1 Introduction: biotechnology in pharmaceutical sciences
The rapid developments in biotechnology and the applications of genetic engineering to practical human problems have allowed the advancement of pharmaceutical biotechnology at a staggering pace. Furthermore, the release of the human genome sequence has also been key for the identification of human genetic diseases and the design of revolutionary approaches for their treatment.
Genetic engineering involves altering DNA molecules outside an organism, making the resultant DNA molecules function in living cells. Many of these cells have been genetically engineered to produce substances that are medically useful to humans. Pharmaceutical biotechnology involves the use of living organisms such as microorganisms to create new pharmaceutical products, or safer and more effective versions of conventionally produced pharmaceuticals, more cost-effectively.
Since the manufacture of the first recombinant pharmaceutical, insulin, there has been a burst in the generation of new recombinant drugs, some of which will be covered later on in this chapter. Furthermore, the use of recombinant DNA technology has spread further allowing the development not only of subunit vaccines, such as the one used in the prevention of hepatitis B, but also of attenuated vaccines, vector vaccines and DNA vaccines. One of pharmaceutical biotechnology’s great potentials lies in gene therapy, which consists in the modification of the genetic material of living cells to prevent, control or cure disease. It encompasses repairing or replacing defective genes and, for example, making tumours more susceptible to other kinds of treatment.
This chapter aims to describe some essential genetic manipulation techniques and to illustrate, with some key examples, their use for the generation of recombinant pharmaceutical drugs. Applications of recombinant DNA techniques in the diagnosis of diseases will also be covered.
To understand how recombinant pharmaceuticals are manufactured we first need to review some of the essential DNA manipulation techniques used to generate these products. We will start by looking at ways to cut and join fragments of DNA and then examine step by step how these techniques can be exploited to clone and express genes from eukaryotic and prokaryotic cells.
2.1 Cutting and joining DNA molecules
DNA isolated from any type of cell can be fragmented using restriction endonucleases. These are enzymes produced by microorganisms which cut foreign DNA and can restrict the proliferation of infecting viruses. Some of these enzymes cut at specific points known as restriction sites which are palindromic sequences (complementary sequences with identical nucleotide sequences when read in the 5′ to 3′ direction) of various lengths. For example, EcoRI (Escherichia coli restriction enzyme I) has specificity for the sequence GAATTC and hydrolyses the G-A phosphodiester bond. Enzymes which recognize and cut at restriction sites of 4–8 base pairs (bp) are particularly useful, as the probability of a site appearing in a random DNA fragment is inversely proportional to its length
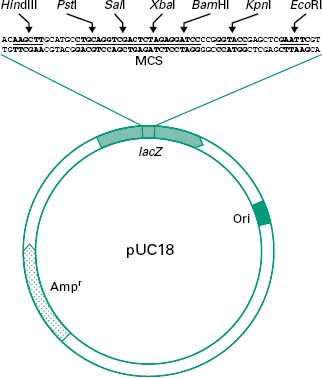
There are two different types of DNA ends that can be generated using restriction enzymes: cohesive or sticky and blunt ends (Table 25.1). DNA fragments obtained by restriction enzyme digestion can be covalently joined together using the enzyme DNA ligase. There is a limitation, however, with regards to the type of ends this enzyme is able to bond together. Only blunt ends generated by some restriction enzymes (e.g. DraI and EcoRV) or compatible sticky ends generated by either the same restriction enzymes (e.g. Eco RI) or by enzymes that generate complementary overhanging ends (e.g. Sau3AI and BamHI) will be bonded by the DNA ligase (Table 25.1).
2.2 Cloning vectors
Genes present in DNA fragments that have been excised with restriction endonucleases can be maintained (replicated) and expressed by inserting them (cloning) into vectors after ligation. These cloning vectors are of different types and are relatively small DNA molecules that have the ability to self-replicate in a host cell. The main type of vectors used for gene cloning are plasmids, cosmids and bacteriophages, which are normally used according to the size of the DNA fragments that need to be cloned.
2.2.1 Cloning of small fragments of DNA
To allow the cloning of small DNA fragments, generally up to 10 kb, plasmids are the main vectors of choice.
2.2.1.1 Plasmids
Plasmids are circular extrachromosomal DNA molecules that replicate independently within cells using their own origin of replication. There are a number of features generally found in plasmids used as cloning vectors:
• Antibiotic resistance markers. These are genes coding for proteins that confer resistance to specific antibiotics. These markers therefore allow the selection of hosts carrying the plasmids.
• Multiple cloning sites (MCS). These are stretches of DNA designed to contain a number of unique different restriction sites, providing a choice of possible restriction enzymes to be used for the cloning of DNA fragments.
• Origin of replication. Required for plasmid replication in a specific host. These DNA sequences also determine the number of copies at which the plasmid will replicate, from just one to several hundreds per cell. Plasmids having more than one host-specific origin of replication are known as shuttle vectors.
• Insertional inactivation markers. These facilitate the selection of recombinant from non-recombinant plasmids. The most commonly used is the lacZ gene coding for β-galactosidase.This enzyme cleaves 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), an artificial substrate that mimics galactose, which results in the generation of an insoluble blue product. Insertion of a recombinant DNA fragment in lacZ will result in the disruption of β-galactosidase production and the inability to cleave X-Gal. Hence colonies from bacteria carrying an intact lacZ gene, i.e. from non-recombinant plasmids, appear blue on agar plates containing X-Gal. In contrast, those with a successful insertion in lacZ, i.e. harbouring recombinant plasmids, appear white.
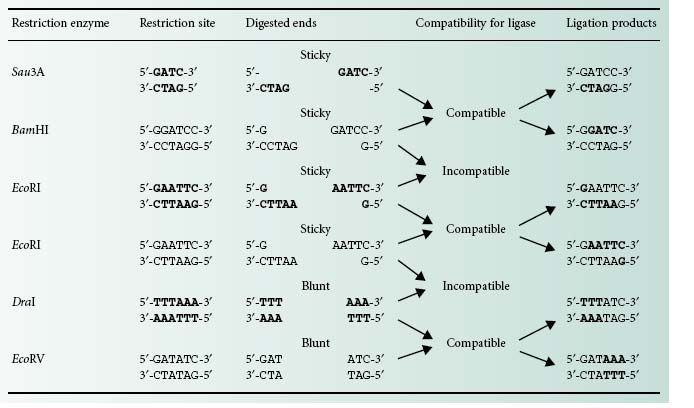
Figure 25.1 shows all these features in a simplified diagram of the cloning vector pUC18.
Figure 25.1 Simplified diagram of the plasmid pUC18. lacZ represents the insertional inactivation marker coding for β-galactosidase activity. A multiple cloning site (MCS) is present within the lacZ gene, allowing the cloning of DNA fragments. ‘Ori’ represents the origin of replication which, in this case, is functional in Escherichia coli, and Ampr represents an ampicillin resistance marker.
An ideal plasmid for cloning should be small (2–10 kb) and conjugation-defective, i.e. non-self-mobilizable from cell to cell, and produce a selectable phenotype in host cells. It should also contain a large MCS and replicate at a high copy number (>10 copies per cell).
2.2.2 Cloning of large fragments of DNA
Sometimes there is a need to clone large fragments of DNA, for example for the isolation of complete gene clusters. An example would be the cloning of large eukaryotic genes or genes required for the synthesis of a certain molecule. Sometimes the synthesis of a compound requires more than 10 different genes and these are frequently organized in operons cotranscribed in a single mRNA molecule. To enable the cloning of large genes or full-length operons, vectors such as bacteriophages, cosmids or BACs need to be used.
2.2.2.1 Bacteriophages
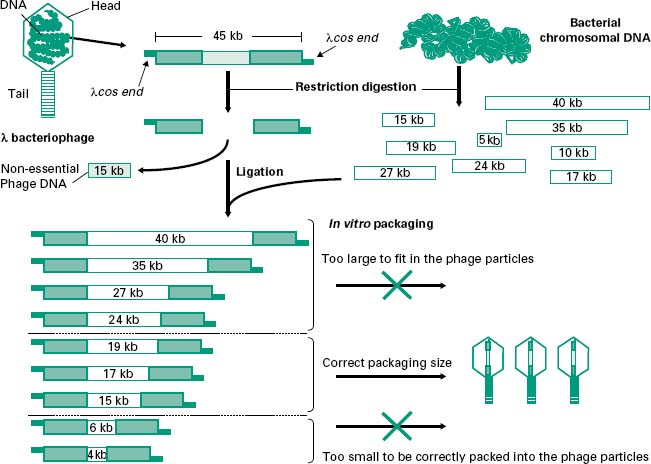
The most popular has been the E. coli λ (lambda) bacteriophage, which is composed of a tubular protein tail and a protein head packed with approximately 50 kb of double-stranded (ds) DNA (Figure 25.2). After injection of the viral DNA into E. coli, bacteriophage λ can multiply and enter a lytic cycle leading to the lysis of the host cell and the subsequent release of a large number of phage particles. Alternatively, injection of the DNA can lead to a lysogenic cycle in which the phage DNA is integrated into the E. coli chromosome where it is maintained until the environmental conditions change and is then excised, entering a lytic cycle. Out of the 50 kb that make the λ bacteriophage less than half are essential for its propagation and therefore around 20 kb can be replaced by recombinant DNA, hence their name λ replacement vectors. For this reason these are very useful vectors for the generation of genomic libraries. DNA in λ bacteriophages contain at each end small single-stranded (ss) complementary DNA fragments called λ cohesive ends (λcos ends). Recombinant phages can be assembled in a test tube into phage-like particles by enzymes which recognize and process the λcos ends, provided that they are 35–45 kb apart. These enzymes as well as the head and tails required for the assembly process are commercially available as part of in vitro packaging kits. The in vitro packaging results in the formation of recombinant phages that can be transduced to E. coli cells. Transduced E. coli will be identified by the formation of lysis plaques in agar plates seeded with a mixture of E. coli and recombinant λ bacteriophages.
Figure 25.2 Cloning of DNA into λ replacement vectors. The DNA from the λ bacteriophage can be purified from the phage particles and digested with restriction enzymes to remove an internal fragment of around 15 kb which is not required for the life cycle of the bacteriophage. This fragment can be replaced with other fragments of DNA such as those coming from digested bacterial chromosomal DNA. As the λ bacteriophage can only pack DNA fragments flanked by λcos ends which are 35–45kb apart, any recombinant fragments larger or smaller than that will not be successfully packed.
2.2.2.2 Cosmids and fosmids
Cosmids are plasmids maintained in E. coli that have been engineered to carry λcos sequences. This allows their packaging in vitro into λ phage particles to be transduced into E. coli cells for replication. Once inside the bacterial host the cosmid DNA is circularized by the joining of the λcos ends and thereafter it replicates as a normal plasmid. This implies that the cloned DNA will be available as E. coli colonies and not as plaques like with the λ bacteriophages. As less DNA is required for plasmid than for bacteriophage replication, cosmids can carry up to 40 kb of cloned DNA still enabling the packaging into λ phage particles.
Eukaryotic and more particularly mammalian genomic DNA rich in multiple repeated elements can be subject to deletions and rearrangements when cloned in standard multicopy cosmids and replicated in bacteria. This inconvenient can be reduced by using special cosmids in which the multicopy origin of replication has been replaced by the single-copy origin of replication from the E. coli F′ factor. These cloning vectors are known as fosmids.
2.2.2.3 Bacterial artificial chromosomes
Although cosmids and fosmids enable the cloning of relatively large DNA fragments, the amount of DNA that can be packed into a λ bacteriophage head is limited to around 50 kb. To clone even larger DNA fragments cloning vectors derived from the single-copy E. coli F′ factor, similar to the fosmids, have been designed. In this case the λcos ends are absent or not used for packaging and instead the recombinant plasmids are introduced directly into the E. coli cells by transformation. This allows the cloning of DNA fragments of several hundreds of kilobases resulting in what are then called bacterial artificial chromosomes (BACs).
2.3 Introduction of vector into hosts
For the expression and maintenance of recombinant genes the recombinant vectors harbouring them need to be introduced into suitable hosts. The four main methods used to achieve this are transformation, electroporation, conjugation and transduction.
• Transformation is the direct incorporation of DNA into host cells. Bacteria such as E. coli can uptake recombinant plasmid DNA when treated with ice-cold CaCl2 until they reach a ‘competent’ state in which they are ready to take up DNA. These cells are then mixed with the recombinant plasmid and exposed briefly to a heat shock of 42°C which causes them to take up the DNA.
• Electroporation is, however, the most efficient way of introducing DNA not only in bacteria but also in eukaryotic cells. This technique is based on the induction of free DNA uptake by the cells after subjecting them to a strong electric field.
• In some cases conjugation can be used as a natural transmission of plasmid DNA from a donor cell to a recipient cell by direct contact through cell–cell junctions. Only plasmid cloning vectors containing conjugative elements can be transferred by conjugation. This procedure requires direct contact between the donor and the recipient cell. Conjugation is not as frequently used as electroporation as most plasmid vectors used for the cloning of recombinant DNA lack conjugative functions, preventing these plasmids from being passed to other cells inadvertently.
• Finally, in transduction (prokaryotes) and transfection (eukaryotes) the transfer of recombinant non-viral DNA to a cell is achieved by a virus. This is the method of choice for the introduction of recombinant λ bacteriophages, cosmids and fosmids into E. coli cells.
2.4 Construction of genomic libraries
Before we study how genomic libraries are made we first need to understand the differences between the genetic organization in eukaryotic and prokaryotic cells. Bacterial genes are uninterrupted sequences of nucleotides encoding the genetic information required for the synthesis of a protein. These genes can sometimes be cotranscribed with adjacent genes of related function into the same mRNA molecule. This set of cotranscribed genes is called an operon (Figure 25.3). The mRNA in bacteria does not generally need to be processed before translation, and transcription and translation occur simultaneously.
Figure 25.3 Genetic organization in bacteria (prokaryotes) and eukaryotes. In prokaryotes genes can sometimes be grouped in operons and hence transcribed together in a single molecule of mRNA. In these organisms the whole process of transcription and translation takes place simultaneously in the cytoplasm. In contrast, in eukaryotes, genes are organized in single transcriptional units incorporating introns. Upon transcription in the nucleus, eukaryotic mRNA is firstly modified by the addition of a CAP and a poly(A) tail and then by the splicing out of the introns. The mature mRNA is then exported into the cytoplasm where it is translated into proteins. P, promoter; T.T., transcriptional terminator; RBS, ribosome binding site.
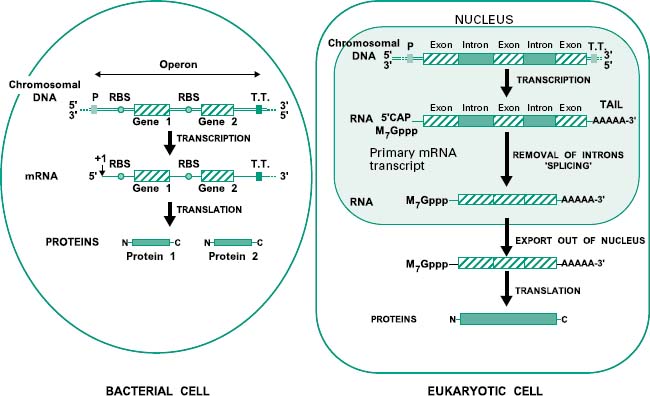
In contrast, genes from eukaryotic cells contain non-coding sequences called introns and coding sequences called exons. The former are removed after transcription by a process called ‘splicing’ that occurs in the nucleus of the cell. In addition the mRNA is subjected to further processing involving the addition of a methylated guanine (M7Gppp) called CAP on its 5′ end, required for translation, and a polyadenine (poly(A))tail on its 3′ end (Figure 25.3). Mature mRNA is then exported from the nucleus into the cytoplasm, where it is translated into proteins. Eukaryotic genes appear to be transcribed individually, as operons have not been described in eukaryotes.
To enable the cloning and isolation of a specific gene(s) from a cell several steps are required. The first consists of choosing the source of genetic material which, in prokaryotic cells, is normally the chromosomal DNA. In contrast, in eukaryotic cells this is more often the mature mRNA, as it is not interrupted by introns and consequently codes for complete, active proteins. The second step consists of the preparation of the purified DNA or RNA for cloning. This step is more straightforward when using prokaryotic DNA (section 2.4.1) than eukaryotic RNA (section 2.4.2). The result will be the construction of a collection of cloned DNA fragments propagated in bacteria that is called the genomic library. This library should ideally contain representatives of every sequence in the chromosome of a prokaryotic cell and every expressed gene in the case of a eukaryotic cell. The final step consists of the screening of the recombinant clones to identify the required gene(s).
2.4.1 Prokaryotic gene libraries: shotgun cloning
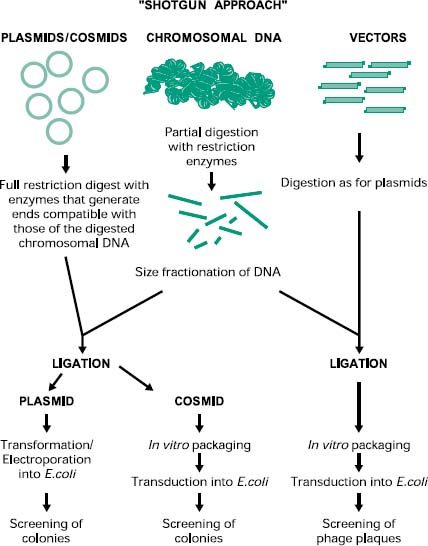
The construction of a prokaryotic gene library can be achieved by a technique called ‘shotgun cloning’ (Figure 25.4). This involves the purification and partial digestion of the genomic (chromosomal) DNA from a prokaryotic organism with restriction endonucleases to produce a random mixture of fragments of different sizes. Chromosomal DNA can also be mechanically sheared and in this case the extremities must be repaired and made blunt with DNA polymerase in the presence of deoxynucleotides (dNTPs). These fragments are then fractionated into different sizes and ligated into a cloning vector appropriately digested. The recombinant vectors are then transformed, in the case of plasmids, or transfected, in the case of bacteriophages and cosmids, into the host cell of choice. The resulting genomic library can then be screened for the presence of the recombinant gene of interest by a number of methods (see section 2.5).
Figure 25.4 Construction of prokaryotic genomic libraries by shotgun cloning. The shotgun approach for the construction of genomic libraries involves the purification and digestion of chromosomal DNA from the prokaryotic organisms, followed by the cloning into a digested suitable vector using DNA ligase. The recombinant vector is then introduced into the host cell using the appropriate method and then the colonies or plaques are screened for the presence of the recombinant gene of interest.
2.4.2 Eukaryotic cDNA gene libraries
The shotgun approach cannot be applied for the construction of eukaryotic gene libraries because of the presence of introns in the DNA, which prevents the direct cloning of functional genes from digested chromosomal DNA. Instead, mature mRNA from the cytoplasm of cells expressing the desired gene is used as the source of genetic material. For example, to make a genomic library containing the insulin gene, RNA from pancreatic cells expressing this gene have to be isolated. Remember that cells show distinct differentiation in different tissues, and express only a small percentage of the whole genome according to their role in the tissue of which they form part. Consequently, it is not possible to purify RNA coding for insulin from, for example, cells of the pituitary gland. Therefore, the cells expressing the gene of interest have to be isolated first, and then their mRNA purified. As mentioned earlier, virtually every eukaryotic mRNA has a poly(A) tail on its 3′ end. This provides a convenient way to isolate mature mRNA from total cellular RNA, most of which (98%) is ribosomal RNA (rRNA) and transfer RNA (tRNA). The total RNA purified from a cell can be passed through an affinity column packed with cellulose linked to deoxythymidine oligonucleotides (oligo(dT)). As the total RNA passes through the column, only the mRNA molecules which have poly(A) tails will bind to the oligo(dT) while the rest will flow through the column to be discarded. The purified mRNA then has to be converted into ds cDNA (complementary DNA) to enable its cloning into a suitable vector.
2.4.2.1 Synthesis and cloning of cDNA
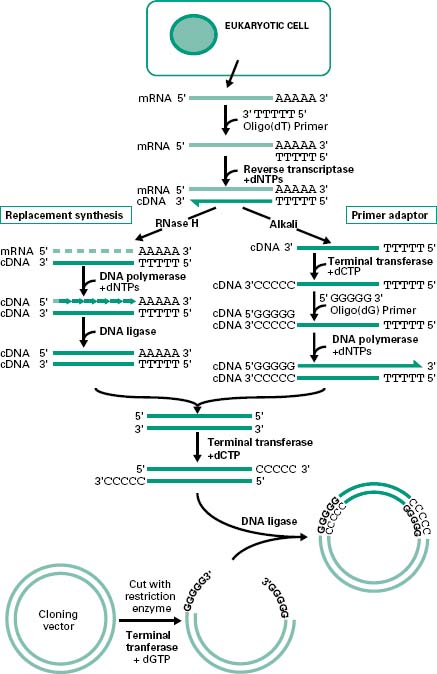
There are generally two main strategies used for the synthesis of cDNA from mRNA: replacement synthesis and primer adaptor synthesis. For both strategies the first strand cDNA synthesis is based on the priming of the mRNA with an oligo-dT which anneals to the poly(A) tail of the mRNA molecule and, consequently, with the action of the enzyme reverse transcriptase and in the presence of dNTPs, the synthesis of the first cDNA strand takes place. This results in the formation of a mRNA/cDNA heteroduplex hybrid. The second stage is different for the two strategies mentioned. The most commonly used is the replacement synthesis, which is based on the use of ribonuclease H (RNaseH), an enzyme that cleaves the RNA moiety of RNA/DNA hybrids and has 5′ to 3′ and 3′ to 5′ direction exonuclease activities. This results in partial digestion of the RNA in both directions. The resulting RNA fragments can serve as primers for DNA synthesis using DNA polymerase I. This enzyme, with its 5′ to 3′ direction exonuclease and polymerase activities will fill the nicks and effectively remove the RNA primers. The cDNA fragments synthesized will be joined using DNA ligase. This method causes the loss of some nucleotides at the 5′ end of the mRNA, including the CAP region.
The primer adaptor method for the synthesis of the second strand of cDNA starts with the removal of the RNA strand from the mRNA/cDNA hybrid, by treatment with alkali. This is followed by the addition of a poly(C) tail to the 3′ end of the DNA strand using an enzyme called terminal transferase. This enables the hybridization of a complementary poly(G) primer that will be the starting point for the synthesis of the second cDNA strand by the DNA polymerase (Figure 25.5). This method, in contrast to the replacement synthesis, generates cDNA molecules with a complete 5′ CAP region. However, it requires more steps and the terminal transferase step is difficult to control.
Figure 25.5 Synthesis and cloning of cDNA. Cloning of eukaryotic genes involves the isolation of mRNA from the cytoplasm of the cells expressing the gene of interest. To allow the cloning of the mRNA the synthesis of ds cDNA is first required. This involves the synthesis of the first cDNA strand by reverse transcriptase using an oligo(dT) primer. To generate the second strand of cDNA there are two main methods. The replacement synthesis method involves the generation of nicks in the mRNA strand by the RNaseH followed by the synthesis of the complementary strand, using the RNA fragments generated by the RNaseH as primers, by the DNA polymerase. The DNA fragments generated are then joined together by the DNA ligase. In contrast, the primer adaptor method requires the degradation of the mRNA strand by alkali followed by the addition of a poly(C) tail at the 3′ end of the cDNA strand by the terminal transferase. For the synthesis of the second strand of cDNA, addition of an oligo(dG) primer and DNA polymerase are required. The ds cDNA generated by either method can be cloned into any vector upon addition of poly(C) sticky ends by the terminal transferase, provided that the vector has complementary poly(G) ends created by this enzyme.