will cause a strong immune response without causing the disease. Hepatitis A virus and influenza vaccines rely on chemically inactivated (formaldehyde, propiolactone) virus particles or components (surface antigens). Hepatitis B virus vaccine is a recombinant vaccine where the viral DNA encoding for a virus surface antigen (HBsAg) is expressed in yeast ( Saccharomyces cerevisiae) or mam-malian cells (Chinese hamster ovary cells or other suitable cell lines). The surface antigen is then purified. Human papillomavirus (HPV) vaccine contains virus-like particles and recombinant capsid protein expressed in yeast or using a baculovirus as an expression system.
It should be noted that most viral vaccines contain one or more adjuvants, e.g. aluminium salts (antigens are absorbed to the aluminium salts), monophosphoryl lipid A, to increase or modulate the host immune response to the antigens.
Immunoglobulin may play a role in the protection of patients with a compromised immunity against viral infections. Human normal immunoglobulin (HNIG) is prepared from a pool of donated human plasma that has been checked to be non-reactive for hepatitis B surface antigen, hepatitis C virus and HIV (types 1 and 2) but contains immunoglobulin G (IgG) and antibodies against viruses that are prevalent in the general population including hepatitis A, measles, mumps, rubella and vari-cella. Intramuscular normal immunoglobulin is thus used to protect against hepatitis A virus in immunocompromised patients visiting areas where the disease is highly endemic and to protect against or attenuate measles infection in immunocompromised patients. It can also be used for pregnant women against rubella virus, where the risk of termination of pregnancy is unacceptable.
Disease-specific immunoglobulins are prepared from a pool of plasma obtained from specific human donors who have high-levels of the specific antibody required. Disease-specific hepatitis B immunoglobulin is used following accidental inoculation by a risk material (e.g. needlestick injury) or for infants born from mothers infected with the virus. Disease-specific rabies immu-noglobulin is available following the bite of an animal suspected of carrying the disease or originating from an area where the disease is endemic. Disease-specific varicella zoster immunoglobulin is indicated for individuals who are at a high risk such as neonates whose mothers develop chickenpox, or for those exposed to the virus while requiring intensive care or prolonged special care, and for immunocompromised individuals.
6.3 Viricidal effects of chemical and physical agents on viruses
Viruses are generally transmitted via surfaces and therefore the use of viricidal disinfectants on hard surfaces and viricidal antiseptics on skin is important. In addition, viruses are often associated with organic materials, such as secretions from the host, blood, faeces etc., which enable them to persist on surfaces for longer periods of time (weeks, months, and rarely years) and to survive better viricidal challenges.
In general, viruses are not particularly resistant to chemical or physical agents, although some exceptions exist. In terms of susceptibility to viricidal agents, small non-enveloped viruses (e.g. poliovirus) are more resistant than large enveloped viruses (e.g. HIV, influenza), the lipid-rich envelope being damaged easily by chemical and physical agents. The susceptibility of large non-enveloped viruses varies, but it is generally considered to be between that of the small naked and large enveloped viruses, however some rotaviruses are proving particularly difficult to destroy.
The viricidal activity of biocides (antiseptics, disinfectants) varies and not all biocides show a strong viricidal activity against non-enveloped viruses (e.g. cationic biocides, phenolics and alcohols). In addition, biocidal activity depends upon a number of factors, such as concentration, contact time, presence of soiling, and formulation (Chapters 18 and 19). Soiling is particularly an issue with water-and food-borne viruses. Indeed, even enveloped viruses can survive for many days on a soiled surface even when exposed to biocides. The interactions between biocides and viruses have been poorly studied. In general, viruses present only a few target sites to biocides, mainly the envelope (when present), glycoproteins, the capsid and viral nucleic acid (Chapter 20). Some biocides (e.g. cationic) are likely to interact with the envelope and glycoproteins, inhibiting viral infectivity, without altering the viral capsid and genome. The main target site is most probably the capsid which has been shown to be severely damaged in the presence of highly reactive bio-cides such as aldehydes (e.g. glutaraldehyde) and oxidizing agents (e.g. peracetic acid, hydrogen peroxide). Less reactive biocides, such as quaternary ammonium compounds (QACs) and biguanides (e.g. chlorhexidine) have been shown to damage viral capsid to a lesser extent, explaining the limited activity of these agents against non-enveloped viruses. The viral genome is the infectious part of the virus and is the ideal target for biocides. The destruction/alteration of the viral nucleic acid would ensure complete viral inactivation. However, only a limited number of reactive biocides, mainly oxidizing agents (e.g. hydrogen peroxide, chlorine dioxide) have been shown to penetrate within the capsid and damage viral nucleic acid, or to damage viral nucleic acid released from a damaged capsid. The nature of the association of the viral nucleic acid with the capsid also plays a role in the susceptibility of the virus to chemical and physical agents. Viruses with a helical capsid structure might be more susceptible since the destruction/alteration of capsid is more likely to cause damage to the viral nucleic acid which is closely associated to this type of structure.
Physical agents such as heat and irradiation are viricidal and play an important role in the control of viral contaminants in pharmaceutical products. Most viruses are susceptible to exposure to temperatures above 60°C for 30 minutes. Such susceptibility is used for the inactivation of viral contaminants, such as HIV, in blood products. However, other viruses such as the hepatitis B virus are less susceptible and appropriate assurance of the absence of such a virus is needed. Viruses survive well at low temperatures and they can be routinely stored at −40°C to − 70°C. In addition to thermal processes, UV irradiation and ionizing radiation (γ-ray and accelerated electrons) are viricidal mainly following the destruction of the viral nucleic acid. Ionizing irradiation and thermal processes are used for terminal sterilization processes applied to medical and pharmaceutical products (Chapter 21).
6.4 Control of viruses in pharmaceutical products
The presence of certain viruses needs to be controlled in pharmaceutical products derived from human and animal origin. This includes blood products such as human plasma for fractionation intended for the manufacture of human antithrombin III, human coagulation factor VII, VIII, IX, XI, dried prothrombin complex, dried fibrinogen, normal immunoglobulin, human α1-proteinase inhibitor, and human von Willebrand factor, for which tests for the presence of antibodies against HIV-1 and HIV-2, hepatitis B surface antigen (HBsAg), and antibodies against hepatitis C virus are required. For the urine-derived urofollitropin (obtained from postmenopausal women), tests for hepatitis virus antigens and HIV antigen are needed.
The risk of a pharmaceutical product being contaminated by viruses depends mainly on the origin of the product component (e.g. species, organ, tissue), the history of the donor, the amount of material used, the manufacturing process and its capacity to remove/ destroy any contaminants. In addition, the infectivity and pathogenicity of possible viral contaminants must be taken into consideration, notably when considering the route of administration of the medicinal product (i.e. transdermal delivery would carry less risk than an injection). A risk assessment is generally carried out for these products containing a component from human or animal origin, which takes into consideration factors affecting the potential level of infectious particles and those related to the use of the product.
Thus stringent controls are applied to the raw materials, in process samples and to the final product. In addition, one or more validated procedures to remove or destroy viruses can be applied. The type of inactivation measures used must be validated against a range of representative viruses (i.e. enveloped, non-enveloped, DNA, and RNA viruses) with different degrees of resistance to that type of treatment. Furthermore, early inactivation limits the extent of contamination. For the preparation of vaccines, the inactivation process must ensure that it does not affect the antigenicity while killing the virus and other potential contaminants such as mycoplasmas; for example for the preparation of influenza vaccine, the inactivation process chosen must cause minimum alteration of the haemagglutinin and neuraminidase antigens.
Certain viruses or virus components are now used as vectors for the delivery of genes to targeted cells. A number of viruses are being used in gene transfer medicinal products (GTMP) and these include adenoviruses (AAV), poxviruses, retroviruses, lentiviruses, adenoassociated viruses and herpesviruses. Viral vectors for human use are freeze-dried or liquid preparations of recombinant viruses, genetically modified to transfer genetic material to human somatic cells in vivo (i.e. injected directly into the patient’s body) or ex vivo (i.e. transferred into host cells before administration).
There are different approaches for the design and construction of viral vectors. The chosen approach depends uon the type of virus used. The procedure aims to minimize the risk of generating replicating viruses or to eliminate helper viruses when used during production. In addition, a number of stringent controls are performed ensuring the complete genetic and phenotypic characterization of the viral vector is carried out. These include the complete sequence of the genome of the viral vector, verification of genomic integrity of the vector, determination of the concentration of the infectious vector, residual host-cell protein and DNA, and residual reagents including antibiotics.
For retroviridae-derived vectors, genetic modification aims to ensure that the recombinant retroviruses are rendered replication-incompetent. Adeno-associated virus vectors (rAAV) are deficient adenovirus in which certain genes (i.e. cap and rep) necessary for viral replication have been replaced. A helper virus is thus needed during production of the rAAV and needs to be eliminated from the final GTMP. As with other viral vectors, the sequence integrity of the viral genes and expression cassette as well as genetic stability need to be controlled, and the absence of wild-type virus (e.g. AAV) verified.
8.1 Bacteriophages
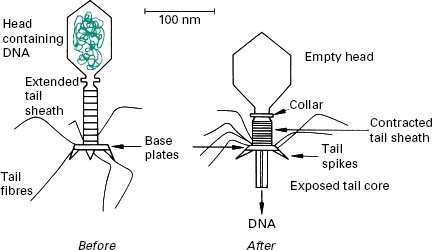
Bacteriophages (phages) are viruses that infect only bacteria. They were first described at the end of the 19th century. They are typically 20–200 nm in size and are highly diverse in their structure and host range and it is likely that all bacterial species can be infected by a phage. Phages are extremely specific in their host range and some will only infect a specific bacterial strain. Such a high specificity is used for bacterial typing as discussed below. The most studied phages are complex ones (e.g. T-phage) sometimes referred to as ‘tadpole-shaped’ consisting of a head (often icosahedral) that contains the viral genome and a tail which function is to recognize the host receptor, attach and subsequently serve as a nucleic acid injection device (Figure 5.5). Indeed, one of the main differences between such phages and common mammalian viruses is that these phages inject their viral genome inside the host cell.
< div class='tao-gold-member'>