27
Skeletal Muscle Relaxants
CASE STUDY
A 30-year-old woman is rushed to the emergency department at a major trauma center after a motor vehicle crash. Although in significant pain, she is awake, alert, and able to give a brief history. She states that she was the driver and was wearing a seatbelt. There were no passengers in the car. Her past medical history is significant only for asthma, for which she has been intubated once in the past. She has no allergies to medications. There are multiple lacerations on her face and extremities and a large open fracture of her right femur. An orthopedic surgeon has scheduled immediate operative repair of the femur fracture, and the plastic surgeon wants to suture the facial lacerations at the same time. You decide to intubate the patient for the procedure. What muscle relaxant would you choose? Would you choose the same agent if she had experienced a 30% total body burn in a fire at the time of the accident? What if the past medical history included right-sided hemiparesis of 10 years’ duration?
Drugs that affect skeletal muscle function include two different therapeutic groups: those used during surgical procedures and in the intensive care unit (ICU) to produce muscle paralysis (ie, neuromuscular blockers), and those used to reduce spasticity in a variety of painful conditions (ie, spasmolytics). Neuromuscular blocking drugs interfere with transmission at the neuromuscular end plate and lack central nervous system (CNS) activity. These compounds are used primarily as adjuncts during general anesthesia to optimize surgical conditions and to facilitate endotracheal intubation in order to ensure adequate ventilation. Drugs in the spasmolytic group have traditionally been called “centrally acting” muscle relaxants and are used primarily to treat chronic back pain and painful fibromyalgic conditions. Dantrolene, a spasmolytic agent that has no significant central effects and is used primarily to treat a rare anesthetic-related complication, malignant hyperthermia, is also discussed in this chapter.
NEUROMUSCULAR BLOCKING DRUGS
History
During the 16th century, European explorers found that natives in the Amazon Basin of South America were using curare, an arrow poison that produced skeletal muscle paralysis, to kill animals. The active compound, d-tubocurarine, and its modern synthetic analogs have had a major influence on the practice of anesthesia and surgery and have proved useful in understanding the basic mechanisms involved in neuromuscular transmission.
Normal Neuromuscular Function
The mechanism of neuromuscular transmission at the motor end plate is similar to that described for preganglionic cholinergic nerves in Chapter 6. The arrival of an action potential at the motor nerve terminal causes an influx of calcium and release of the neurotransmitter acetylcholine. Acetylcholine then diffuses across the synaptic cleft to activate nicotinic receptors located on the motor end plate, present at a density of 10,000/μm*. As noted in Chapter 7, the adult NM receptor is composed of five peptides: two alpha peptides, one beta, one gamma, and one delta peptide (Figure 27–1). The binding of two acetylcholine molecules to receptors on the α-β and δ-α subunits causes opening of the channel. The subsequent movement of sodium and potassium through the channel is associated with a graded depolarization of the end plate membrane (see Figure 7–4, panel B). This change in voltage is termed the motor end plate potential. The magnitude of the end plate potential is directly related to the amount of acetylcholine released. If the potential is small, the permeability and the end plate potential return to normal without an impulse being propagated from the end plate region to the rest of the muscle membrane. However, if the end plate potential is large, the adjacent muscle membrane is depolarized, and an action potential will be propagated along the entire muscle fiber. Muscle contraction is then initiated by excitation-contraction coupling. The released acetylcholine is quickly removed from the end plate region by both diffusion and enzymatic destruction by the local acetylcholinesterase enzyme.
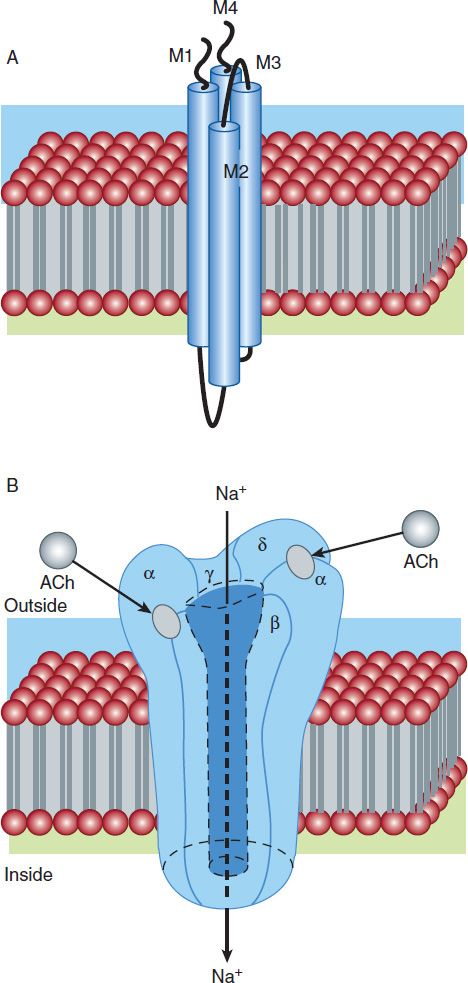
FIGURE 27–1 The adult nicotinic acetylcholine receptor (nAChR) is an intrinsic membrane protein with five distinct subunits (α2 β δ γ). A: Cartoon of the one of five subunits of the AChR in the end plate surface of adult mammalian muscle. Each subunit contains four helical domains labeled M1 to M4. The M2 domains line the channel pore. B: Cartoon of the full nAChR. The N termini of two subunits cooperate to form two distinct binding pockets for acetylcholine (ACh). These pockets occur at the α-β and the δ-α subunit interfaces. Binding of one molecule of ACh enhances the receptor’s affinity for the second molecule, followed by multiple intermediate steps leading to channel opening. These steps are the subject of intense investigation.
At least two additional types of acetylcholine receptors are found within the neuromuscular apparatus. One type is located on the presynaptic motor axon terminal, and activation of these receptors mobilizes additional transmitter for subsequent release by moving more acetylcholine vesicles toward the synaptic membrane. The second type of receptor is found on extrajunctional cells and is not normally involved in neuromuscular transmission. However, under certain conditions (eg, prolonged immobilization, thermal burns), these receptors may proliferate sufficiently to affect subsequent neuromuscular transmission. This proliferation of extrajunctional acetylcholine receptors may be clinically relevant when using depolarizing or nondepolarizing skeletal muscle relaxant drugs and is described later.
Skeletal muscle relaxation and paralysis can occur from interruption of function at several sites along the pathway from the CNS to myelinated somatic nerves, unmyelinated motor nerve terminals, nicotinic acetylcholine receptors, the motor end plate, the muscle membrane, and the intracellular muscular contractile apparatus itself.
Blockade of end plate function can be accomplished by two basic mechanisms. First, pharmacologic blockade of the physiologic agonist acetylcholine is characteristic of the antagonist neuromuscular blocking drugs (ie, nondepolarizing neuromuscular blocking drugs). These drugs prevent access of the transmitter to its receptor and thereby prevent depolarization. The prototype of this nondepolarizing subgroup is d-tubocurarine. The second mechanism of blockade can be produced by an excess of a depolarizing agonist, such as acetylcholine. This seemingly paradoxical effect of acetylcholine also occurs at the ganglionic nicotinic acetylcholine receptor. The prototypical depolarizing blocking drug is succinylcholine. A similar depolarizing block can be produced by acetylcholine itself when high local concentrations are achieved in the synaptic cleft (eg, by cholinesterase inhibitor intoxication) and by nicotine and other nicotinic agonists. However, the neuromuscular block produced by depolarizing drugs other than succinylcholine cannot be precisely controlled and is of no clinical value.
 BASIC PHARMACOLOGY OF NEUROMUSCULAR BLOCKING DRUGS
BASIC PHARMACOLOGY OF NEUROMUSCULAR BLOCKING DRUGS
Chemistry
All of the available neuromuscular blocking drugs bear a structural resemblance to acetylcholine. For example, succinylcholine is two acetylcholine molecules linked end-to-end (Figure 27–2). In contrast to the single linear structure of succinylcholine and other depolarizing drugs, the nondepolarizing agents (eg, pancuronium) conceal the “double-acetylcholine” structure in one of two types of bulky, semirigid ring systems (Figure 27–2). Examples of the two major families of nondepolarizing blocking drugs—the isoquinoline and steroid derivatives—are shown in Figures 27–3 and 27–4. Another feature common to all currently used neuromuscular blockers is the presence of one or two quaternary nitrogens, which makes them poorly lipid soluble and limits entry into the CNS.
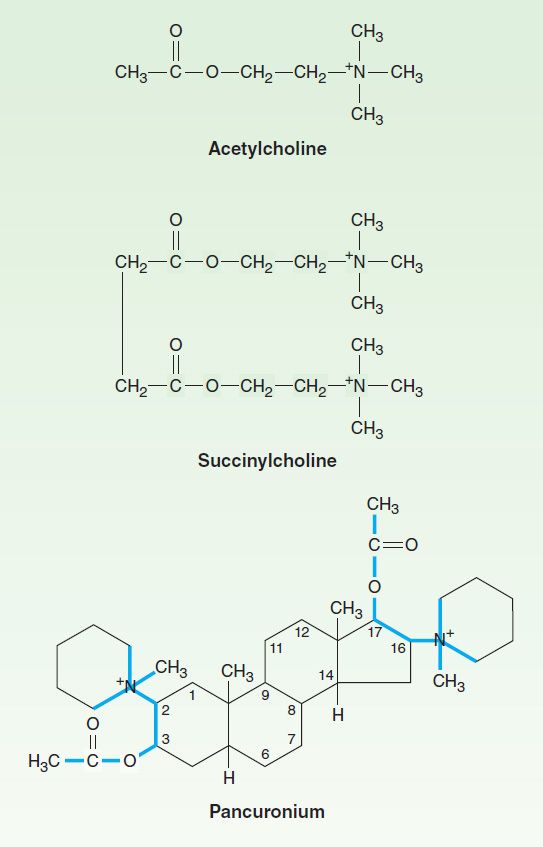
FIGURE 27–2 Structural relationship of succinylcholine, a depolarizing agent, and pancuronium, a nondepolarizing agent, to acetylcholine, the neuromuscular transmitter. Succinylcholine, originally called diacetylcholine, is simply two molecules of acetylcholine linked through the acetate methyl groups. Pancuronium may be viewed as two acetylcholine-like fragments (outlined in color) oriented on a steroid nucleus.
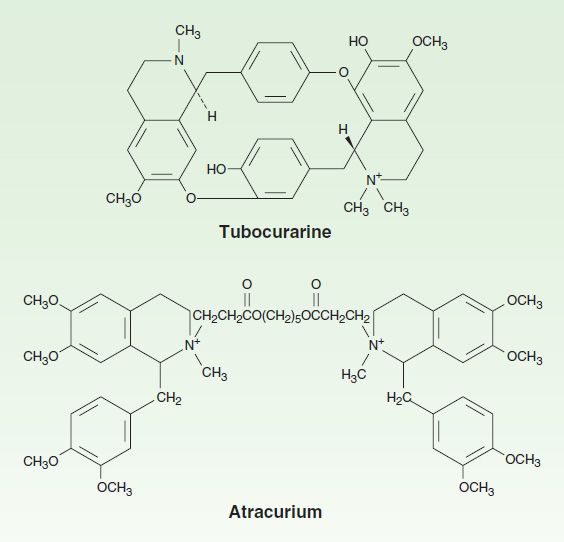
FIGURE 27–3 Structures of two isoquinoline neuromuscular blocking drugs. These agents are nondepolarizing muscle relaxants.
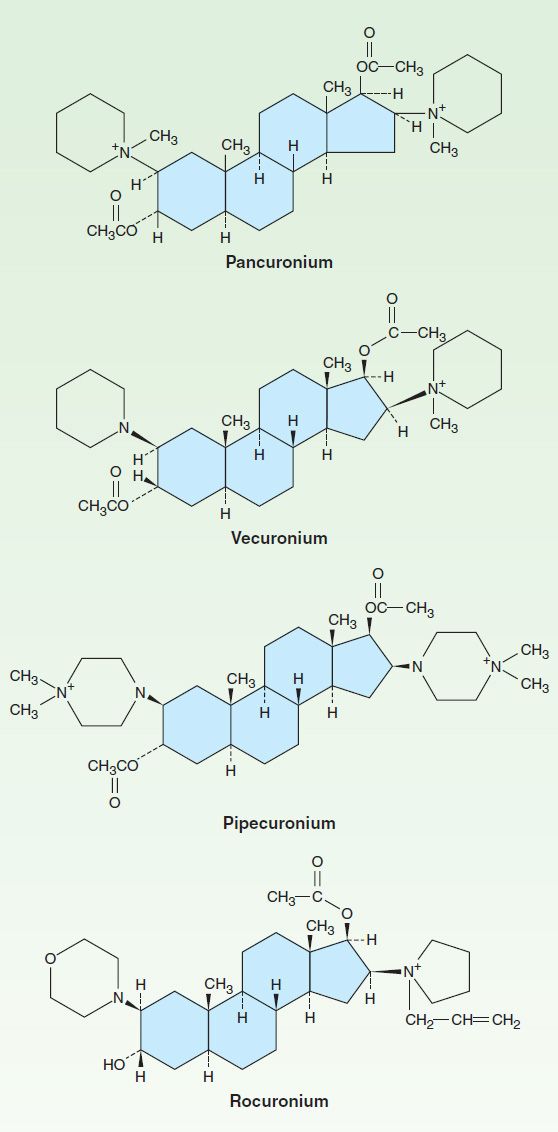
FIGURE 27–4 Structures of steroid neuromuscular blocking drugs (steroid nucleus in color). These agents are all nondepolarizing muscle relaxants.
Pharmacokinetics of Neuromuscular Blocking Drugs
All of the neuromuscular blocking drugs are highly polar compounds and inactive orally; they must be administered parenterally.
A. Nondepolarizing Relaxant Drugs
The rate of disappearance of a nondepolarizing neuromuscular blocking drug from the blood is characterized by a rapid initial distribution phase followed by a slower elimination phase. Neuromuscular blocking drugs are highly ionized, do not readily cross cell membranes, and are not strongly bound in peripheral tissues. Therefore, their volume of distribution (80–140 mL/kg) is only slightly larger than the blood volume.
The duration of neuromuscular blockade produced by nondepolarizing relaxants is strongly correlated with the elimination half-life. Drugs that are excreted by the kidney typically have longer half-lives, leading to longer durations of action (> 35 minutes). Drugs eliminated by the liver tend to have shorter half-lives and durations of action (Table 27–1). All steroidal muscle relaxants are metabolized to their 3-hydroxy, 17-hydroxy, or 3,17-dihydroxy products in the liver. The 3-hydroxy metabolites are usually 40–80% as potent as the parent drug. Under normal circumstances, metabolites are not formed in sufficient quantities to produce a significant degree of neuromuscular blockade during or after anesthesia. However, if the parent compound is administered for several days in the ICU setting, the 3-hydroxy metabolite may accumulate and cause prolonged paralysis because it has a longer half-life than the parent compound. The remaining metabolites possess minimal neuromuscular blocking properties.
TABLE 27–1 Pharmacokinetic and dynamic properties of neuromuscular blocking drugs.
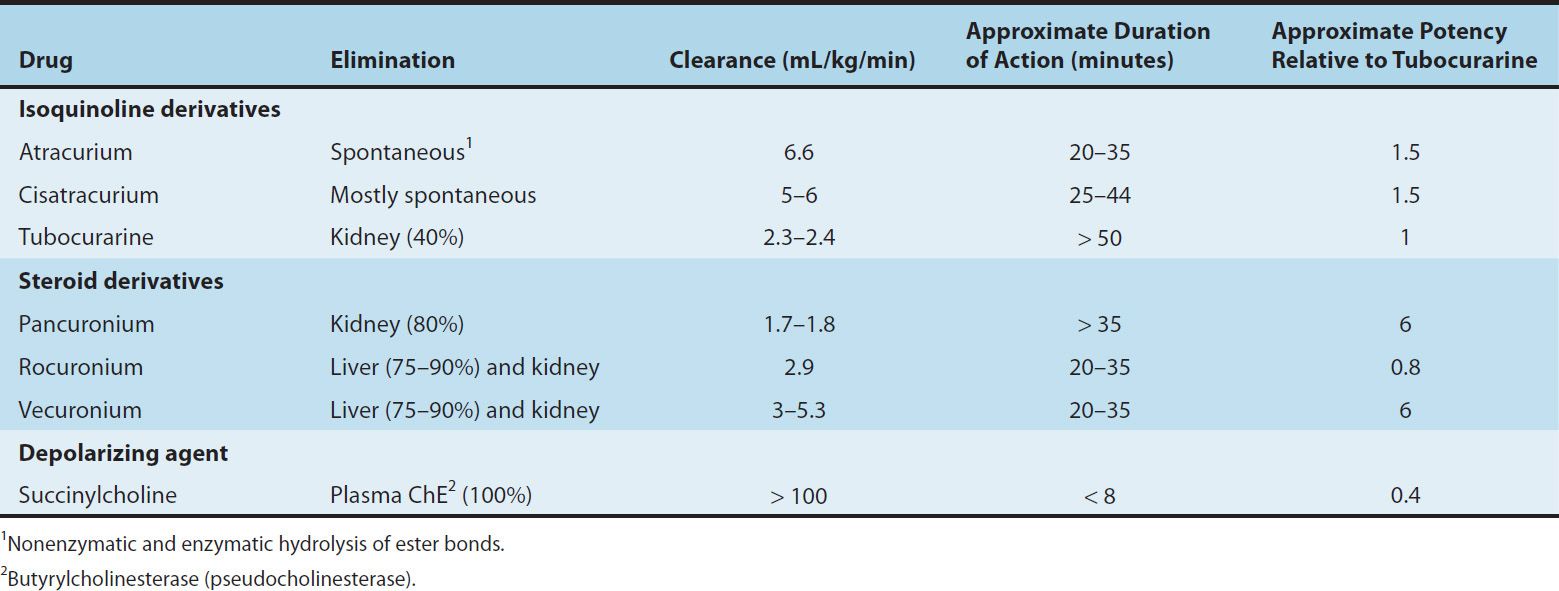
The intermediate-acting steroid muscle relaxants (eg, vecuronium and rocuronium) tend to be more dependent on biliary excretion or hepatic metabolism for their elimination. These muscle relaxants are more commonly used clinically than the long-acting steroid-based drugs (eg, pancuronium). The duration of action of these relaxants may be prolonged significantly in patients with impaired liver function.
Atracurium (Figure 27–3) is an intermediate-acting isoquinoline nondepolarizing muscle relaxant that is no longer in widespread clinical use. In addition to hepatic metabolism, atracurium is inactivated by a form of spontaneous breakdown known as Hofmann elimination. The main breakdown products are laudanosine and a related quaternary acid, neither of which possesses neuromuscular blocking properties. Laudanosine is slowly metabolized by the liver and has a longer elimination half-life (ie, 150 minutes). It readily crosses the blood-brain barrier, and high blood concentrations may cause seizures and an increase in the volatile anesthetic requirement. During surgical anesthesia, blood levels of laudanosine typically range from 0.2 to 1 mcg/mL; however, with prolonged infusions of atracurium in the ICU, laudanosine blood levels may exceed 5 mcg/mL.
Atracurium has several stereoisomers, and the potent isomer cisatracurium has become one of the most common muscle relaxants in use today. Although cisatracurium resembles atracurium, it has less dependence on hepatic inactivation, produces less laudanosine, and is much less likely to release histamine. From a clinical perspective, cisatracurium has all the advantages of atracurium with fewer adverse effects. Therefore, cisatracurium has virtually replaced atracurium in clinical practice.
Gantacurium represents a new class of nondepolarizing neuromuscular blockers, called asymmetric mixed-onium chlorofumarates. It is degraded nonenzymatically by adduction of the amino acid cysteine and ester bond hydrolysis. Gantacurium is currently in phase 3 clinical trials and not yet available for widespread clinical use. Preclinical and clinical data indicate gantacurium has a rapid onset of effect and predictable duration of action (very short, similar to succinylcholine) that can be reversed with neostigmine or more quickly (within 1–2 minutes), with administration of L-cysteine. At doses above three times the ED95, cardiovascular adverse effects (eg, hypotension) have occurred, probably due to histamine release. No bronchospasm or pulmonary vasoconstriction has been reported at these higher doses.
B. Depolarizing Relaxant Drugs
The extremely short duration of action of succinylcholine (5–10 minutes) is due to its rapid hydrolysis by butyrylcholinesterase and pseudocholinesterase in the liver and plasma, respectively. Plasma cholinesterase metabolism is the predominant pathway for succinylcholine elimination. The primary metabolite of succinylcholine, succinylmonocholine, is rapidly broken down to succinic acid and choline. Because plasma cholinesterase has an enormous capacity to hydrolyze succinylcholine, only a small percentage of the original intravenous dose ever reaches the neuromuscular junction. In addition, because there is little if any plasma cholinesterase at the motor end plate, a succinylcholine-induced blockade is terminated by its diffusion away from the end plate into extracellular fluid. Therefore, the circulating levels of plasma cholinesterase influence the duration of action of succinylcholine by determining the amount of the drug that reaches the motor end plate.
Neuromuscular blockade produced by succinylcholine can be prolonged in patients with an abnormal genetic variant of plasma cholinesterase. The dibucaine number is a measure of the ability of a patient to metabolize succinylcholine and can be used to identify at-risk patients. Under standardized test conditions, dibucaine inhibits the normal enzyme by 80% and the abnormal enzyme by only 20%. Many genetic variants of plasma cholinesterase have been identified, although the dibucaine-related variants are the most important. Given the rarity of these genetic variants, plasma cholinesterase testing is not a routine clinical procedure but may be indicated for patients with a family history of plasma cholinesterase deficiency. Another reasonable strategy is to avoid the use of succinylcholine where practical in patients with a possible family history of plasma cholinesterase deficiency.
Mechanism of Action
The interactions of drugs with the acetylcholine receptor-end plate channel have been described at the molecular level. Several modes of action of drugs on the receptor are illustrated in Figure 27–5.
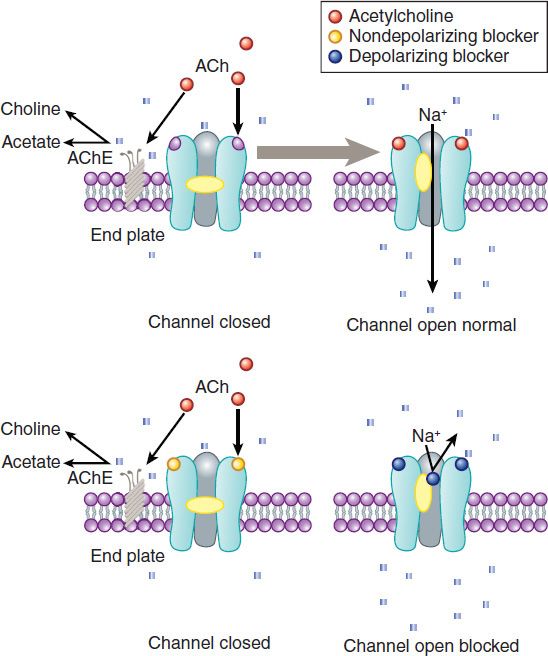
FIGURE 27–5 Schematic diagram of the interactions of drugs with the acetylcholine receptor on the end plate channel (structures are purely symbolic). Top: The action of the normal agonist, acetylcholine (red) in opening the channel. Bottom, left: A nondepolarizing blocker, eg, rocuronium (yellow), is shown as preventing the opening of the channel when it binds to the receptor. Bottom, right: A depolarizing blocker, eg, succinylcholine (blue), both occupying the receptor and blocking the channel. Normal closure of the channel gate is prevented and the blocker may move rapidly in and out of the pore. Depolarizing blockers may desensitize the end plate by occupying the receptor and causing persistent depolarization. An additional effect of drugs on the end plate channel may occur through changes in the lipid environment surrounding the channel (not shown). General anesthetics and alcohols may impair neuromuscular transmission by this mechanism.
A. Nondepolarizing Relaxant Drugs
All the neuromuscular blocking drugs in current use in the USA except succinylcholine are classified as nondepolarizing agents. Although it is no longer in widespread clinical use, d-tubocurarine is considered the prototype neuromuscular blocker. When small doses of nondepolarizing muscle relaxants are administered, they act predominantly at the nicotinic receptor site by competing with acetylcholine. The least potent nondepolarizing relaxants (eg, rocuronium) have the fastest onset and the shortest duration of action. In larger doses, nondepolarizing drugs can enter the pore of the ion channel (Figure 27–1) to produce a more intense motor blockade. This action further weakens neuromuscular transmission and diminishes the ability of the acetylcholinesterase inhibitors (eg, neostigmine, edrophonium, pyridostigmine) to antagonize the effect of nondepolarizing muscle relaxants.
Nondepolarizing relaxants can also block prejunctional sodium channels. As a result of this action, muscle relaxants interfere with the mobilization of acetylcholine at the nerve ending and cause fade of evoked nerve twitch contractions (Figure 27–6, and described below). One consequence of the surmountable nature of the postsynaptic blockade produced by nondepolarizing muscle relaxants is the fact that tetanic stimulation (rapid delivery of electrical stimuli to a peripheral nerve) releases a large quantity of acetylcholine and is followed by transient posttetanic facilitation of the twitch strength (ie, relief of blockade). An important clinical consequence of this principle is the reversal of residual blockade by cholinesterase inhibitors. The characteristics of a nondepolarizing neuromuscular blockade are summarized in Table 27–2 and Figure 27–6.
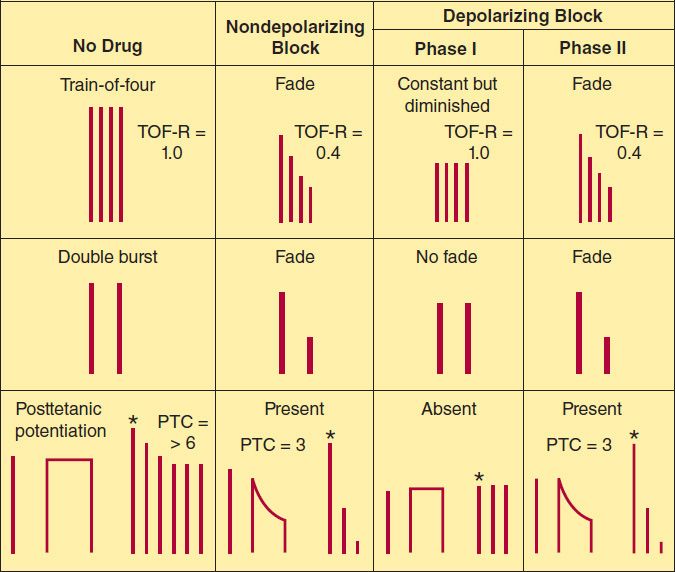
FIGURE 27–6 Muscle contraction responses to different patterns of nerve stimulation used in monitoring skeletal muscle relaxation. The alterations produced by a nondepolarizing blocker and depolarizing and desensitizing blockade by succinylcholine are shown. In the train-of-four (TOF) pattern, four stimuli are applied at 2 Hz. The TOF ratio (TOF-R) is calculated from the strength of the fourth contraction divided by that of the first. In the double-burst pattern, three stimuli are applied at 50 Hz, followed by a 700 ms rest period and then repeated. In the posttetanic potentiation pattern, several seconds of 50 Hz stimulation are applied, followed by several seconds of rest and then by single stimuli at a slow rate (eg, 0.5 Hz). The number of detectable posttetanic twitches is the posttetanic count (PTC).*, first posttetanic contraction.
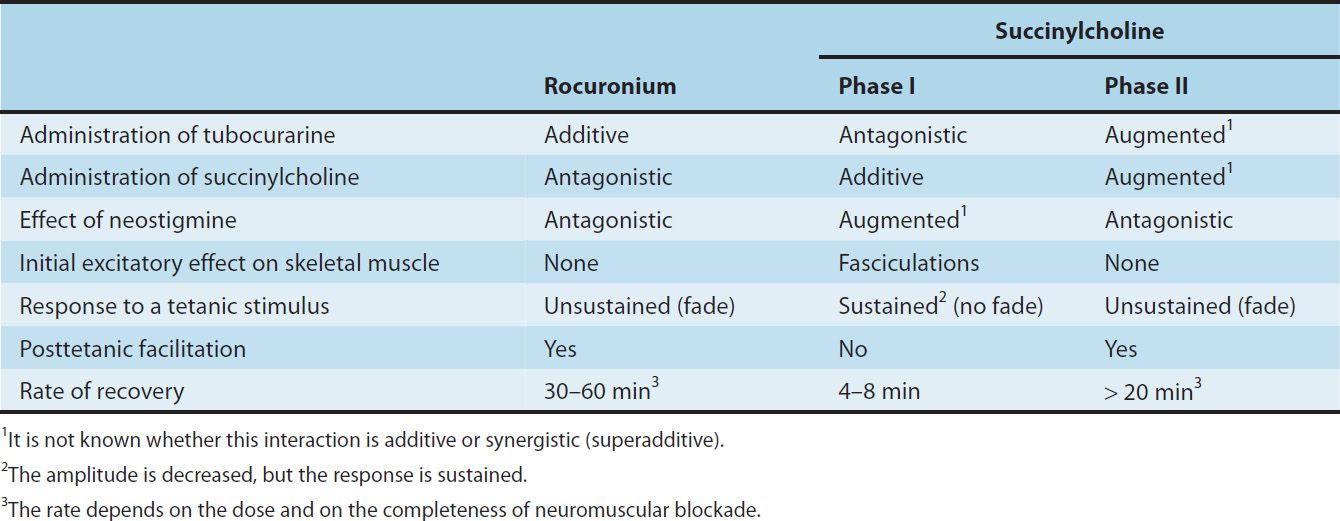
TABLE 27–2 Comparison of a typical nondepolarizing muscle relaxant (rocuronium) and a depolarizing muscle relaxant (succinylcholine).
B. Depolarizing Relaxant Drugs
1. Phase I block (depolarizing)—Succinylcholine is the only clinically useful depolarizing blocking drug. Its neuromuscular effects are like those of acetylcholine except that succinylcholine produces a longer effect at the myoneural junction. Succinylcholine reacts with the nicotinic receptor to open the channel and cause depolarization of the motor end plate, and this in turn spreads to the adjacent membranes, causing contractions of muscle motor units. Data from single-channel recordings indicate that depolarizing blockers can enter the channel to produce a prolonged “flickering” of the ion conductance (Figure 27–7). Because succinylcholine is not metabolized effectively at the synapse, the depolarized membranes remain depolarized and unresponsive to subsequent impulses (ie, a state of depolarizing blockade). Furthermore, because excitation-contraction coupling requires end plate repolarization (“repriming”) and repetitive firing to maintain muscle tension, a flaccid paralysis results. In contrast to the nondepolarizing drugs, this so-called phase I (depolarizing) block is augmented, not reversed, by cholinesterase inhibitors.
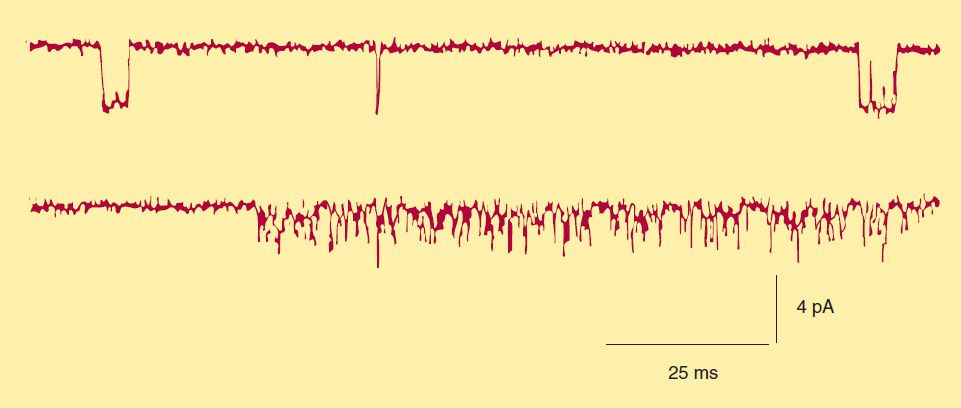
FIGURE 27–7 Action of succinylcholine on single-channel end plate receptor currents in frog muscle. Currents through a single AChR channel were recorded using the patch clamp technique. The upper trace was recorded in the presence of a low concentration of succinylcholine; the downward deflections represent openings of the channel and passage of inward (depolarizing) current. The lower trace was recorded in the presence of a much higher concentration of succinylcholine and shows prolonged “flickering” of the channel as it repetitively opens and closes or is “plugged” by the drug. (Reproduced, with permission, from Marshall CG, Ogden DC, Colquhoun D: The actions of suxamethonium (succinyldicholine) as an agonist and channel blocker at the nicotinic receptor of frog muscle. J Physiol [Lond] 1990;428:155.)
The characteristics of a depolarizing neuromuscular blockade are summarized in Table 27–2 and Figure 27–6.
2. Phase II block (desensitizing)—With prolonged exposure to succinylcholine, the initial end plate depolarization decreases and the membrane becomes repolarized. Despite this repolarization, the membrane cannot easily be depolarized again because it is desensitized
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


