1 Antibiotic development, past and present
An antibiotic was originally defined in the 1940s as a substance produced by one microorganism which, in low concentrations, inhibited the growth of other microorganisms. This definition necessarily meant that an antibiotic was a naturally occurring substance, a microbial metabolite, so any antimicrobial agent that was manufactured by chemical synthesis fell outside the definition. The use of the term has changed over the years, both because an increasing number of synthetic analogues or derivatives of ‘true’ antibiotics have come on to the market, and because several agents that bear little or no resemblance to natural microbial metabolites have also been developed which, for all practical purposes, mimic traditional antibiotics in their potency, low toxicity and, crucially, systemic action. Consequently, drugs like trimethoprim, metronidazole, imidazole derivatives and fluoroquinolones are commonly referred to as antibiotics, and the term ‘antibiotic’ will be used in this chapter to include these and other agents with systemic antimicrobial activity.
By this broader definition, sulphonamides, introduced into therapy in the 1930s, were the first significant antibiotics and predated penicillin by about 10 years. Although Fleming received the credit for discovering penicillin in 1929, much of the development work was undertaken at Oxford University over the next 10 years, and the onset of the Second World War was the impetus for the American pharmaceutical industry to convert the discovery into a medicine that could be manufactured on a large scale. Benzylpenicillin was the original antibiotic in this class, but in the late 1940s and early 1950s it was joined both by other penicillins and by several other classes of antibiotic that are still in widespread use today, such as tetracyclines, macrolides and aminoglycosides. All of these were ‘true’ antibiotics in the traditional sense, i.e. they were extracted from largevolume cultures of Streptomyces bacteria or fungi (in the case of penicillins), but the 1960s saw the advent of semisynthetic antibiotics—penicillins particularly—in which the naturally occurring substance was extracted from the microbial culture, purified and then structurally modified by conventional chemical means. Since then, an increasing number of antibiotics have been totally synthesized.
Although bacterial resistance was a problem that was recognized from the start of the antibiotic era, the international pharmaceutical industry developed new antibiotics steadily throughout the period 1950–1970, so that new drugs regularly became available to replace those to which resistance developed; this ability to keep ahead of the problem lead to a degree of complacency and a belief that the industry would maintain its supremacy indefinitely. The naivety of this assumption became steadily apparent during the remaining years of the century as antibiotic resistance became a major problem and hospital ‘superbugs’ like MRSA (meticillin-resistant Staphylococcus aureus) and ‘C. diff’ (Clostridium difficile) rose to prominence, whilst at the same time the industry diverted research resources away from antibiotics.
Most of the antibiotics developed during the period 1970–2000 were structural modifications of existing ones, and it was not until the new millennium that genuinely new antibiotics like linezolid and quinupristin/ dalfopristin (Synercid) came into use. Unfortunately, though, despite the need for new drugs to treat the superbugs mentioned above and others that have more recently come into prominence, such as multiply resistant tuberculosis (MRTB) and vancomycin-resistant enterococci (VRE), the short duration of therapy and the likelihood of eventual resistance and falling sales mean that the commercial incentive to develop new antibiotics is still much lower than that for drugs treating chronic diseases like diabetes or hypertension, or ‘lifestyle’ problems like obesity. Consequently, the supply of new antibiotics is likely to remain limited, and the use and distribution of those that are developed will be carefully managed to avoid their indiscriminate use which predisposes to resistance development (see Chapter 15 on antimicrobial stewardship). The effects of short durations of therapy (typically 5–10 days for many infections), antimicrobial stewardship policies limiting their prescribing, relatively short patent life, and the likelihood of some degree of resistance eventually developing anyway, all combine to make modern antibiotics very expensive drugs. This trend of escalating cost is starkly illustrated by comparing the UK price differential of more than 700-fold between trimethoprim (from the 1960s) and linezolid (marketed in 2000) (based on a single day’s treatment using current British National Formulary prices).
1.1 Antibiotic usage
Despite the fact that few new classes of antibiotics have been developed in recent years, there are, nevertheless, a large number of different antibiotics in current use. Some of these are only available in particular countries; and even within a single country the list of available antibiotics changes year by year as new products replace old ones that are withdrawn for commercial reasons or as a result of toxicity concerns. Currently in the UK there are approximately 70 antibacterial antibiotics, 20 antifungals and 40 antiviral agents on the market, and numbers are similar in other countries, so it is not the intention in this chapter to describe, or necessarily even to mention, all of these drugs. Instead, the approach will be to consider the important constituents of the major antibiotic classes and refer the reader to more detailed sources for information about some of the less frequently used agents.
The various classes of antibacterial antibiotics will be considered in their order of importance in terms of usage, which will be taken to mean the frequency with which they are prescribed. Surprisingly perhaps, this does not necessarily coincide with their rank order in terms of the amount manufactured by the pharmaceutical industry each year, because a significant fraction of the annual production is used in animal feed supplements. According to one estimate, the amount used in this way in the USA has been as much as eight times that used to treat human infections.
Table 11.1 shows the relative frequency of prescribing of the major antibiotic classes as determined by recent data from an ongoing Europewide survey of hospital outpatient prescriptions. The relative numbers would be likely to change somewhat if the survey were to be conducted on inpatient prescriptions or on those issued by community medical practitioners, but some general trends are clear. β-Lactam antibiotics are, by far, the most frequently prescribed class, and of these, prescriptions for penicillins substantially outnumber those for cephalosporins. Trimethoprim features strongly, and as an individual drug would rank very highly even in comparison to penicillins given that the value of 5.854 represents total prescriptions for nearly 10 different penicillins (see section 2 below). Antibiotics that are available primarily as parenteral products, e.g. aminogly-cosides and glycopeptides, are much less likely to be prescribed for outpatients than for inpatients because of the problems of administration, so they appear low in the rank order on this particular survey. It is worth emphasizing that this is part of a European survey, and the popularity of particular antibiotics and, consequently, the rank order of the table, is likely to vary from one country to another.
Table 11.1 Relative frequency of prescribing of antibiotics in the UK
UK antibiotic use in defined daily doses per 1000 inhabitants per day based on ambulatory outpatients. Based on data from the European Surveillance of Antimicrobial Consumption project: http://www.esac.ua.ac.be/esac_service/applet/eidb.xhtml
| β-Lactams (total) | 7.582 |
| Penicillins | 5.854 |
| Penicillins with β-lactamase inhibitors | 0.967 |
| Cephalosporins | 0.761 |
| Monobactams and carbapenems | <0.001 |
| Tetracyclines | 3.352 |
| Macrolides | 2.226 |
| Trimethoprim | 1.042 |
| Quinolones | 0.477 |
| Sulphonamides | 0.034 |
| Aminoglycosides | 0.005 |
| Glycopeptides | 0.001 |
| Chloramphenicol | <0.001 |
| Others | 0.246 |
For 20 years after their introduction, the penicillins were the only category of β-lactam antibiotics. They were joined, in the mid 1960s, by the cephalosporins, and towards the end of the 20th century by carbapenems and monobactams. These antibiotics all have the same mechanism of antibacterial action and all possess the β-lactam ring as an integral part of their structure, but they differ widely in other characteristics.
2.1 Penicillins
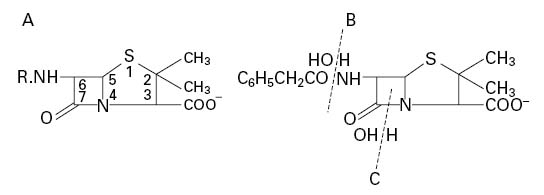
The penicillins (general structure shown in Figure 11.1A) may be considered as being of the following types:
Figure 11.1 (A) General structure of penicillins. (B) Removal of the side chain from benzylpenicillin. (C) Site of action of β-lactamases.
• Naturally occurring. For example, those produced by fermentation of moulds such as Penicillium notatum and P. chrysogenum. The most important examples are benzylpenicillin (penicillin G) and phenoxy-methylpenicillin (penicillin V).
• Semisynthetic. In 1959, scientists at Beecham Research Laboratories succeeded in isolating the penicillin ‘nucleus’, 6-aminopenicillanic acid (6-APA; Figure 11.1A: R represents H). During the commercial production of benzylpenicillin, phenylacetic (phenylethanoic) acid (C6H5.CH2.COOH) is added to the medium in which the Penicillium mould is growing. This substance is a precursor of the side chain (R; see Figure 11.2) in benzylpenicillin. Growth of the organism in the absence of phenylacetic acid led to the isolation of 6-APA; this has a different RF value from benzylpenicillin, which allowed it to be detected chromatographically.
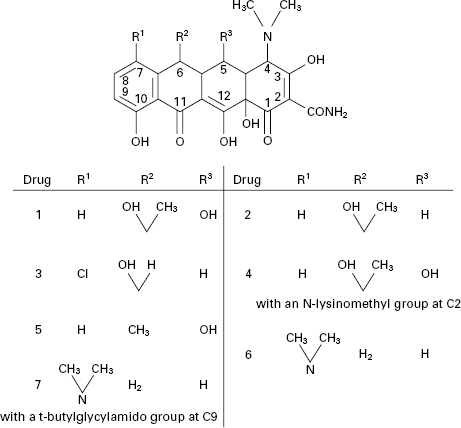
Figure 11.2 Examples of the side chain R in various penicillins (the numbers 1–9 correspond to those in Table 11.2). Number 8 (temocillin) has a methoxy (-OCH3) group at position 6α. Number 9 (pivmecillinam) is a 6-β-amidinopenicillanic acid.
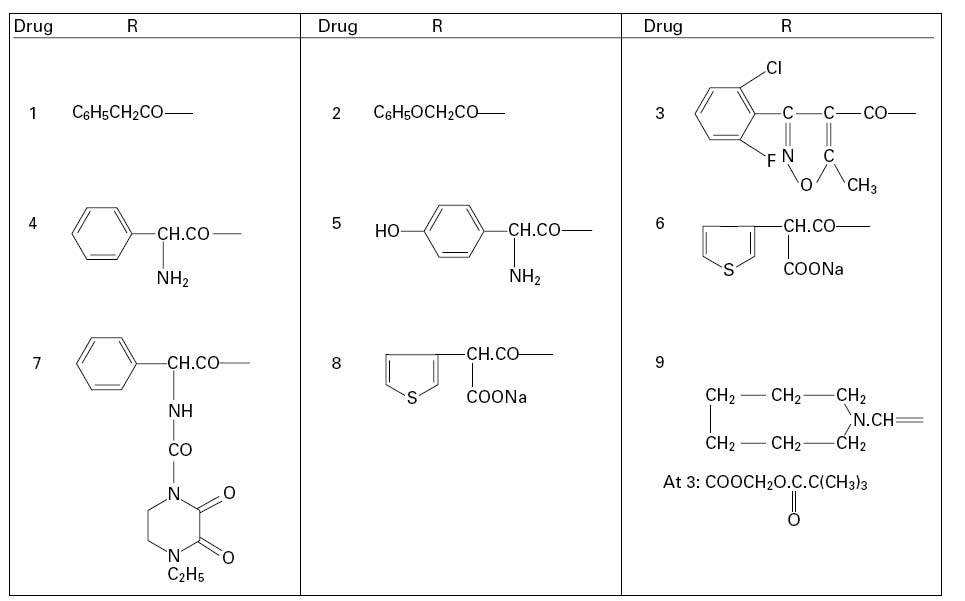
A second method of producing 6-APA came with the discovery that certain microorganisms produce enzymes, penicillin amylases (amylases), which catalyse the removal of the side chain from benzylpenicillin (Figure 11.1B). Acylation of 6APA with appropriate substances results in new penicillins being produced which differ only in the nature of the side chain (Table 11.2; Figure 11.2). Some of these penicillins have considerable activity against Gram-negative as well as Gram-positive bacteria, and are thus broad-spectrum antibiotics. Pharmacokinetic properties may also be altered.
Table 11.2 Properties of the common penicillins
+, applicable; −, inapplicable; NR, not relevant: pivmecillinam has no effect on Gram-positive bacteria; ±, variable result depending on species and strain.
Notes
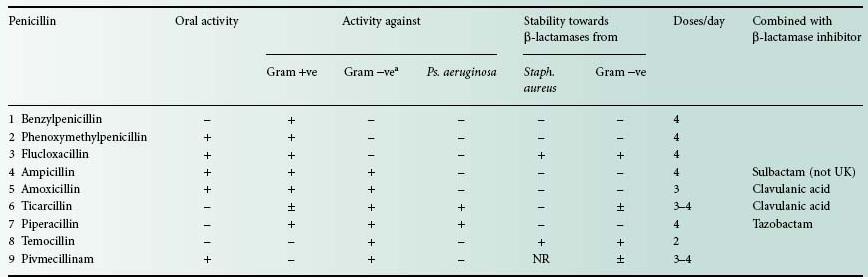
1 For additional information on β-lactamase-mediated resistance, see Chapter 1 3.
2 Most penicillins are active against Gram-positive bacteria, although in the case of Staph. aureus this may depend upon the resistance of the antibiotic to β-lactamase.
3 The table is not intended to list every penicillin available worldwide; for encyclopaedic data, see the books edited by Bryskier (2005) and Finch et al. (2003).
aExcept Ps. aeruginosa. All penicillins show some degree of activity against Gram-negative cocci.
The sodium and potassium salts are very soluble in water, but they are hydrolysed in solution at a temperature-dependent rate to the corresponding penicilloic acid (Figure 11.3A) which is not antibacterial. Penicilloic acid is produced at alkaline pH or (via penicillenic acid; Figure 11.3B) at neutral pH, but at acid pH a molecular rearrangement occurs, giving penillic acid (Figure 11.3C). Susceptibility to hydrolysis means that penicillins cannot be formulated as aqueous products, so oral syrups and mixtures must be manufactured as dry granules for resuspension in water, and injections freeze dried in vials or ampoules. Typically, aqueous solutions of penicillins lose 10% or more of their activity in 24 hours at room temperature. Instability in acid medium logically precludes oral administration, since the antibiotic may be destroyed in the stomach; for example at pH 1.3 and 35°C benzylpenicillin has a half-life of less than 5 minutes and is therefore not administered orally, whereas ampicillin, with a half-life of 600 minutes, is obviously suitable for oral use. Benzylpenicillin is also rapidly excreted, but this can be overcome by the use of sparingly soluble salts (benzathine, benethamine and procaine) which slowly release penicillin into the circulation over a period of time, thus giving a continuous high concentration in the blood.
Figure 11.3 Degradation products of benzylpenicillin in solution: (A) penicilloic acid; (B) penicillenic acid; (C) penillic acid.
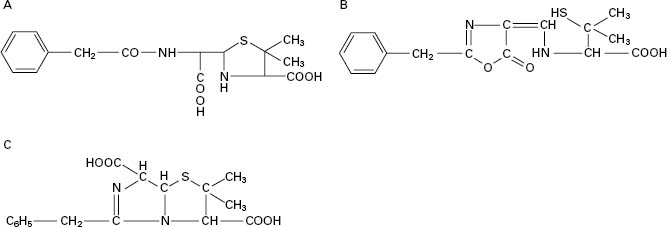
Many bacteria produce enzymes, β-lactamases (formerly called penicillinase; see Chapter 13), which may inactivate a penicillin by opening the β-lactam ring, as in Figure 11.1C. The only clinically significant β-lactamase produced by a Grampositive species is that of Staph. aureus, but most, if not all, Gram-negative bacteria have the potential to produce the enzymes, albeit in such small amounts in some cases that they are of no clinical importance. The Gram-negative β-lactamases exhibit small interspecies, and even interstrain, differences in chemical structure which can have profound effects on their ability to hydrolyse the various β-lactam antibiotics. Some penicillins (Table 11.2) are considerably more enzymeresistant than others, and consequently may be extremely valuable in the treatment of infections caused by β-lactamase-producing bacteria.
The older penicillins, benzylpenicillin and phenoxymethylpenicillin, are primarily active against Gram-positive bacteria; they are also β-lactamase sensitive. These shortcomings were overcome to varying degrees by the semisynthetic penicillins developed in the 1960s. Ampicillin remained β-lactamase sensitive, but its possession of an amino group on the benzyl side chain gave the molecule a much broader spectrum of activity than its parent, benzylpenicillin. The oral absorption of ampicillin was found to be adequate rather than good; a figure of less than 50% is commonly quoted. This was significantly improved by the inclusion of a p-hydroxyl group on the benzene ring of the ampicillin side chain, thus creating amoxicillin, which has largely superseded the older antibiotic. Both ampicillin and amoxicillin are effective against many Gram-negative bacteria including Haemophilus influenzae, Escherichia coli, Salmonella, Shigella and Proteus species, though not Pseudomonas aeruginosa. This last organism has represented a problem in antibiotic therapy for many years, and carbenicillin, also developed in the early 1960s, was the first penicillin showing antipseudomonal activity, although it has now been largely replaced by ticarcillin. Piperacillin, an acyl derivative of ampicillin, also possesses activity against Pseudomonas, and, like ticarcillin, is moderately susceptible to β-lactamases, so both antibiotics are normally used as combination products with β-lactamase inhibitors (see section 2.3).The property of resistance to Gram-negative β-lactamases, strongly exhibited by temocillin, is conferred by the possession of a 6α-methoxy group,although this causes the molecule to exhibit an antibacterial spectrum confined almost exclusively to Gramnegative species. A similar spectrum arises with pivmecillinam, an amidinopenicillin, but in this case the enzyme resistance is much weaker.
Penicillins possess a carboxylic acid group on C3 which can be esterified to create lipophilic prodrugs with enhanced absorption from the gastrointestinal tract, after which tissue esterases hydrolyse the ester to release the active antibiotic. This strategy has been particularly successful in remedying the poor oral absorption of ampicillin and resulted in the development of bacampicillin, pivampicillin and talampicillin; the first two of these, in particular, are widely available elsewhere, but not currently in the UK.
The problem of sensitivity to staphylococcal β-lactamase was overcome by the development of meticillin in which the bulky substituent groups of the side chain largely prevented enzyme binding. Meticillin, which was only available as an injection, has largely been replaced with other β-lactamase-stable penicillins, particularly the orally active flucloxacillin, although there are several related drugs whose availability varies from country to country. The great majority of Staph. aureus strains remained sensitive to meticillin for about 15 years after its introduction in 1960, but the emergence of MRSA gathered momentum from the mid 1970s; the incidence in the USA was 2.4% in 1975 but rose to 35% by 1996.
Penicillins generally are of low toxicity, with allergic reactions as the only serious problem; these arise more commonly with benzylpenicillin and ampicillin than the rest. All penicillins, but particularly those administered orally, can cause diarrhoea, and, rarely, pseudomembranous colitis; this is more of a problem with ampicillin because a higher proportion of an oral dose remains in the colon to disturb the natural flora. Penicillins are excreted primarily in the urine, in which they achieve much higher levels than in the blood, and accumulation of sodium and potassium may arise with high-dose injections in patients with poor kidney function.
2.2 Cephalosporins
There are quite a number of similarities between cephalosporins and penicillins, including their early history. In both cases, the academic research was conducted at Oxford University and the American pharmaceutical companies converted that early work into a marketed product. The discovery of the first cephalosporin was made in 1948 in Sardinia, the research was conducted at the William Dunn School of Pathology during the 1950s and the first antibiotics in the class, cephalothin and cephaloridine, became available in the mid 1960s. Since then many cephalosporins have been synthesized, and more than 60 have been marketed in various countries over the last 40 years, although not all of them are still available. As with the penicillins, the policy in this chapter is to consider the general properties of this class of antibiotics and to provide more detail about selected important cephalosporins, rather than provide a comprehensive listing.
Cephalosporins consist of a six-membered dihydrothiazine ring fused to a β-lactam ring (top structure in Figure 11.4). The position of the double bond in Δ3-cephalosporins is important, since Δ2-cephalosporins (double bond between 2 and 3) are not antibacterial, irrespective of the composition of the side chains. The similarity to the basic penicillin structure is immediately apparent, but the crucial difference is that there is much greater scope for structural modification of the cephalosporins because of the presence of two side chains (on carbons 3 and 7) in contrast to the single side chain of penicillins. Again, the fundamental properties of acid stability (and hence oral availability), antimicrobial spectrum, resistance to β-lactamases and pharmacokinetics can all be substantially varied by sidechain modifications. Several of the cephalosporins act as good inducers of β-lactamases.
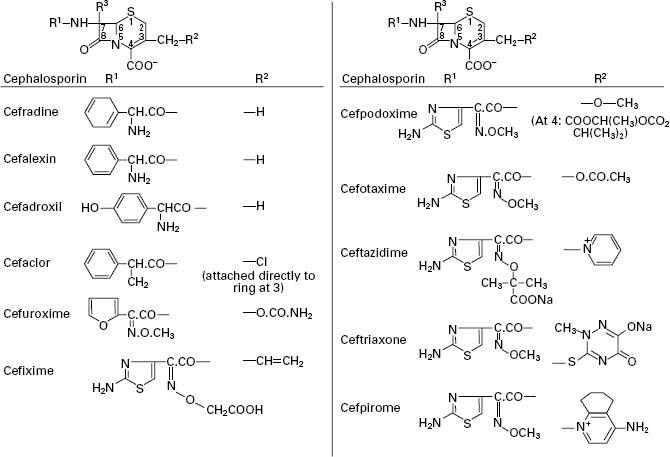
Figure 11.4 General structure of cephalosporins and examples of side chains R1 and R2 (R3 is H in all examples in this figure).
The many cephalosporins have been classified into ‘g enerations’, although the usefulness of such a classification has been questioned. There is general acceptance of four generations, but ceftobiprole has been claimed as the first member of the fifth. The assignment of cephalosporins into the first four generations has broadly followed the time course of their introduction, but there has been some overlap, so certain drugs that have been classified into one particular generation were marketed in some countries after the earliest ones of the next. The situation is further complicated by the fact that there is not universal agreement on the generation to which some cephalosporins belong; cefaclor, for example, is considered a first-generation antibiotic in Japan, but is regarded as second-generation in most other countries.
The general trends have been for an increase in activity towards Gramnegative species (usually with a corresponding loss of antistaphylococcal action), and increased resistance to β-lactamase as the development of cephalosporins has progressed through the generations. First-generation drugs are those that have moderate antimicrobial activity and resistance to staphylococcal, but not Gramnegative, β-lactamases. These were originally used primarily as alternative antibiotics for the treatment of staphylococcal infections, and are rarely first-choice therapy. Like the oral penicillins, the oral cephalosporins may disturb the gut flora and give rise to diarrhoea. The possession of good resistance to both staphylococcal and Gram-negative β-lactamases is the principal characteristic distinguishing the secondfrom the first-generation antibiotics, although improved potency, particularly towards H. influenzae and enterobacteria, is also a feature. Yet higher activity towards Gram-negative bacteria is displayed by thirdgeneration drugs, to an extent that some of them have little or no value in the treatment of staphylococcal infections. The parenterally administered thirdgeneration cephalosporins, e.g. cefotaxime and ceftazidime, are sometimes used in combination with gentamicin or other aminoglycosides with the intention of achieving synergy. The usefulness of the thirdgeneration drugs has diminished somewhat since their introduction as a consequence of the spread of strains capable of producing extended-spectrum β-lactamases (see Chapter 13) and it was this deficiency that the fourth-generation cephalosporins were intended to remedy. Cefpirome and cefepime exhibit extremely good enzyme resistance, but otherwise have much the same antibacterial spectrum as ceftazidime and other third-generation molecules.
2.2.1 Structure-activity relationships
The activity of cephalosporins (and other β-lactams) against Grampositive bacteria depends on antibiotic affinity for penicillin-sensitive enzymes (PSEs) also known as penicillin binding proteins (PBPs). Resistance results from altered PBPs or, more commonly, from β-lactamases. Activity against Gram-negative bacteria depends on penetration of β-lactams through the outer membrane, resistance to β-lactamases found in the periplasmic space and binding to PBPs. (For further information on mechanisms of action and bacterial resistance, see Chapters 12 and 13). Modification of the cephalosporin nucleus (Figure 11.4) at 7α (i.e. R3) by addition of a methoxy group increases β-lactamase stability but decreases activity against Gram-positive bacteria because of reduced affinity for PBPs; molecules possessing a 7α-methoxy group, e.g. cefoxitin, are termed cephamycins.
Side chains containing a 2-aminothiazolyl group at R1, e.g. cefotaxime, ceftriaxone and ceftazidime, yield cephalosporins with enhanced affinity for PBPs of Gram-negative bacteria and streptococci. An iminomethoxy group (-C=N.OCH3) in, for example, cefuroxime, provides β-lactamase stability against common plasmid-mediated β-lactamases. A propylcarboxy group ((CH3)2-C-COOH) as in ceftazidime increases β-lactamase resistance and also provides activity against Ps. aeruginosa, while at the same time reducing β-lactamase induction capabilities. In cephalosporins susceptible to β-lactamases, opening of the β-lactam ring occurs with concomitant loss of the substituent at R2 (except in cefalexin, where R2 represents H; see Figure 11.4). This is followed by fragmentation of the molecule.
The nature of the R2 substituent influences both the pharmacokinetic properties of the molecule and its ability to enter bacterial cells—particularly to cross the outer membrane of Gramnegative bacteria via porins. For good oral absorption: (1) the R2 substituent must be small, non-polar and stable; a methyl group is considered desirable but might decrease antibacterial activity; and (2) the 7-acyl group (R1) must be based on phenylglycine and the amino group must remain unsubstituted. Esterification of the carboxylic acid group at C4 can, as with the penicillins, result in enhanced oral absorption provided that the ester is rapidly hydrolysed by tissue esterases; this is exemplified in both cefuroxime axetil and cefpodoxime proxetil. The possession of a quaternary nitrogen on the side chain at position 3 has two benefits: it reduces the affinity of the cephalosporin for Gramnegative β-lactamases, in other words, makes it more resistant to enzyme attack, and it makes the molecule zwitterionic which increases the rate at which it can pass through the porin channels into the Gram-negative cell.
Side chains of the various cephalosporins, including those most recently developed, are presented in Figure 11.4 and a summary of the properties of these antibiotics in Table 11.3.
Table 11.3 Properties of selected cephalosporins
+, active; I, injection; O, oral; R, resistant; V, varies between strains, or weak activity.
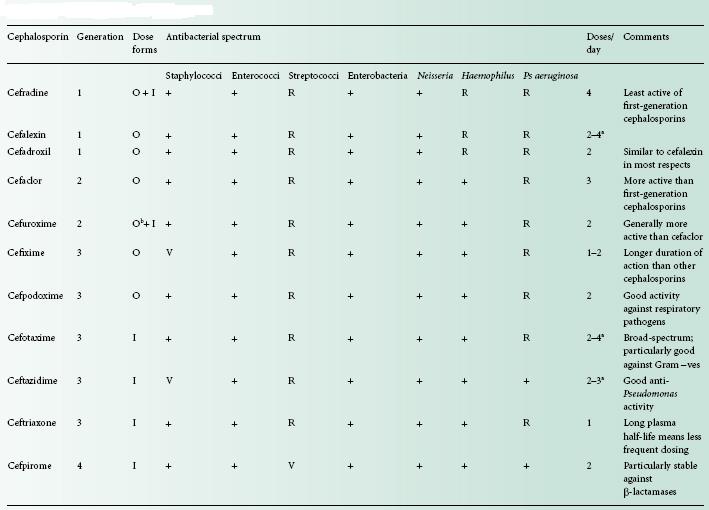
a Depending on dose.
b As axetil ester.
2.3 β-Lactamase inhibitors
The strategy of protecting vulnerable penicillins against enzymemediated hydrolysis by combining them with a β-lactamase inhibitor resulted in the first such combination product, coamoxiclav, in 1981. Table 11.1 shows that penicillins with β-lactamase inhibitors represent approximately 6.5% of the antibiotics prescribed to UK outpatients in the European hospital survey, and coamoxiclav, which consists of amoxicillin plus clavulanic acid, is by far the most important combination available. Clavulanic acid has also been used to protect ticarcillin from β-lactamase attack, and two penicillanic acid sulphones, sulbactam and tazobactam, have been used to protect ampicillin and piperacillin respectively. In each case the protecting molecule is itself a β-lactam antibiotic, but one possessing little antimicrobial activity in its own right. The benefit afforded is an extension of the antimicrobial spectrum of the antibiotic receiving protection; this is achieved by negating the effects of β-lactamases produced by staphylococci and some Gram-negative species, which would otherwise be resistant. In the case of coamoxiclav for example, the combination exhibits activity not only against many strains of Staph. aureus, but also against strains of E. coli, H. influenzae and Klebsiella species, against which amoxicillin alone would be ineffective. This means that coamoxiclav should, in theory, be reserved for infections known, or likely, to be due to amoxicillin-resistant β-lactamase-producing strains, but unfortunately it is not always used in this prudent manner.
Clavulanic acid was isolated from Streptomyces clavuligerus and belongs to a class of β-lactams termed clavems, which differ from penicillins in two respects: namely the replacement of sulphur in the penicillin thiazolidine ring (Figure 11.1) with oxygen in the clavam oxazolidine ring (Figure 11.5A), and the absence of the side chain at position 6. Clavulanic acid also affords some protection against enzymes that are primarily active against cephalosporins rather than penicillins, but this protection is quite modest compared with its activity against ‘penicillinases’.
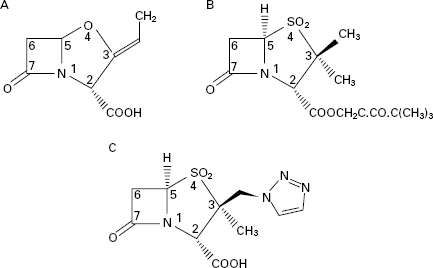
Sulbactam and tazobactam (Figure 11.5B and C) can be regarded as β-lactam molecules that resemble penicillins except that the sulphur atom of the thiazolidine ring is converted to a sulphone, and again, there is no side chain at position 6. Sulbactam is effective against a similar range of β-lactamases to clavulanic acid, although it is not quite as potent. In both cases, the range of enzymes does not normally include those manufactured by Ps. aeruginosa and other problem Gramnegative organisms. Sulbactam has been combined with both ampicillin and cefoperazone, but in both products the two ingredients were separate entities; this poses the potential problem that their pharmacokinetics might not perfectly match, so that the two agents might not appear at the infection site in the optimal concentration ratio at the same time. In the case of sulbactam and ampicillin this problem was partly overcome by covalently linking the two molecules to create sultamicillin which is well absorbed following oral administration and then hydrolysed to liberate equimolar proportions of the individual components.
2.4 Carbapenems and aztreonam
The spread of organisms that had developed various forms of resistance to the early penicillins and cephalosporins lead to a search for other β-lactam antibiotics that avoided these resistance problems. Many naturally occurring and synthetic compounds were examined but only a few were developed to become marketed products, and the most useful of these were the carbapenems. This group may be considered as penicillin or cephalosporin derivatives in which the sulphur atom has been replaced with a carbon. The nomenclature is confusing because some reference sources used terms like carbapenems, olivanic acids and thienamycins as if they were synonymous. In fact, carbapenems is the generic term for the group which includes olivanic acids (of which there are no products in therapeutic use) and the thienamycins. The earliest thienamycins were discovered in the 1970s but proved difficult to develop because of their poor stability. The N-formimidoyl derivative of thienamycin, which was named imipenem (Figure 11.6A), proved to combine the desirable properties of in vitro stability, a broad spectrum of antimicrobial activity and good resistance to almost all of the thenknown β-lactamases. Its only shortcoming was poor in vivo stability because it was vulnerable to hydrolysis by mammalian renal dipeptidase, but this was solved by the development of a renal dipeptidase inhibitor, cilastatin, with which imipenem was marketed. Meropenem, marketed more recently, is more stable than imipenem to dipeptidase and may thus be administered without cilastatin; its chemical structure is depicted in Figure 11.6B. Ertapenem (Figure 11.6C) has properties similar to those of meropenem but affords the additional advantage of once daily dosing.
Figure 11.6 Carbapenems and aztreonam: (A), imipenem; (B), meropenem; (C), ertapenem; (D), aztreonam.
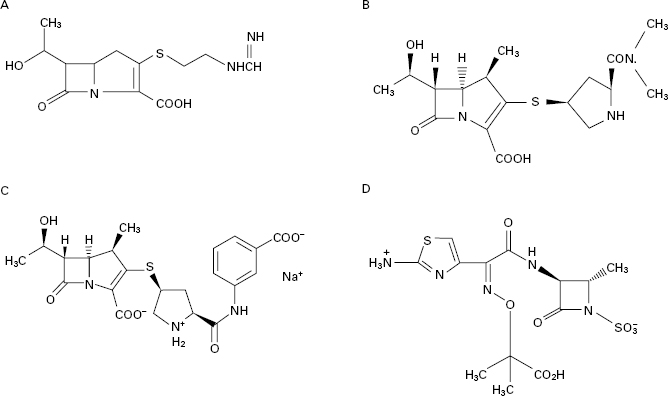
Examination of the structure-activity relationships of the early β-lactam antibiotics led to an expectation that molecules possessing only the β-lactam ring without a second ring fused to it would have no antimicrobial activity. This proved not to be so when such naturally occurring antibiotics, termed monobactams, were discovered, and found not only to possess activity, but to exhibit good resistance to β-lactamases. The naturally occurring monobactams were not developed for clinical use, but an analogue, aztreonam, produced totally by conventional chemical synthesis, was marketed in 1986. It is highly active against most Gramnegative bacteria and stable to most types of β-lactamases, although its resistance to staphylococcal β-lactamases is irrelevant because it is inactive against all strains of Staph. aureus as well as other Grampositive species and anaerobes. Like the carbapenems, it is formulated as an intravenous injection and, as such, it tends to be limited to use in hospitals for the treatment of serious Gramnegative infections, including those due to Ps. aeruginosa. As with the carbapenems and thirdgeneration cephalosporins, it exhibits synergy with aminoglycosides like gentamicin and tobramycin, and these combinations are employed for the treatment of Pseudomonas lung infections in cystic fibrosis.
2.5 Hypersensitivity
Although penicillins are among the safest antimicrobial agents available, allergic reactions, particularly skin allergies, may occur in 1–10% of patients receiving them, and the much more serious, and potentially fatal, anaphylactic reactions occur in 0.005–0.05% of cases. Patients sensitive to one penicillin will be sensitive to all others because it is the basic penicillin structure that is responsible for the hypersensitivity, but benzylpenicillin and ampicillin are the ones most likely to cause anaphylaxis and skin allergy respectively. Although hypersensitivity reactions can occur after administration via any route, the more severe reactions are most likely to arise after intravenous injection. About 10% of patients allergic to penicillins will also be sensitive to cephalosporins.
The tetracyclines are a group of broad-spectrum antibiotics that are declining in use as a result of increasing bacterial resistance. Despite that, they remain important antibiotics for several dangerous, but relatively rare, infections due to chlamydia (e.g. trachoma), rickettsia (e.g. typhus and Q-fever) and spirochaetes (e.g. Lyme disease) as well as those caused by ‘typical’ bacteria (e.g. brucellosis and bubonic plague). They also represent useful alternatives to macrolides (see section 4 of this chapter) and to β-lactams (particularly in cases of allergy) for the treatment of more common infections, including those of the respiratory tract.
The tetracyclines (Figure 11.7) were first developed during the 1940s and 1950s and several that are still in use date from that time, e.g. tetracycline itself, oxytetracycline and chlortetracycline. Doxycycline and minocycline are more potent semisynthetic analogues discovered in 1966 and 1972 respectively, after which there were no significant developments until the introduction, in 2005, of tigecycline (a glycylglycine derivative), which is generally more potent again than other tetracyclines and maintains activity against some organisms that have become resistant to earlier members of the group.
Figure 11.7 Tetracycline antibiotics: (1) oxytetracycline; (2) tetracycline; (3) demeclocycline; (4) lymecycline; (5) doxycycline; (6) minocycline; (7) tigecycline.