17
Vasoactive Peptides
CASE STUDY
During a routine check, a 45-year-old man was found to have high blood pressure (165/100 mm Hg). Blood pressure remained high on two follow-up visits. His physician initially prescribed hydrochlorothiazide, a diuretic commonly used to treat hypertension. Although his blood pressure was reduced by hydrochlorothiazide, it remained at a hypertensive level (145/95 mm Hg), and he was referred to the university hypertension clinic. Your evaluation revealed that the patient had elevated plasma renin activity and aldosterone concentration. Hydrochlorothiazide was therefore replaced with enalapril, an angiotensin-converting enzyme inhibitor. Enalapril lowered the blood pressure to almost normotensive levels. However, after several weeks on enalapril, the patient now returns complaining of a persistent cough. In addition, some signs of angioedema are detected. How does enalapril lower blood pressure? Why does it occasionally cause coughing and angioedema? What other drugs could be used to inhibit renin secretion or suppress the renin-angiotensin system, and decrease blood pressure, without the adverse effects of enalapril?
Peptides are used by most tissues for cell-to-cell communication. As noted in Chapters 6 and 21, they play important roles as transmitters in the autonomic and central nervous systems. Several peptides exert important direct effects on vascular and other smooth muscles. These peptides include vasoconstrictors (angiotensin II, vasopressin, endothelins, neuropeptide Y, and urotensin) and vasodilators (bradykinin and related kinins, natriuretic peptides, vasoactive intestinal peptide, substance P, neurotensin, calcitonin gene-related peptide, and adrenomedullin). This chapter focuses on the smooth muscle actions of the peptides and on drugs that alter their biosynthesis or actions.
 ANGIOTENSIN
ANGIOTENSIN
BIOSYNTHESIS OF ANGIOTENSIN
The pathway for the formation and metabolism of angiotensin II (ANG II) is summarized in Figure 17–1. The principal steps include enzymatic cleavage of angiotensin I (ANG I) from angiotensinogen by renin, conversion of ANG I to ANG II by converting enzyme, and degradation of ANG II by several peptidases.
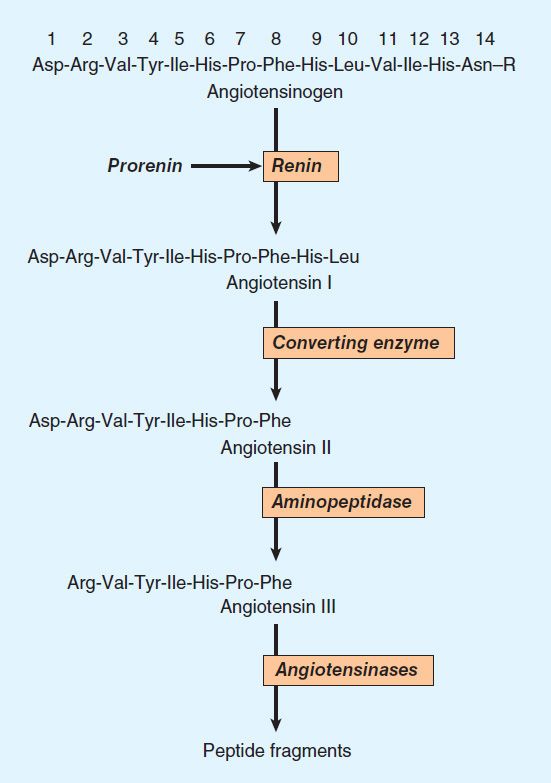
FIGURE 17–1 Chemistry of the renin-angiotensin system. The amino acid sequence of the amino terminal of human angiotensinogen is shown. R denotes the remainder of the protein molecule. See text for additional steps in the formation and metabolism of angiotensin peptides.
Renin
Renin is an aspartyl protease enzyme that specifically catalyzes the hydrolytic release of the decapeptide ANG I from angiotensinogen. It is synthesized as a prepromolecule that is processed to prorenin, which has poorly understood actions, and then to active renin, a glycoprotein consisting of 340 amino acids.
Renin in the circulation originates in the kidneys. Enzymes with renin-like activity are present in several extrarenal tissues, including blood vessels, uterus, salivary glands, and adrenal cortex, but no physiologic role for these enzymes has been established. Within the kidney, renin is synthesized and stored in the juxtaglomerular apparatus of the nephron. Specialized granular cells called juxtaglomerular cells are the site of synthesis, storage, and release of renin. The macula densa is a specialized segment of the nephron that is closely associated with the vascular components of the juxtaglomerular apparatus. The vascular and tubular components of the juxtaglomerular apparatus, including the juxtaglomerular cells, are innervated by the sympathetic nervous system.
Control of Renin Release
The rate at which renin is released by the kidney is the primary determinant of activity of the renin-angiotensin system. Active renin is released by exocytosis immediately upon stimulation of the juxtaglomerular apparatus. Prorenin is released constitutively, usually at a rate higher than that of active renin, thus accounting for the fact that prorenin can constitute 80–90% of the total renin in the circulation. The significance of circulating prorenin is discussed at the end of this section. Active renin release is controlled by a variety of factors, including the macula densa, a renal vascular receptor, the sympathetic nervous system, and ANG II.
A. Macula Densa
Renin release is controlled in part by the macula densa, a structure that has a close anatomic association with the afferent arteriole. The initial step involves the detection of some function of NaCl concentration in, or delivery to, the distal tubule, possibly by the Na+/K+/2Cl− cotransporter. The macula densa then signals changes in renin release by the juxtaglomerular cells such that there is an inverse relationship between NaCl delivery or concentration and renin release. Potential candidates for signal transmission include prostaglandin E2 (PGE2) and nitric oxide, which stimulate renin release, and adenosine, which inhibits it. Because the sodium intake in the general population is high, macula densa-mediated renin secretion is usually at basal levels, increasing only when sodium intake decreases.
B. Renal Baroreceptor
The renal vascular baroreceptor mediates an inverse relationship between renal artery pressure and renin release. The mechanism is not completely understood but it appears that the juxtaglomerular cells are sensitive to stretch and that increased stretch results in decreased renin release. The decrease may result from influx of calcium which, somewhat paradoxically, inhibits renin release. The paracrine factors PGE2, nitric oxide, and adenosine have also been implicated in the baroreceptor control of renin release. At normal blood pressure, renal baroreceptor-mediated renin secretion is low; it increases in hypotensive states.
C. Sympathetic Nervous System
Norepinephrine released from renal sympathetic nerves stimulates renin release indirectly by α-adrenergic activation of the renal baroreceptor and macula densa mechanisms, and directly by an action on the juxtaglomerular cells. In humans, the direct effect is mediated by β1 adrenoceptors. Through this mechanism, reflex activation of the sympathetic nervous system by hypotension or hypovolemia leads to activation of the renin-angiotensin system.
D. Angiotensin
Angiotensin II inhibits renin release. The inhibition results from increased blood pressure acting by way of the renal baroreceptor and macula densa mechanisms, and from a direct action of the peptide on the juxtaglomerular cells. The direct inhibition is mediated by increased intracellular Ca2+ concentration and forms the basis of a short-loop negative feedback mechanism controlling renin release. Interruption of this feedback with drugs that inhibit the renin-angiotensin system results in stimulation of renin release.
E. Intracellular Signaling Pathways
The release of renin by the juxtaglomerular cells is controlled by interplay among three intracellular messengers: cAMP, cyclic guanosine monophosphate (cGMP), and free cytosolic Ca2+ concentration (Figure 17–2). cAMP plays a major role; maneuvers that increase cAMP levels, including activation of adenylyl cyclase, inhibition of cAMP phosphodiesterases, and administration of cAMP analogs, increase renin release. In experimental studies, selective deficiency of Gsα in the juxtaglomerular cells is associated with a marked reduction in basal renin secretion and in the response to several stimuli to renin secretion.
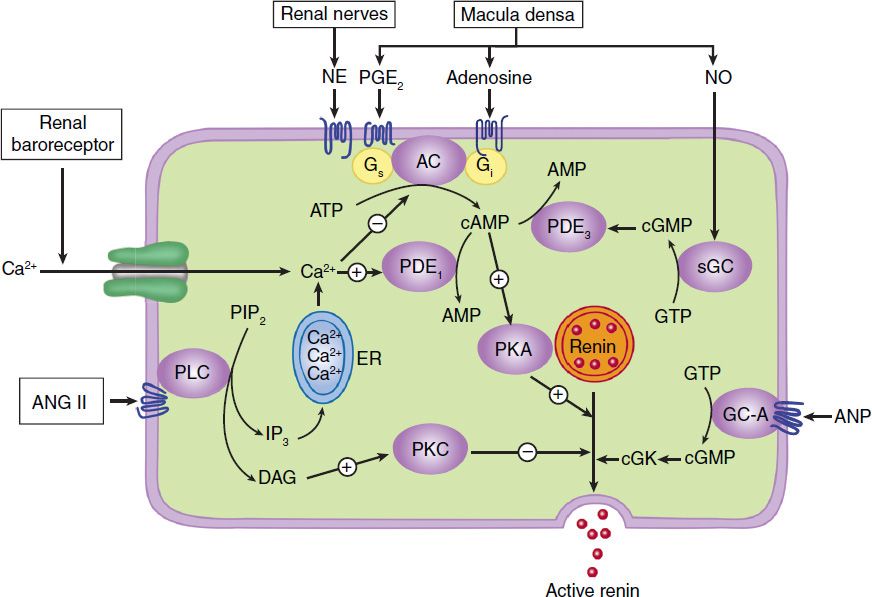
FIGURE 17–2 Major physiologic inputs to renin release and proposed integration with signaling pathways in the juxtaglomerular cell. AC, adenylyl cyclase; ANG II, angiotensin II; ANP, atrial natriuretic peptide; cGK, protein kinase G; DAG, diacylglycerol; GC-A, particulate guanylyl cyclase; ER, endoplasmic reticulum; IP3, inositol trisphosphate; NE, norepinephrine; NO, nitric oxide; PDE, phosphodiesterase; PKA, protein kinase A; PLC, phospholipase C; sGC, soluble guanylyl cyclase. (Adapted, with permission, from Castrop H et al: Physiology of kidney renin. Physiol Rev 2010;90:607.)
Increases in intracellular Ca2+ can result from increased entry of extracellular Ca2+ or mobilization of Ca2+ from intracellular stores, while increases in cGMP levels can result from activation of soluble or particulate guanylyl cyclase. Ca2+ and cGMP appear to alter renin release indirectly, primarily by changing cAMP levels.
F. Pharmacologic Alteration of Renin Release
The release of renin is altered by a wide variety of pharmacologic agents. Renin release is stimulated by vasodilators (hydralazine, minoxidil, nitroprusside), β-adrenoceptor agonists, α-adrenoceptor antagonists, phosphodiesterase inhibitors (eg, theophylline, milrinone, rolipram), and most diuretics and anesthetics. This stimulation can be accounted for by the control mechanisms just described. Drugs that inhibit renin release are discussed below.
Many of the peptides reviewed in this chapter also alter renin release. Release is stimulated by adrenomedullin, bradykinin, and calcitonin gene-related peptide, and inhibited by atrial natriuretic peptide, endothelin, substance P, and vasopressin.
Angiotensinogen
Angiotensinogen is the circulating protein substrate from which renin cleaves ANG I. It is synthesized in the liver. Human angiotensinogen is a glycoprotein with a molecular weight of approximately 57,000. The 14 amino acids at the amino terminal of the molecule are shown in Figure 17–1. In humans, the concentration of angiotensinogen in the circulation is less than the Km of the renin-angiotensinogen reaction and is therefore an important determinant of the rate of formation of angiotensin.
The production of angiotensinogen is increased by corticosteroids, estrogens, thyroid hormones, and ANG II. It is elevated during pregnancy and in women taking estrogen-containing oral contraceptives. The increased plasma angiotensinogen concentration is thought to contribute to the hypertension that may occur in these situations.
Angiotensin I
Although ANG I contains the peptide sequences necessary for all of the actions of the renin-angiotensin system, it has little or no biologic activity. Instead, it must be converted to ANG II by converting enzyme (Figure 17–1). ANG I may also be acted on by plasma or tissue aminopeptidases to form [des-Asp1]angiotensin I; this in turn is converted to [des-Asp1]angiotensin II (commonly known as angiotensin III) by converting enzyme.
Converting Enzyme (ACE, Peptidyl Dipeptidase, Kininase II)
Converting enzyme is a dipeptidyl carboxypeptidase with two active sites that catalyzes the cleavage of dipeptides from the carboxyl terminal of certain peptides. Its most important substrates are ANG I, which it converts to ANG II, and bradykinin, which it inactivates (see Kinins, below). It also cleaves enkephalins and substance P, but the physiologic significance of these effects has not been established. The action of converting enzyme is prevented by a penultimate prolyl residue in the substrate, and ANG II is therefore not hydrolyzed by converting enzyme. Converting enzyme is distributed widely in the body. In most organs, converting enzyme is located on the luminal surface of vascular endothelial cells and is thus in close contact with the circulation.
A homolog of converting enzyme known as ACE2 was recently found to be highly expressed in vascular endothelial cells of the kidneys, heart, and testes. Unlike converting enzyme, ACE2 has only one active site and functions as a carboxypeptidase rather than a dipeptidyl carboxypeptidase. It removes a single amino acid from the C-terminal of ANG I forming ANG 1-9 (Figure 17–3), which is inactive but is converted to ANG 1-7 by ACE. ACE2 also converts ANG II to ANG 1-7. ANG 1-7 has vasodilator activity, apparently mediated by the orphan heterotrimeric guanine nucleotide-binding protein-coupled receptor (Mas receptor). This vasodilation may serve to counteract the vasoconstrictor activity of ANG II. ACE2 also differs from ACE in that it does not hydrolyze bradykinin and is not inhibited by converting enzyme inhibitors (see below). Thus, ACE2 more closely resembles an angiotensinase than a converting enzyme.
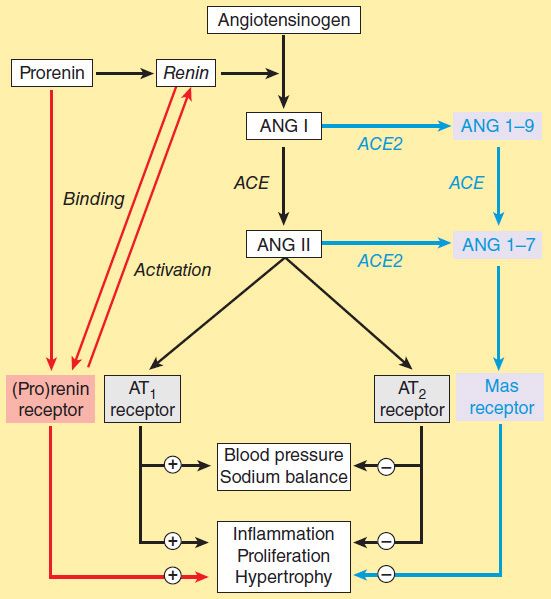
FIGURE 17–3 The renin-angiotensin system showing the established system (black) and recently discovered pathways involving the (pro)renin receptor (red) and ANG 1-7 (blue). (Adapted, with permission, from Castrop H et al: Physiology of kidney renin. Physiol Rev 2010;90:607.)
Angiotensinase
Angiotensin II, which has a plasma half-life of 15–60 seconds, is removed rapidly from the circulation by a variety of peptidases collectively referred to as angiotensinase. It is metabolized during passage through most vascular beds (a notable exception being the lung). Most metabolites of ANG II are biologically inactive, but the initial product of aminopeptidase action—[des-Asp1]angiotensin II—retains considerable biologic activity.
ACTIONS OF ANGIOTENSIN II
Angiotensin II exerts important actions at vascular smooth muscle, adrenal cortex, kidney, heart, and brain via the receptors described below. Through these actions, the renin-angiotensin system plays a key role in the regulation of fluid and electrolyte balance and arterial blood pressure. Excessive activity of the renin-angiotensin system can result in hypertension and disorders of fluid and electrolyte homeostasis.
Blood Pressure
Angiotensin II is a potent pressor agent—on a molar basis, approximately 40 times more potent than norepinephrine. The pressor response to intravenous ANG II is rapid in onset (10–15 seconds) and sustained during long-term infusions. A large component of the pressor response is due to direct contraction of vascular—especially arteriolar—smooth muscle. In addition, however, ANG II can also increase blood pressure through actions on the brain and autonomic nervous system. The pressor response to ANG II is usually accompanied by little or no reflex bradycardia because the peptide simultaneously acts on the brain to reset the baroreceptor reflex control of heart rate to a higher pressure.
Angiotensin II also interacts with the autonomic nervous system. It stimulates autonomic ganglia, increases the release of epinephrine and norepinephrine from the adrenal medulla, and most important, facilitates sympathetic transmission by an action at adrenergic nerve terminals. The latter effect involves both increased release and reduced reuptake of norepinephrine. Angiotensin II also has a less important direct positive inotropic action on the heart.
Adrenal Cortex & Kidney
Angiotensin II acts directly on the zona glomerulosa of the adrenal cortex to stimulate aldosterone synthesis and release. At higher concentrations, ANG II also stimulates glucocorticoid synthesis. Angiotensin II acts on the kidney to cause renal vasoconstriction, increase proximal tubular sodium reabsorption, and inhibit the release of renin.
Central Nervous System
In addition to its central effects on blood pressure, ANG II acts on the central nervous system to stimulate drinking (dipsogenic effect) and increase the secretion of vasopressin and adrenocorticotropic hormone (ACTH). The physiologic significance of these effects is not known.
Cell Growth
Angiotensin II is mitogenic for vascular and cardiac muscle cells and may contribute to the development of cardiovascular hypertrophy. It also exerts a variety of important effects on the vascular endothelium. Indeed, overactivity of the renin-angiotensin system has been implicated as one of the most significant factors in the development of hypertensive vascular disease. Considerable evidence now indicates that ACE inhibitors and ANG II receptor antagonists (see below) slow or prevent morphologic changes (remodeling) following myocardial infarction that would otherwise lead to heart failure. The stimulation of vascular and cardiac growth by ANG II is mediated by other pathways, probably receptor and nonreceptor tyrosine kinases such as the Janus tyrosine kinase Jak2, and by increased transcription of specific genes (see Chapter 2).
ANGIOTENSIN RECEPTORS & MECHANISM OF ACTION
Angiotensin II receptors are widely distributed in the body. Like the receptors for other peptide hormones, ANG II receptors are G protein-coupled and located on the plasma membrane of target cells, and this permits rapid onset of the various actions of ANG II. Two distinct subtypes of ANG II receptors, termed AT1 and AT2, have been identified on the basis of their differential affinity for antagonists and their sensitivity to sulfhydryl-reducing agents. AT1 receptors have a high affinity for the inhibitor losartan and a low affinity for PD 123177 (an experimental nonpeptide antagonist), whereas AT2 receptors have a high affinity for PD 123177 and a low affinity for losartan. Angiotensin II binds equally to both subtypes. The relative proportion of the two subtypes varies from tissue to tissue: AT1 receptors predominate in vascular smooth muscle. Most of the known actions of ANG II are mediated by the AT1 receptor, a Gq protein-coupled receptor. Binding of ANG II to AT1 receptors in vascular smooth muscle results in activation of phospholipase C and generation of inositol trisphosphate and diacylglycerol (see Chapter 2). These events, which occur within seconds, result in smooth muscle contraction.
The AT2 receptor has a structure and affinity for ANG II similar to those of the AT1 receptor. In contrast, however, stimulation of AT2 receptors causes vasodilation that may serve to counteract the vasoconstriction resulting from AT1 receptor stimulation. AT2 receptor-mediated vasodilation appears to be nitric oxide-dependent and may involve the bradykinin B2 receptor-nitric oxide-cGMP pathway. AT2 receptors are present at high density in all tissues during fetal development, but they are much less abundant in the adult where they are expressed at high concentration only in the adrenal medulla, reproductive tissues, vascular endothelium, and parts of the brain. AT2 receptors are up-regulated in pathologic conditions including heart failure and myocardial infarction. The functions of the AT2 receptor appear to include fetal tissue development, inhibition of growth and proliferation, cell differentiation, apoptosis, and vasodilation.
INHIBITION OF THE RENIN-ANGIOTENSIN SYSTEM
In view of the importance of the renin-angiotensin system in cardiovascular disease, considerable effort has been directed to developing drugs that inhibit the system. A wide variety of agents that block the formation or action of ANG II is now available. Some of these drugs block renin release, but most inhibit the conversion of ANG I to ANG II, block angiotensin AT1 receptors, or inhibit the enzymatic action of renin.
Drugs that Block Renin Release
Drugs that interfere with the sympathetic nervous system inhibit the release of renin. Examples are propranolol and other β-adrenoceptor-blocking drugs, which act by blocking the renal β receptors involved in the sympathetic control of renin release.
Angiotensin-Converting Enzyme Inhibitors
An important class of orally active ACE inhibitors, directed against the active site of ACE, is now extensively used. Captopril and enalapril are examples of the many ACE inhibitors that are available. These drugs differ in their structure and pharmacokinetics, but they are interchangeable in clinical use. ACE inhibitors decrease systemic vascular resistance without increasing heart rate, and they promote natriuresis. As described in Chapters 11 and 13, they are effective in the treatment of hypertension, decrease morbidity and mortality in heart failure and left ventricular dysfunction after myocardial infarction, and delay the progression of diabetic nephropathy.
ACE inhibitors not only block the conversion of ANG I to ANG II but also inhibit the degradation of other substances, including bradykinin, substance P, and enkephalins. The action of ACE inhibitors to inhibit bradykinin metabolism contributes significantly to their hypotensive action (see Figure 11–5) and is apparently responsible for some adverse side effects, including cough and angioedema. These drugs are contraindicated in pregnancy because they cause fetal kidney damage.
Angiotensin Receptor Blockers
Peptide antagonists of the action of ANG II are available for research use. However, the nonpeptide ANG II receptor blockers (ARBs) are of much greater interest. Losartan, valsartan, and several others are orally active, potent, and specific competitive antagonists of angiotensin AT1 receptors. The efficacy of these drugs in hypertension is similar to that of ACE inhibitors, but they are associated with a lower incidence of cough. Like ACE inhibitors, ARBs slow the progression of diabetic nephropathy and valsartan has been reported to decrease the incidence of diabetes in patients with impaired glucose tolerance. The antagonists are also effective in the treatment of heart failure and provide a useful alternative when ACE inhibitors are not well tolerated. ARBs are generally well tolerated but should not be used by patients with nondiabetic renal disease or in pregnancy. In addition, some ARBs may cause a syndrome known as sprue-like enteropathy.
Marfan syndrome is a connective tissue disorder associated with aortic disease and other abnormalities involving increased transforming growth factor (TGF)-β signaling. Since ANG II increases TGF-β levels, it was reasoned that blockade of the renin-angiotensin system might be beneficial in Marfan syndrome. Promising initial results have been obtained with losartan, and clinical trials are underway.
The currently available ARBs are selective for the AT1 receptor. Since prolonged treatment with the drugs disinhibits renin release and increases circulating ANG II levels, there may be increased stimulation of AT2 receptors. This may be significant in view of the evidence that activation of the AT2 receptor causes vasodilation and other beneficial effects. AT2 receptor antagonists such as PD 123177 are available for research but have no clinical applications at this time. However, a selective AT2 agonist, compound 21, lowers blood pressure in hypertensive animals and may be beneficial in human hypertension. The clinical benefits of ARBs are similar to those of ACE inhibitors, and it is not clear if either group has significant advantages over the other.
Renin Inhibitors
Cleavage of angiotensinogen by renin (Figures 17–1 and 17–3) is the rate-limiting step in the formation of ANG II and thus represents a logical target for inhibition of the renin-angiotensin system. Drugs that inhibit renin have been available for many years but have been limited by low potency, poor bioavailability, and short duration of action. However, a new class of nonpeptide, low-molecular-weight, orally active inhibitors is now available.
Aliskiren is the first nonpeptide renin inhibitor to be approved for the treatment of hypertension. In healthy subjects, aliskiren produces dose-dependent reductions in plasma renin activity and ANG I and II and aldosterone concentrations. In patients with hypertension, many of whom have elevated plasma renin levels, aliskiren suppresses plasma renin activity and causes dose-related reductions in blood pressure similar to those produced by ACE inhibitors and ARBs. The safety and tolerability of aliskiren appear to be comparable to angiotensin antagonists and placebo. Thus, renin inhibition may be an important approach to the treatment of hypertension. Aliskiren is contraindicated in pregnancy.
Inhibition of the renin-angiotensin system with ACE inhibitors or ARBs may be incomplete because the drugs disrupt the negative feedback action of ANG II on renin release and thereby increase plasma renin activity. Other antihypertensive drugs, notably hydrochlorothiazide and other diuretics, also increase plasma renin activity. Aliskiren not only decreases baseline plasma renin activity in hypertensive subjects but also eliminates the rise produced by ACE inhibitors, ARBs, and diuretics, and thereby results in a greater antihypertensive effect. For this reason, aliskiren has been used in combination with an ACE inhibitor or ARB. However, such dual blockade may not produce significant clinical benefit and may even be associated with adverse effects including hypokalemia, although this point is controversial.
Prorenin Receptors
For many years, prorenin was considered to be an inactive precursor of renin, with no function of its own. Thus the observation noted above in the section on renin that prorenin circulates at high levels was surprising. Recently, however, a receptor that preferentially binds prorenin has been identified. Since it also binds active renin, the receptor is referred to as the (pro)renin receptor.
The receptor is a 350-amino acid protein with a single transmembrane domain. When prorenin binds to the (pro)renin receptor, the prorenin undergoes a conformational change and becomes enzymatically active without cleavage of the prosegment. The catalytic activity of active renin also increases further when it binds to the receptor. The activated prorenin and renin interact with circulating angiotensinogen to form angiotensin (Figure 17–3). However, binding of prorenin to the receptor also activates intracellular signaling pathways that differ depending on the cell type. For example, in mesangial and vascular smooth muscle cells, prorenin binding activates MAP kinases and expression of profibrotic molecules. Thus, elevated prorenin levels (as occur, for example, in diabetes mellitus) could produce a variety of adverse effects via both angiotensin-dependent and independent pathways. Recent research indicates that the (pro)renin receptor is functionally linked to the vacuolar proton-ATPase (ATP6ap2) and is necessary for Wnt signaling pathways involved (independently of renin) in stem cell biology, embryology, and cancer.
The renin activity of the receptor-bound forms of prorenin and renin can be inhibited by aliskiren. A synthetic peptide named handle region peptide (HRP), which consists of the amino acid sequence corresponding to the “handle” region of the prorenin prosegment, has been synthesized and shown to competitively inhibit binding of prorenin to the (pro)renin receptor. HRP has beneficial effects in the kidneys of diabetic rats and so there is considerable interest in developing noncompetitive antagonists of the (pro)renin receptor.
This novel receptor could be important in cardiovascular and other diseases, but at the present time its role in human pathology is far from clear.
 KININS
KININS
BIOSYNTHESIS OF KININS
Kinins are potent vasodilator peptides formed enzymatically by the action of enzymes known as kallikreins acting on protein substrates called kininogens. The kallikrein-kinin system has several features in common with the renin-angiotensin system.
Kallikreins
Kallikreins are serine proteases present in plasma (plasma kallikrein) and in several organs (tissue kallikrein), including the kidneys, pancreas, intestine, sweat glands, and salivary glands. The two groups are secreted as zymogens and are activated by proteolytic cleavage. Plasma prekallikrein is activated by activated blood coagulation factor XII (FXIIa). The two groups differ in their gene structure, molecular weight, substrate specificity, and kinin produced. Kallikreins can convert prorenin to active renin, but the physiologic significance of this action is not known.
Kininogens
Kininogens—the substrates for kallikreins and precursors of kinins—are present in plasma, lymph, and interstitial fluid. Two kininogens are present in plasma: a low-molecular-weight form (LMW kininogen) and a high-molecular-weight form (HMW kininogen). The two forms result from differential splicing of the kininogen gene to generate proteins that differ at the C-terminus. About 15–20% of the total plasma kininogen is in the HMW form. It is thought that LMW kininogen crosses capillary walls and serves as the substrate for tissue kallikreins, whereas HMW kininogen is confined to the bloodstream and serves as the substrate for plasma kallikrein.
FORMATION & METABOLISM OF KININS
The pathway for the formation and metabolism of kinins is shown in Figure 17–4. The two major kinins in humans are bradykinin and Lys-bradykinin or kallidin. Bradykinin is released from HMW kininogen by plasma kallikrein, whereas kallidin is released from LMW kininogen by tissue kallikrein. Kallidin can be converted to bradykinin by an arginine aminopeptidase. The two kinins are present in plasma and urine. Bradykinin is the predominant kinin in plasma, whereas Lys-bradykinin is the major urinary form.
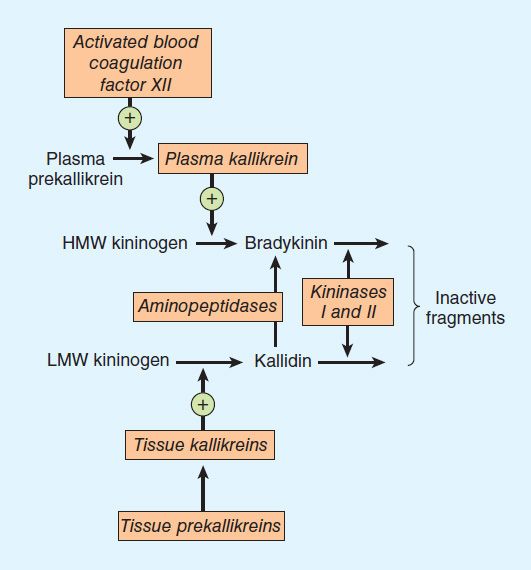
FIGURE 17–4 The kallikrein-kinin system. Kininase II is identical to converting enzyme peptidyl dipeptidase (ACE).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


