Virus Evolution
Edward C. Holmes
Although Charles Darwin preempted many of the great questions in evolutionary biology, he wrote little about viral infections. Of course, viruses were not formally identified until a full decade after Darwin’s death, and, aside from some brief discussion of the origins of yellow fever, his writings make scant reference to what we now know are diseases caused by viruses. This is a great historical shame because it seems certain that Darwin would have held viruses up as some of the best exemplars of evolution by natural selection, and in the case of RNA viruses the evolutionary process is so rapid that it can be effectively followed in “real time”.123,204
Although evolutionary analysis arrived relatively late in the science of virology, the study of virus evolution has become one of the most rapidly growing and successful aspects of modern microbiology. The blossoming of evolutionary virology is largely due to two developments. First, viruses, and especially those that possess RNA genomes, have become remarkably powerful research tools for the study of evolutionary processes. The utility of RNA viruses in this respect is a function of the fact that they evolve extremely rapidly, are easy to manipulate in vitro and sometimes in vivo, often have large and measurable effects on phenotype, and possess such small genomes that the mutations associated with any phenotype change can be determined relatively easily.60 It is therefore no surprise that a growing number of evolutionary researchers are turning to viruses as model systems. For example, studies of viruses represent one of the few cases in which biologists have been able to achieve two of the great aims of modern evolutionary genetics: to measure the fitness effects of individual mutations195 and to determine the nature of the epistatic interactions between these mutations.197 Second, the rapidity of RNA virus evolution has acted as a direct stimulus for the development of phylogenetic and coalescent methods that are able to incorporate information on the exact time of sampling of the sequences in question, in turn revolutionizing molecular epidemiology.49,132 Indeed, many of the computer programs designed for the evolutionary analysis of gene sequence data were first applied to viruses, such that determining the origin and pathways of spread of specific viruses over epidemiologic time has become a relatively exact science with a myriad of potential applications. The advent of next-generation sequencing promises even more rapid advances in this area, potentially enabling the analysis of many thousands of sequences with detailed associated metadata.95 An important spinoff from these studies has been new insights into the patterns and processes of virus evolution.79,92 In sum, although their focus is very different, the combination of experimental analyses of model viruses as a means to understand the intricacies of the evolutionary process and studies of molecular epidemiology based on the comparative analysis of virus gene sequence data to document patterns of virus spread has told us a great deal about the nature of virus evolution. Evolutionary virology has blossomed into a well-developed science.
Despite advances on multiple fronts, some fundamental aspects of viral evolution remain unknown, contentious, or both, which will be highlighted in this chapter. For example, there are still major debates over some of the key mechanisms of evolutionary change in RNA viruses, particularly whether their populations routinely form quasispecies, which reflects a wider uncertainty on the roles of mutation, natural selection, and genetic drift as forces of evolutionary change.92 For example, it is striking that precise estimates of mutation rates are absent from some important groups of viruses even though they represent a sort of “ground zero” in studies of evolutionary change. Similarly, although we know a great deal more about the origin of viruses than we did 10 years ago, particularly since the discovery and analysis of highly conserved protein structures, exactly when and how viruses first evolved, whether this occurred before or after the appearance of the first cellular organisms, what a precellular world might have looked like, and even if viruses should be classified as living are sources of major debate.71,121,138,159 In part, this debate highlights our
profound ignorance of the virosphere. For example, does the apparent absence of RNA viruses in Archaea mean that they never existed in these species, that they have been selectively removed, or that we have simply not looked hard enough? Another important issue is that in some respects our knowledge of DNA virus evolution lags behind what we know of the evolution of RNA viruses. We remain ignorant of a number of key aspects of the patterns and processes of DNA virus evolution, particularly whether common principles can be applied to DNA viruses that differ so greatly in size and genome structure. It is hoped that the rise of metagenomics, such that we will sample far more of the virosphere, likely leading to the discovery of a multitude of diverse viruses, will stimulate many advances in this area. Finally, the time scale of evolutionary history in many viruses is unclear, with very different inferences drawn from either the study of endogenous viruses, which are usually indicative of ancient origins, or the molecular clock analysis of recently sampled and often rapidly evolving viral genomes, which usually paints a picture of very recent origins.80,94 Advances in this area may require the development of a new class of analytical methods.
profound ignorance of the virosphere. For example, does the apparent absence of RNA viruses in Archaea mean that they never existed in these species, that they have been selectively removed, or that we have simply not looked hard enough? Another important issue is that in some respects our knowledge of DNA virus evolution lags behind what we know of the evolution of RNA viruses. We remain ignorant of a number of key aspects of the patterns and processes of DNA virus evolution, particularly whether common principles can be applied to DNA viruses that differ so greatly in size and genome structure. It is hoped that the rise of metagenomics, such that we will sample far more of the virosphere, likely leading to the discovery of a multitude of diverse viruses, will stimulate many advances in this area. Finally, the time scale of evolutionary history in many viruses is unclear, with very different inferences drawn from either the study of endogenous viruses, which are usually indicative of ancient origins, or the molecular clock analysis of recently sampled and often rapidly evolving viral genomes, which usually paints a picture of very recent origins.80,94 Advances in this area may require the development of a new class of analytical methods.
Aside from their ability to inform on evolutionary processes, there are a number of practical reasons that the study of virus evolution deserves attention. It is likely that a better understanding of the exact processes of evolutionary change in viruses will assist in the development of improved strategies for their treatment and control and for predicting the spread of newly emerged pathogens. For example, knowing whether natural selection or genetic drift largely controls how mutations spread through a population is essential to understanding the likelihood and rate that a specific drug resistance mutation will become established,131 while a knowledge of the factors that control how viruses diffuse at the epidemiologic scale represents useful information for any emergent virus.95 Similarly, it is likely that a better understanding of the origin of viruses will be essential to obtaining a more precise picture of the earliest events in the early history of life on earth, including the genesis of both RNA and DNA, as it seems reasonable to suppose that some of the earliest replicators resemble what we now know as viruses.
The future of evolutionary virology appears strong. The development and continued refinement of next-generation sequencing methods will doubtless provide unprecedented amounts of data for evolutionary study and stimulate the development of new analytical methods for use in all genetic systems. Innovations in experimental studies of virus evolution will continue, addressing ever more intricate questions, increasingly considering in vivo systems, and providing broad-scale evolutionary insights. Metagenomic studies of the virosphere will provide a powerful new perspective on virus biodiversity, in turn bringing important new information on virus ecology, origins, and cross-species transmission and emergence. It therefore seems easy to predict that our understanding of viral evolution 10 years from now will be very different, and more complete, than it is today.
The Origin and Time Scale of Virus Evolution
The Origins of Viruses
Of all topics in the study of virus evolution, determining exactly how and when viruses first evolved is perhaps the most difficult. The main hindrance to progress in this area is that viruses likely originated so long ago, and perhaps even before the first cellular species, that the signal of ancient evolutionary history that can be recovered through phylogenetic analysis has largely been eroded. This is particularly true for RNA viruses where rapid rates of evolutionary change ensure that the phylogenetic signal is quickly lost. Accordingly, each individual amino acid and nucleotide site in a viral genome has accumulated so many substitutions since its origin that accurate phylogenetic inference becomes an impossible task. For this reason sequence-based phylogenies have proven to be blunt tools for the study of viral origins, although signs of common ancestry may still reside in aspects of protein structure.
Despite the inherent limitations to understanding viral origins, a number of important theories have been proposed for the genesis of both RNA and DNA viruses, which continue to be debated to this day. Currently two such theories dominate discussions in this area; first, that viruses have a precellular origin, such that they are billions of years old, and may have even contributed to some of the fundamental architecture of the first cells; second, that viruses evolved after the first cellular organisms as “escaped genes” that acquired capsid proteins and the ability to replicate autonomously (Fig. 11.1). Although a third hypothesis—that viruses are regressed copies of cellular species that have shed those genes whose functions are provided by the host—has also been proposed, most notably in the case of the giant mimivirus and megavirus of amoeba,9,128 it does appear to be of general applicability. For example, the gene contents of RNA viruses and cellular species have almost no overlap, whereas under the regressive theory virus genes should have their ancestries in cellular genomes. In addition, although often discussed as such, these theories of viral origins are not mutually exclusive, and it is plausible that while some viruses predate the appearance of the first cells, others appeared more recently.
For many years the escaped gene theory dominated discussions on virus origins.160 Support for this theory was often based on the idea that as viruses are obligate parasites of host cells now, they must have always been so in the past, such that cells must have evolved before viruses. However, this idea is easy to refute. Because it is commonly thought that the first replicating molecules resided in an “RNA world” that existed before the evolution of DNA, it is easy to believe that modern RNA viruses originated from such ancient self-replicating RNA molecules and parasitized cells at a later date. Important recent evidence for the existence of an RNA world was the demonstration that ribonucleotides could be synthesized de novo under conditions that might replicate those of early earth.179 In most cases the escaped gene theory was also taken to mean that viruses could have escaped from host cells on multiple occasions. This is an attractive idea given the huge phenotypic diversity seen in viruses and that there is no one gene that characterizes all viruses. For example, an early idea was that eukaryotic viruses had escaped from eukaryotic cells, while bacteriophages had escaped from bacterial cells.180 Similarly, it is possible that RNA, DNA, and perhaps retroviruses represent independent episodes of host gene escape, as could the single- (ss) and double-stranded (ds) versions of RNA and DNA viruses, particularly as small ssDNA and large dsDNA viruses clearly have little in common.
However many different origins are postulated, the same general mechanisms are thought to have occurred: that a host
gene that possessed or acquired the ability to self-replicate escaped from the cell, acquiring a protein coat on the way, eventually evolving into an autonomously replicating entity. For example, single-strand positive-sense RNA (ssRNA+) viruses might be descended from escaped cellular messenger RNA (mRNA) molecules that either possessed or evolved RNA polymerase activity, while DNA viruses could be descended from DNA transposable elements or bacterial plasmids. It is also the case that all forms of the escaped gene theory make two important predictions: first, that most virus genes, including the capsid and replicase proteins, ultimately have their ancestries in cellular genomes, and second, because escape events could have occurred multiple times, viruses do not have a single (i.e., monophyletic) origin. In other words, there is no single phylogeny linking all types of virus, as is easily argued from the huge diversity of viruses described today. In theory, both of these predictions are testable, although in practice this is greatly inhibited by the enormous sequence divergence among viruses.
gene that possessed or acquired the ability to self-replicate escaped from the cell, acquiring a protein coat on the way, eventually evolving into an autonomously replicating entity. For example, single-strand positive-sense RNA (ssRNA+) viruses might be descended from escaped cellular messenger RNA (mRNA) molecules that either possessed or evolved RNA polymerase activity, while DNA viruses could be descended from DNA transposable elements or bacterial plasmids. It is also the case that all forms of the escaped gene theory make two important predictions: first, that most virus genes, including the capsid and replicase proteins, ultimately have their ancestries in cellular genomes, and second, because escape events could have occurred multiple times, viruses do not have a single (i.e., monophyletic) origin. In other words, there is no single phylogeny linking all types of virus, as is easily argued from the huge diversity of viruses described today. In theory, both of these predictions are testable, although in practice this is greatly inhibited by the enormous sequence divergence among viruses.
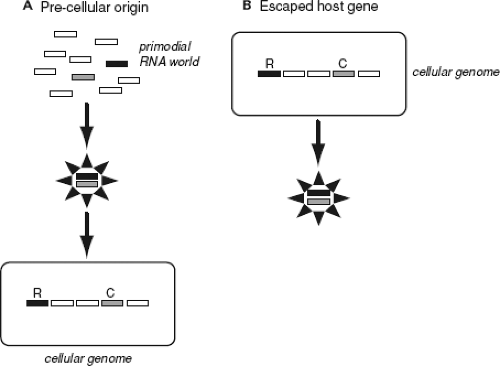 Figure 11.1. Schematic representation of two competing models for the origin of viruses. A: The precellular origin theory (in this case depicting the origin of RNA viruses). B: The escaped gene theory. Cellular genomes are represented by rounded rectangles, and the simplest model virus is shown here to comprise replicase (R) and capsid (C) genes only. (From Holmes EC. The Evolution and Emergence of RNA Viruses. Oxford: Oxford University Press, 2009, by permission of Oxford University Press.) |
A number of pieces of data have been used to support the idea that viruses had multiple origins after the appearance of cells. At the level of primary amino acid sequence, there is no robust sequence-based phylogeny for either RNA or DNA viruses, nor any gene that contains statistically significant sequence similarity at such vast evolutionary distances. Although there have been attempts to infer the evolutionary history of RNA viruses based on phylogenetic analyses of the RNA-dependent RNA polymerase (RdRp), the phylogenies in question are highly uncertain at the interfamily level where there is often no more sequence similarity than expected by chance alone.92,236 However, lack of phylogenetic resolution is not the same thing as an absence of common ancestry, and it is more likely that the inability to accurately infer the evolutionary history of all RNA viruses simply reflects extreme levels of sequence divergence. Indeed, it is striking that the RdRp sequences assigned to different RNA virus families still share a number of short, signature, amino acid motifs (such as a highly conserved GDD motif), some of which are also found in the reverse transcriptase (RT) protein used by retroviruses.81,120 Such conservation, albeit fragmentary, suggests that these replicatory proteins are distantly related. Unfortunately, these motifs are too short to allow the inference of reliable phylogenetic trees. Even more notable is that recent analyses of protein structure have revealed strong similarities between viruses that exhibit no primary sequence similarity, including between RNA and DNA viruses (see later).
In the case of RNA viruses, early phylogenetic analyses of RdRp sequences combined with information on gene order and content were used to construct supergroup classification schemes encompassing multiple viral families. For example, one such study suggested that RNA viruses be classified into the alpha-like, carmo-like, corona-like, flavi-like, picorna-like, and sobemo-like supergroups, each of which is characterized by a conserved gene order, distinctive 5′ and 3′ genome structures, as well as a putative clustering in RdRp phylogenies.81 However, as noted earlier, extreme sequence divergence means that these RdRp phylogenies are of debatable validity,236 and it is difficult to construct trees on gene order and content when these differ so dramatically among viral families and in genomes as small as those of RNA viruses. As a consequence, these deep interfamily phylogenies have in reality told us little about virus origins. However, a number of higher-order viral groupings, usually referred to as orders, do receive strong phylogenetic support, such that some aspects of the early evolutionary history of RNA viruses can be resolved. These groupings are (a) the Mononegavirales, which comprises four families of unsegmented ssRNA− viruses—the Bornaviridae, Filoviridae, Paramyxoviridae, and Rhabdoviridae (and the Mononegavirales clearly cluster together in RdRp trees); (b) the Nidovirales, comprising the Arteriviridae, Coronaviridae, and Roniviridae families of ssRNA+ viruses; and (c) the Picornavirales, comprising the Picornaviridae, Comoviridae, Dicistroviridae, Marnaviridae, and Sequiviridae families of ssRNA+ viruses.
Interfamily phylogenetic analyses of DNA viruses have generally proven more successful, in large part because the reliance on high-fidelity DNA polymerases for replication means that dsDNA viruses exhibit lower rates of nucleotide substitution and hence preserve the phylogenetic signal for longer time periods. For example, a number of families of large dsDNA viruses (i.e., those with genomes greater than 100 kb) clearly possess common ancestry such that they can be classified as nucleocytoplasmic large DNA viruses (NCLDVs), comprising ascoviruses, asfarviruses, iridoviruses, phycodnaviruses, and poxviruses.104 More recent analyses extended the NCLDV group to include the giant amoebal mimivirus, which is most closely related to the phycodnaviruses,105 as well as the recently described Marseillevirus, which was isolated from the same amoebal host as mimivirus.235 In other DNA viruses elements of capsid protein structure have been used to link herpesviruses with tailed bacteriophages,149 while there are clear evolutionary links between the Papillomaviridae and Polyomaviridae families of small dsDNA viruses.232 However, there is no phylogeny that encompasses both single- and double-stranded DNA viruses, which again reflects the great divergence between these very different types of virus (which differ massively in genome size) that share no genes in common. More starkly, it is even difficult to infer phylogenetic trees that link all the large dsDNA viruses that infect eukaryotes.105
The second prediction of the escaped gene theory—that most virus proteins ultimately have a host origin—is equally difficult to resolve. The two most important proteins in this respect, as they essentially define viruses, are the polymerase (a defining feature of all RNA viruses that carry an RdRp or RT) and those that make up the capsid (a defining feature of viruses). The case of the DNA polymerases used by DNA viruses is the easiest to discuss in this context as these enzymes are of the same form, and hence ancestry, as those used by cellular species (i.e., they are classified within the same polymerase families), and small DNA viruses utilize the host DNA polymerases for replication. However, while it is clear that these host and virus DNA polymerases are related,65,103 the position of the root, and therefore the direction of evolutionary change, in phylogenetic trees of DNA polymerases is uncertain. Hence, it is difficult to determine whether DNA polymerases are ultimately of host or viral origin,200 particularly as DNA polymerases may also have been involved in ancient lateral gene transfer events.65
A similar discussion can be mounted in the case of reverse transcriptase. Proteins that function as reverse transcriptase are a common component of cellular genomes in the form of telomerase, the group II (self-replicating) introns observed in a variety of bacterial species, not to mention the abundant retroelements found in many cellular species, as well as a variety of other genetic elements. Importantly, there are recognizable sequence similarities between the RTs of viruses and those that reside in host genomes such that it is possible to infer phylogenetic trees containing both.32,54 These trees have revealed a number of interesting features, including a major division between the long terminal repeat (LTR) and non-LTR retrotransposons, with retroviruses most closely related to the LTR retrotransposons, and that hepadnaviruses and caulimoviruses (small dsDNA viruses that utilize RT) have independent origins and are probably from LTR retrotransposons. However, as with the case of the DNA polymerases, the lack of an outgroup makes the rooting of these phylogenies uncertain, so whether viral RT genes preceded those present in cells or vice versa is difficult to determine.55
The situation is far more complex when it comes to the origin of the RdRp used by RNA viruses. Although the cells of some eukaryotic species contain proteins that function as RdRps, particularly those involved in the production of microRNAs, these exhibit little similarity with the RdRps encoded by viruses, even at the structural level.106 Similarly, cellular DNA polymerase (Pol) II, which catalyzes the synthesis of RNA from DNA, possesses RdRp activity130 yet shares little similarity with the RdRp utilized by RNA viruses, such that their evolutionary origins are currently impossible to resolve. Clearly, determining the evolutionary relationships among these highly diverse polymerase proteins represents a major technical challenge.
While the evidence from phylogenetic trees is ambiguous at best, other pieces of data do provide some support for the escaped gene theory. The most compelling of these is that there is at least one example of a virus whose origins likely lie with a host cellular protein, demonstrating that this mode of viral genesis is possible. The case in point involves hepatitis delta virus (HDV) agent, the ribozyme of which is related to the CPEB3 ribozyme found in a human intron sequence.192 That HDV is only found in humans and requires hepatitis B virus (HBV) for replication strongly suggests that its origins lie with the human genome.192
There has also been considerable debate over the significance of the giant amoebal mimivirus for theories of viral origins and evolution. Although phylogenetic analysis has shown that a small proportion (less than 1%) of mimivirus genes are of host origin, which has been used as support for the idea that viruses are “gene pickpockets” that originated after cellular species,158,159 at least 25% of the approximately 1,000 genes in mimivirus clearly link it to the NCLDV group of large DNA viruses,105 while an even larger set of genes (∼70% at the time of writing) have no known homologs, in either viral or cellular genomes, such that they can be regarded as orphans.64
Finally, it is striking that, at the time of writing, no RNA viruses have been discovered in Archaea. This could mean that either RNA viruses arose as escaped genes after the divergence of Archaea from other cellular species or that temperature constraints have led to a major reduction in the frequency of RNA viruses in hyperthermophilic Archaea,237 although this does not explain their absence in nonthermophiles. An alternative, and perhaps more likely, explanation is that RNA viruses do exist in Archaea but have simply not been detected as yet.
The competing theory for the origin of viruses, and one that is growing in popularity, is that they originated before the last universal cellular ancestor (LUCA) and represent the modern descendants of the earliest time in earth’s history. Hence, modern RNA viruses would be descendants of replicating elements from the RNA world, while DNA viruses would be remnants of the first DNA replicators, and retroviruses perhaps descendants of the first molecules that made the transition from RNA to DNA. For example, because they lack protein-coding regions, possess ribozyme activity, exhibit complex secondary structures, and mutate very rapidly, viroids are potential candidates for extant descendants of the RNA world.59 However, although the earliest RNA replicators may share some features with contemporary viroids, because viroids are only seen in plants and likely replicate with the assistance of host cellular
DNA Pol II makes it more likely that they represent escaped host genes or introns that never acquired protein coats.
DNA Pol II makes it more likely that they represent escaped host genes or introns that never acquired protein coats.
There are a number of theories for what the pre-LUCA world may have looked like, although all reasonably assume that this precellular stage of evolutionary history contained genetic elements less complex than the viruses we see today. One theory is that there was an ancient virus world of primordial replicators that existed before any cellular organisms and that both RNA (first) and DNA (later) viruses originated at this time.121 A version of this view of virus origins is shown in Figure 11.2. These ancient “viruses” may even have provided some of the features that characterized the first cellular organisms. For example, it has been proposed that the eukaryotic cell nucleus is derived from a virus envelope (the so-called viral eukaryogenesis hypothesis).13 An alternative theory for the pre-LUCA world is that RNA cells existed before the LUCA, that RNA viruses parasitized these hypothetical RNA cells, and that DNA evolved later as a way of escaping host cell responses.70 Although fascinating, such theories are unfortunately extremely difficult to test.
As sequence-based phylogenetic trees cannot provide insights into the pre-LUCA world, the main evidence for the precellular theory of virus origins is the presence of conserved genes, and more notably protein structures, among divergent viruses. In fact, arguably one of the most important advances in viral evolution in recent years has been the discovery of protein structures that are conserved among diverse viruses that possess little, if any, primary sequence similarity.11 For example, a conserved palm subdomain protein structure, consisting of a four-stranded antiparallel β-sheet and two α-helices, is found in both RNA-dependent and DNA-dependent polymerases.84 A more important case in point concerns the jelly-roll capsid, a tightly structured protein barrel that forms the major capsid subunit of virions with an icosahedral structure. Remarkably, the jelly-roll capsid is found in the virions of both RNA and DNA viruses, including such diverse groups as herpesviruses (dsDNA), picornaviruses (ssRNA+), and birnaviruses (dsRNA).11,41 Such conservation is strongly suggestive of an ancient common ancestry. Other highly conserved capsid architectures that strongly argue for ancient origins include the PRD1-adenovirus lineage, which is characterized by a double β-barrel fold and found in dsDNA viruses as diverse as bacteriophage PRD1, human adenovirus, and a variety of archaean viruses; the BTV-like lineage, which is found in some dsRNA viruses including members of the Reoviridae and Totiviridae; and the HK97-like lineage, which encompasses tailed dsDNA viruses that infect archaea, bacteria, and eukaryotes.16,17,124 Finally, a common virion architecture has been proposed for some viruses that do not possess an icosahedral capsid, including the archaean virus Halorubrum pleomorphic virus type 1 (HRPV-1).176
Although such structural conservation seems to provide a compelling argument for the antiquity of viruses, it has been proposed that any similarities in protein structure could have arisen more recently due to either strong convergent evolution or lateral gene transfer.159 While it is theoretically possible that convergent evolution may occur relatively frequently in viral capsid proteins that may be subject to strong selection to be small and perhaps of a specific shape, such large-scale convergence seems highly unlikely given that the similarity in capsid structure covers a huge range of viral taxa. As a consequence, multiple convergent events need to be invoked from very different starting points, and the more convergent evolution that is required, the less likely it becomes. Frequent lateral gene transfer also seems unlikely. As noted later, current data suggest that lateral gene transfer is relatively rare in RNA viruses (although commonplace in large DNA viruses), in large part because of major selective pressures against the expansion of genome sizes.92 As the earliest replicating RNAs likely possessed higher error rates than those of contemporary RNA viruses, the first genomes would have been even more restricted in size, such that lateral gene transfer without exact gene replacement must also have been uncommon at this time. Hence, although DNA viruses may be habitual gene pickpockets, RNA viruses do not seem to be. In conclusion, while not conclusive, the presence of structural similarities among highly divergent viruses currently constitutes the strongest evidence that viruses have a precellular origin.
The Time Scale of Virus Evolution
As our understanding of viral origins is vague, so is our knowledge of the antiquity of those families of viruses that circulate today, in part because viruses lack any sort of fossil record. In general, there are three ways in which the evolutionary history of viruses can be placed on a chronological scale. First, if there is a strong match between the phylogenetic tree of viruses and that of their hosts, such that they have co-diverged, then it is possible to use the divergence times of hosts to calibrate the time scale of virus evolution. Second, for viruses that evolve rapidly such that there is measurable evolution (i.e., mutations are fixed in viral populations during the time frame of human observation), which has been clearly demonstrated in both RNA viruses and ssDNA viruses, it is possible to determine the number of substitutions that have occurred between viruses sampled at known times (heterochronous samples) and use this information to calibrate the time scale of virus evolution under the assumption of a molecular clock (i.e., that there is an approximately constant rate of nucleotide or amino acid fixation). Third, for viruses where endogenous genome copies are present in the host, it is possible to use the substitution rate of the host to determine when these genome integration events occurred, especially if the endogenous sequences also co-diverge with their host species. All three approaches have limitations and can lead to wildly different interpretations of evolutionary time scales, the resolution of which has yet to be achieved.
Dating the time scale of virus evolution through the use of host divergence times (i.e., co-divergence) is perhaps the simplest and most robust approach to this form of molecular archaeology. This approach has been particularly successful in the study of DNA virus evolution. Good examples of its utility are the dating of herpesvirus evolution through an examination of the phylogenetic relationships of their vertebrate hosts, in which virus–host co-divergence may extend to some 400 million years148,149; of the animal iridoviruses107; of the baculoviruses of insects90; and of the papillomaviruses sampled from a number of vertebrates including humans.19 Clearly, although each of these virus families can be considered as “ancient,” they are in no way of sufficient age to inform on the question of virus origins. In addition, in some other large DNA viruses, with the poxviruses a good example, frequent host-jumping means that patterns of host–virus co-divergence can be difficult to infer,
so that the times of origin of key human pathogens like variola virus (VARV; the agent of smallpox) are still the source of considerable debate.102,135,206
so that the times of origin of key human pathogens like variola virus (VARV; the agent of smallpox) are still the source of considerable debate.102,135,206
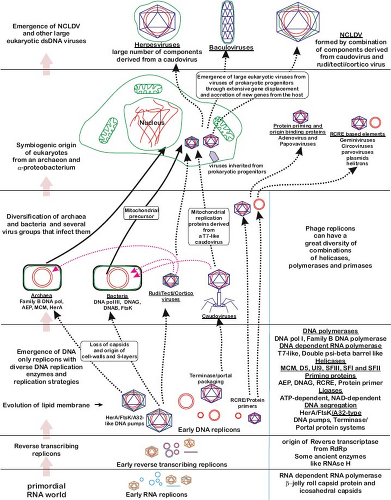 Figure 11.2. A plausible scenario for the origin of viruses. Major evolutionary transitions are shown on the left of the figure, while the innovations that occurred at each of these transitions are listed on the right. Colors are as follows: blue, RNA genomes; red, DNA genomes; green, lipid membranes. (Reprinted from Iyer LM, Balaji S, Koonin EV, et al. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res 2006;117:156–184, with permission from Elsevier. Figure kindly provided by Eugene Koonin.) |
Virus–host co-divergence has also been used to date the origin of a number of RNA viruses and retroviruses, although these estimates have sometimes proven controversial. Perhaps the most compelling case to date concerns the retrovirus simian foamy virus (SFV), where a statistically significant match between the phylogenetic trees of host and virus may extend to at least 30 million years137,217 (Fig. 11.3), and where the analysis of endogenous foamy viruses places their evolutionary history in mammals to over 100 million years.114
While bona fide examples of virus–host co-divergence constitute a powerful way to date the age of specific viruses, it is also the case that co-divergence is sometimes claimed without any associated statistical test, especially when a small number of taxa are involved such that any resemblance between host and virus phylogeny could occur by chance alone. Given that
cross-species transmission is a very common mode of virus macroevolution, it can be dangerous to construct a time scale of virus evolution without statistically significant co-divergence. In addition, there are a number of other evolutionary processes that can lead to a match between host and virus phylogenies that do not entail co-divergence. For example, it could be that cross-species transmission occurs more often among closely related host species.215 This preferential host switching36 may produce phylogenetic patterns that are difficult to distinguish from those of co-divergence. A good example is provided by the primate lentiviruses that infect humans (human immunodeficiency virus [HIV]), chimpanzees (simian immunodeficiency virus [SIV] cpz), and gorillas (SIVgor). That these three viruses are very closely related and infect related hosts might at face value be taken to mean that host and virus have co-diverged for several million years. However, closer inspection of the relevant virus phylogenies revealed that the genetic diversity in each case was in fact due to more recent cross-species transmission. Indeed, cross-species transmission involving very closely related host species appears to be common in viruses.119
cross-species transmission is a very common mode of virus macroevolution, it can be dangerous to construct a time scale of virus evolution without statistically significant co-divergence. In addition, there are a number of other evolutionary processes that can lead to a match between host and virus phylogenies that do not entail co-divergence. For example, it could be that cross-species transmission occurs more often among closely related host species.215 This preferential host switching36 may produce phylogenetic patterns that are difficult to distinguish from those of co-divergence. A good example is provided by the primate lentiviruses that infect humans (human immunodeficiency virus [HIV]), chimpanzees (simian immunodeficiency virus [SIV] cpz), and gorillas (SIVgor). That these three viruses are very closely related and infect related hosts might at face value be taken to mean that host and virus have co-diverged for several million years. However, closer inspection of the relevant virus phylogenies revealed that the genetic diversity in each case was in fact due to more recent cross-species transmission. Indeed, cross-species transmission involving very closely related host species appears to be common in viruses.119
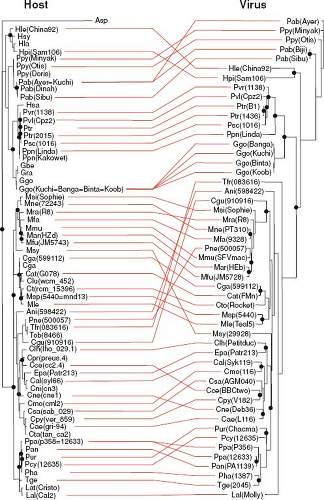 Figure 11.3. Long-term co-divergence between simian foamy virus (SFV) and its primate hosts. The tree on the left comprises 55 primate species and was inferred using mitochondrial COII sequences. The tree on the right contains 44 SFV sequences and was inferred using the viral pol gene. Strong host–virus associations are shown as horizontal lines, while incongruent relationships (i.e., host jumps) are shown as crossed lines (note that some hosts have no SFV associated with them). Host–virus co-divergence events are depicted by solid circles at the nodes. The match between the host and virus trees is far stronger than expected by chance (p = 0.007). Branch lengths are drawn to scale of nucleotide substitutions per site. (From Switzer WM, Salemi M, Shanmugam V, et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005;434:376–380; reprinted by permission from Macmillan Publishers Ltd.) |
Using heterochronous samples to calibrate the virus molecular clock is an extremely powerful and increasingly popular way to study the time scale of virus evolution in the recent past and is the most common method used with RNA viruses where measurable sequence evolution is a routine observation. However, it is also an approach where erroneous conclusions can be drawn if not performed with care. Because large numbers of gene sequences where the precise date of sampling is known are now available, and because virus evolution is often relatively clock-like, it is a straightforward exercise to date the age of samples of genetic diversity.49 In some cases divergence times estimated in this manner can be very accurate. For example, an analysis of heterochronous samples of human influenza A virus was able to accurately reconstruct the seasonal peaks and troughs in the population size of this virus.184 However, while these molecular clock approaches can work well for recent virus evolution—that is, for time scales covering that last few hundred years—they are prone to error at far deeper divergence times, providing a picture of virus evolution that is far too recent. An illustrative example of this effect is provided by the case of the SIVs. While molecular clock studies of SIV evolution using heterochronous samples place this on a time scale of hundreds of years, a calibration based on the biogeographic separation of Bioko Island from the coast of West Africa gave dates of at least 32,000, and perhaps over 100,000 years.233 More generally, there are cases in the literature where sensible preconception says that a specific virus should be a certain age, usually because it is thought to have diverged with a particular host species or is associated with a particular event in human history, yet molecular clock studies present a far more recent depiction of its origin.91
More difficult to explain is precisely why recently calibrated molecular clocks fail so badly in the estimation of ancient divergent times. The most likely explanation is that the statistical models used to estimate the number of nucleotide substitutions separating any two sequences fail to adequately account for all the details of virus evolution, thereby greatly underestimating the true numbers of mutations that have accumulated.91,94 In short, there has been excessive site saturation that leads to erroneous estimates of divergence times. At present there is no clear way to resolve this problem, although a likely path for the future is the development of more sophisticated models of nucleotide and amino acid substitution.
The final, and most recently developed, way to infer the time scale of virus evolution involves the use of endogenous viral sequences that are a common component of eukaryotic genomes. Endogenous genomic copies of exogenous viruses that have entered the germline are particularly commonplace in retroviruses, and it is estimated that approximately 5% to 8% of the human genome is composed of endogenous retrovirus, comprising at least 31 distinct families.115 In addition, there is a growing list of endogenous RNA and small DNA viruses, also referred to as endogenous viral elements (EVEs), usually composed of partial virus genome sequences.94,113
The importance of endogenous viruses is that they represent a sort of “fossil record” of past viral infections; once integrated into host genomes they cease to evolve like viruses and instead assume the low rates of nucleotide substitution that characterize their hosts, replicating using high-fidelity host DNA polymerases and likely experiencing fewer replications per unit time (Fig. 11.4). Consequently, if the mutational differences between endogenous viruses are known to occur postintegration, such as those observed between the LTRs of a single endogenous retrovirus, between duplicated EVEs, or when there is clear evidence for co-divergence, then divergence times can be estimated in a relatively straightforward manner using host substitution rates.
Molecular clock dating in this manner suggests that the exogenous ancestors of some human retroviruses may have diversified relatively early on in mammalian evolution.115 Most dramatic are cases of when both exogenous and endogenous copies of the same virus exist, which have generally resulted in a radically different picture of the time scale of viral evolution than using clock estimates based on heterochronous samples. For example, estimates of the age of primate lentiviruses based on the use of heterochronous sequences generally results in time scales of thousands of years at most,205 while the presence of endogenous lentiviruses in lemurs suggests that these viruses have circulated in primates for at least several million years.116 The same is true of endogenous viruses that are not retroviruses. Perhaps the most compelling of these are the avian hepadnaviruses, in which the observation of EVEs integrated at the same genomic positions in bird species that diverged at least 19 million years ago strongly suggests that hepadnaviruses are at least of the same age.80 This antiquity sits in stark contrast to studies of hepadnavirus evolution based on the use of heterochronous sequences, in which divergence times are measured on scales of a few thousand years.240 The same phenomenon has been proposed for a variety of other RNA viruses, including bornaviruses and filoviruses,14,97,113,218 as well as ssDNA viruses of the families Circoviridae and Parvoviridae.15,111,113,136 In each case, integrated copies of these viruses are observed in diverse host species, for example, comprising both placental and marsupial mammals in the case of the filoviruses, and sometimes showing virus–host co-divergence such that they are clearly millions of years old.113 However, there are also many cases where the phylogenies of the endogenous viruses and their hosts do not match, which complicates estimates of virus divergence times. As more host genomes are sequenced, it is certain that more endogenous viral elements will be discovered, which will undoubtedly shed new light on the true time scale of virus evolution.
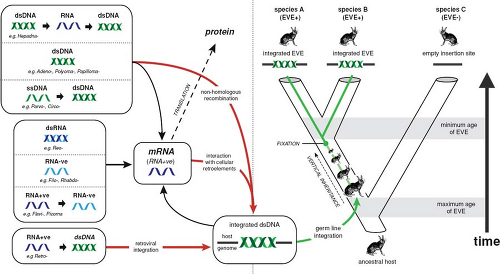 Figure 11.4. How endogenous virus elements (EVEs) are generated and can be used to estimate the age of viral families. Examples of different types of mammalian RNA and DNA viruses are shown. Critically, the presence of EVEs in related host species A and B and integrated into the same genomic position indicates that this integration event occurred prior to the divergence of these two species, such that its minimum age can be estimated if it is known when the host species diverged. (From Katzourakis A, Gifford RJ. Endogenous viral elements in animal genomes. PLoS Genet 2010;6:e1001191. Original figure kindly provided by Rob Gifford.) |
Virus Classification
Finally, and as a brief digression, it is useful to discuss the schemes used for virus classification, ranging from orders to species and genotypes, and what they mean in an evolutionary context, particularly as they indirectly relate to the issue of virus origins. The main points to make here are that although a useful and important aspect of comparative biology, all such classification schemes have a large arbitrary component to them, are not based on a specific evolutionary pattern or process, and do not necessarily mean that the viruses in question exhibit a specific set of biological differences. Indeed, these limitations are likely to apply to most microorganisms. To some extent, this arbitrariness sits in contrast to what is seen in sexually reproducing eukaryotes, where species represents a distinct and clearly definable evolutionary group—a population of interbreeding individuals. Hence, although what are termed virus species may possess phenotypic characteristics that can be defined for each group and form well-supported clusters on phylogenetic trees, this should not be taken as evidence that they have been formed by a distinct evolutionary process that is analogous to reproductive isolation in eukaryotes. The same is also true of the higher-order classifications proposed for viruses (e.g., supergroups, orders, genera) and for those below the level of species (e.g., genotypes or subtypes). All these groupings can be thought of as points in phylogenetic space rather than describing taxonomic groups that have attained a specific level of phenotypic divergence. In other cases viruses are classified simply through estimates of pairwise genetic diversity, with different levels of diversity signifying the division into species, genotypes, and so forth. Although simple, classification schemes constructed in this manner similarly have no basis in evolutionary theory and may be biased if different lineages evolve at different rates. In sum, it is overly simplistic to think that nature will generate clear-cut divisions in phenotypic and genotypic space that can be used to construct meaningful classification schemes, and hence that systematics tells us anything about virus origins and evolution.
Processes of Virus Evolution
Irrespective of debates over whether viruses are alive, it is clear that they are subject to the same forces that shape the evolution of cellular species, that is, mutation, natural selection, genetic drift, recombination (and reassortment), and migration. I will discuss the first four of these processes in this chapter. Migration, in the guise of viral epidemiology, is discussed elsewhere in this volume.
Mutation and Nucleotide Substitution in Viruses
The simplest way to examine the role of mutation in virus evolution is to measure the rate of its occurrence. Indeed, understanding the factors that shape the speed at which genetic variation is generated in viruses is central to understanding many aspects of their evolution. For RNA viruses mutation can in some ways be thought of as their defining evolutionary
feature, as it occurs at a pace that greatly exceeds that observed in other organisms.
feature, as it occurs at a pace that greatly exceeds that observed in other organisms.
Although mutation is the ultimate source of genetic variation, the pace of evolutionary change can in fact be measured in two rather different ways. One method is to estimate, experimentally, the rate at which mutations are generated de novo. Such rates have usually been presented as the number of mutations per nucleotide, per replication or the number of mutations per genome, per replication. However, because of the inherent complexities and biases in making these estimates, it has been suggested that estimates of mutation rate per nucleotide, per cell infection may be more informative.196 For example, one important complicating factor is that some viruses employ so-called stamping machine replication, in which a single virus acts as the template for all progeny genomes, so that mutations accumulate linearly, while others utilize “geometric” replication, in which some of the early progeny genomes are used as templates to produce further progeny, in turn increasing the rate of mutation accumulation.52
The power of mutation rate estimates is that they reveal the intrinsic error dynamics of the RNA or DNA polymerases used in viral replication and in theory allow a count of each type of mutation—advantageous, neutral, or deleterious—before they have been shaped by natural selection, although it is always difficult to accurately count the number of lethal mutations that are rapidly removed by purifying selection. A detailed compilation of mutation rate estimates for 23 viruses, and accounting for many of the complexities inherent in analyses of this kind, revealed that these rates varied from 10−6 to 10-4 mutations/nucleotide/cell infection for RNA viruses to 10−8 to 10−6 mutations/nucleotide/cell infection for DNA viruses196 (Fig. 11.5). For RNA viruses that replicate with RdRp, an enzyme that lacks a proofreading or repair function, this equates to mutation rates that are usually a little below approximately one per genome, per replication.47,48,52 Similarly, the lower mutation rates observed in large DNA viruses clearly reflect the higher fidelity of the DNA polymerases employed in their replication cycle. Of particular note is that mutation rate estimates in ssDNA viruses (a maximum of 1.1 × 10−6, although only two estimates are available) are higher than those of large dsDNA viruses (which range from 5.9 × 10−8 to 5.4 × 10−7), even though ssDNA viruses have such small genomes that they use host DNA polymerases for replication. It is therefore possible that the relatively high mutation rates in ssDNA viruses reflect less efficient proofreading and excision repair on ssDNA and/or frequent deamination.51 The study of Sanjuán et al.196 was also of note in that it revealed that retroviruses such as HIV have error rates that overlapped with those of RdRp-utilizing viruses, even though earlier studies suggested that RT exhibits higher fidelity than RdRp.47,48,143
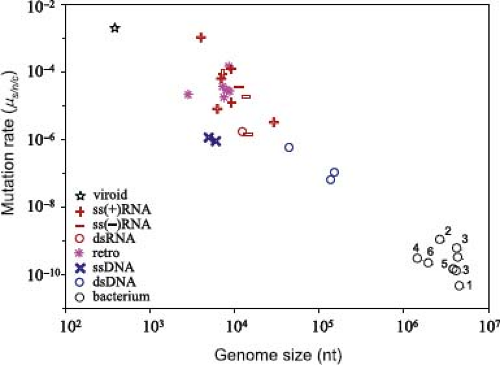 Figure 11.5. Comparative rates of mutation in different types of virus and their relationship to genome size. Comparable values from bacteria are also shown. Mutation rates (y-axis) are given per nucleotide per cell infection (s/n/c). See reference 196 for more details including the taxa analyzed. (From Sanjuán R, Nebot MR, Chirico N, et al. Viral mutation rates. J Virol 2010;84:9733–9748. Figure kindly provided by Rafa Sanjuán.) |
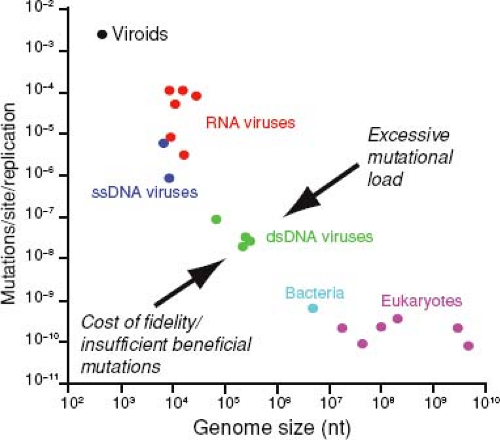 Figure 11.6. Relationship between mutation rate and genome size in diverse organisms including RNA and DNA viruses and viroids. The competing evolutionary forces that might be responsible for the limited range of observed error rates and genome sizes are shown. For details see reference 93. Data from reference 75. (From Holmes EC. What does virus evolution tell us about virus origins? J Virol 2011;85:5247–5251.) |
From a broader perspective these estimates of mutation rate are compatible with the idea that for all living systems there is a strongly inverse relationship between mutation rate and genome size (Fig. 11.6).75,93 It also seems likely that the systematic difference in mutation rate and genome size between RNA and DNA viruses is associated with many of the main evolutionary distinctions between these two types of infectious agents that are discussed throughout this chapter, such that they can be thought of as occupying very different regions of evolutionary parameter space (Table 11.1).
Although they utilize related DNA polymerases, mutation rates in large DNA viruses are higher than those of bacteria and eukaryotes, which may reflect an absence of the full set of repair enzymes and pathways in the former. However, there are two possible exceptions to this rule. First, there is currently no estimate of mutation rates in mimivirus or megavirus, although these are expected to fall within the bacterial range (as the genome sizes of these viruses overlap with those of bacteria), and hence are lower than that observed in any virus to date.
Second, we similarly lack an estimate of mutation rate in the small dsDNA viruses, such as the papillomaviruses. Although the relationship depicted in Figures 11.5 and 11.6 implies that these viruses will mutate rapidly, most estimates of substitution rate in papillomaviruses are in a similar range to those of large DNA viruses.68,187 This implies that papillomaviruses similarly mutate relatively slowly, which would break the simple relationship between mutation rate and genome size.
Second, we similarly lack an estimate of mutation rate in the small dsDNA viruses, such as the papillomaviruses. Although the relationship depicted in Figures 11.5 and 11.6 implies that these viruses will mutate rapidly, most estimates of substitution rate in papillomaviruses are in a similar range to those of large DNA viruses.68,187 This implies that papillomaviruses similarly mutate relatively slowly, which would break the simple relationship between mutation rate and genome size.
Table 11.1 The Differing Evolutionary Parameter Spaces Occupied by RNA and DNA Virusesa | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
The relationship between mutation rate and genome size is of twofold importance. First, because it incorporates genetic systems ranging from viroids to eukaryotes, it covers at least eight orders of magnitude of both genome size and mutation rate, and few things in biology encompass such diversity. Second, it implies that mutation rates that are either too high or too low are selected against.93 With respect to the latter, a popular idea is that viral mutation rates (and particularly the high mutation rates of RNA viruses) are the result of an evolutionary trade-off, either between replication rate and replication fidelity74 or between the rates of deleterious and advantageous mutation.52,213 Data can be cited in support of both relationships.92,144,224 The possible trade-off between the rates of deleterious and advantageous mutation seems particularly compelling. On the one hand, as viruses will be commonly exposed to changing environments (i.e., different hosts, a variety of cell types, frequent immune pressure), the generation of some genetic variation via mutation is likely to be selectively advantageous. This idea also has experimental support; RNA polymerases with lower fidelity are sometimes selectively favored over those with higher fidelity.92,144,224 On the other hand, there must be an upper limit on the mutation rates experienced by viruses (and all living systems), as excessive error will result in major fitness losses. Powerful evidence for this ceiling on mutation rates is provided by experiments invoking lethal mutagenesis (see later), in which artificially increasing error rates with mutagens such as 5-fluorouracil and ribavirin result in an excessive mutational load.26,171 It is therefore likely that RNA viruses exist close to their maximum tolerable mutation rates, which in turn imposes an upper limit on genome size. However, it is also likely that RNA viruses are mechanistically unable to reduce their error rates to the levels associated with DNA polymerases. A higher-fidelity RNA polymerase would need to be more complex, and hence longer, than those that currently exist, yet this cannot evolve because, by increasing genome length, it will result in too many deleterious mutations. This evolutionary conundrum is commonly referred to as Eigen’s paradox and is a key element in theories for the early evolution of genomic complexity.
The second measure of the pace of virus evolution is the rate of nucleotide substitution per nucleotide site, per year (subs/site/year). As this measure reflects the population success of any mutation, it by necessity incorporates the action of natural selection. Accordingly, deleterious mutations that have been removed by purifying selection will not be counted, while advantageous mutations will be fixed more rapidly than neutral ones. Nucleotide substitution rates are usually far easier to estimate than mutation rates and as such provide a simple and powerful means to compare patterns of virus evolution.
As with rates of mutation, rates of nucleotide substitution vary markedly among viruses. Across RNA and DNA viruses as a whole, nucleotide substitution rates vary by over five orders of magnitude, in large part reflecting the differences in background mutation rate described earlier. Hence, most substitution rates in RNA viruses fall within an order of magnitude of a value of 1 × 10−3 subs/site/year,87,110 while the rates in many dsDNA viruses are closer to 1 × 10−8 subs/site/year.89,145,148,187 Undoubtedly, our understanding of viral substitution rates would be improved by measures of evolutionary dynamics in the smallest (i.e., viroids) and largest (i.e., mimivirus) viral systems.
Most of the variance in the substitution rates in both RNA and DNA viruses likely reflects virus-specific differences in either mutation rate, replication rate, or both. Mutation rates have been discussed earlier, and some studies have revealed that substitution rates in RNA viruses are negatively associated with genome size as expected if background mutation is the main determinant of substitution rate.110 Although few direct estimates of replication rate are available, they likewise clearly play a major role in shaping substitution rates. For example, although the retrovirus SFV likely has an RT-associated error rate that is similar to those of other retroviruses, its co-divergence with primates for over 30 million years (Fig. 11.3) leads to estimates of the substitution rate of only 1.7 × 10−8 subs/site/year.217 This most likely reflects a low rate of replication,
although this merits further investigation. Similarly, replication rates appear to be low in papillomaviruses, in which virus replication occurs simultaneously with the division of host epithelial cells, at approximately 10 to 100 generations per year,19 which likely contributes to the low substitution rates estimated in this virus. In contrast, that DNA viruses often replicate more rapidly than their hosts may in part explain why virus substitution rates are higher than host substitution rates even though they utilize similar polymerases. For example, the Bo17 protein of bovine herpesvirus 4 represents a viral capture of the mammalian 2β-1,6-N-acetylglucosaminyltransferase-mucin protein.145 As a phylogenetic analysis revealed that this gene was captured from the host after the split between cattle and African buffalo approximately 1.5 million years ago, it was possible to estimate that the viral gene had evolved 20 to 30 times faster than its cellular homolog.145 Finally, for persistently infecting viruses, substitution rates may also differ between periods of intra- and interhost evolution. For example, HIV-1 substitution rates are higher within than among hosts.142 This may be because intra-host HIV evolution is dominated by the positive selection of immune escape mutations or because some of the mutations that occur within hosts are purged at interhost transmission, thereby reducing the substitution rate.
although this merits further investigation. Similarly, replication rates appear to be low in papillomaviruses, in which virus replication occurs simultaneously with the division of host epithelial cells, at approximately 10 to 100 generations per year,19 which likely contributes to the low substitution rates estimated in this virus. In contrast, that DNA viruses often replicate more rapidly than their hosts may in part explain why virus substitution rates are higher than host substitution rates even though they utilize similar polymerases. For example, the Bo17 protein of bovine herpesvirus 4 represents a viral capture of the mammalian 2β-1,6-N-acetylglucosaminyltransferase-mucin protein.145 As a phylogenetic analysis revealed that this gene was captured from the host after the split between cattle and African buffalo approximately 1.5 million years ago, it was possible to estimate that the viral gene had evolved 20 to 30 times faster than its cellular homolog.145 Finally, for persistently infecting viruses, substitution rates may also differ between periods of intra- and interhost evolution. For example, HIV-1 substitution rates are higher within than among hosts.142 This may be because intra-host HIV evolution is dominated by the positive selection of immune escape mutations or because some of the mutations that occur within hosts are purged at interhost transmission, thereby reducing the substitution rate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


