36
Nonsteroidal Anti-Inflammatory Drugs, Disease-Modifying Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout
CASE STUDY
A 48-year-old man presents with complaints of bilateral morning stiffness in his wrists and knees and pain in these joints on exercise. On physical examination, the joints are slightly swollen. The rest of the examination is unremarkable. His laboratory findings are also negative except for slight anemia, elevated erythrocyte sedimentation rate, and positive rheumatoid factor. With the diagnosis of rheumatoid arthritis, he is started on a regimen of naproxen, 220 mg twice daily. After 1 week, the dosage is increased to 440 mg twice daily. His symptoms are reduced at this dosage, but he complains of significant heartburn that is not controlled by antacids. He is then switched to celecoxib, 200 mg twice daily, and on this regimen his joint symptoms and heartburn resolve. Two years later, he returns with increased joint symptoms. His hands, wrists, elbows, feet, and knees are all now involved and appear swollen, warm, and tender. What therapeutic options should be considered at this time? What are the possible complications?
ACRONYMS

THE IMMUNE RESPONSE
The immune response occurs when immunologically competent cells are activated in response to foreign organisms or antigenic substances liberated during the acute or chronic inflammatory response. The outcome of the immune response for the host may be deleterious if it leads to chronic inflammation without resolution of the underlying injurious process (see Chapter 55). Chronic inflammation involves the release of multiple cytokines and chemokines plus a very complex interplay of immunoactive cells. The whole range of autoimmune diseases (eg, RA, vasculitis, SLE) and inflammatory conditions (eg, gout) derive from abnormalities in this cascade.
The cell damage associated with inflammation acts on cell membranes to release leukocyte lysosomal enzymes; arachidonic acid is then liberated from precursor compounds, and various eicosanoids are synthesized (see Chapter 18). The lipoxygenase pathway of arachidonate metabolism yields leukotrienes, which have a powerful chemotactic effect on eosinophils, neutrophils, and macrophages and promote bronchoconstriction and alterations in vascular permeability. During inflammation, stimulation of the neutrophil membranes produces oxygen-derived free radicals and other reactive molecules such as hydrogen peroxide and hydroxyl radicals. The interaction of these substances with arachidonic acid results in the generation of chemotactic substances, thus perpetuating the inflammatory process.
THERAPEUTIC STRATEGIES
The treatment of patients with inflammation involves two primary goals: first, the relief of symptoms and the maintenance of function, which are usually the major continuing complaints of the patient; and second, the slowing or arrest of the tissue-damaging process. In RA, several validated combined indices are used to define response (eg, Disease Activity Index [DAS], American College of Rheumatology Response Index [ACR Response]). These indices often combine joint tenderness and swelling, patient response, and laboratory data. Reduction of inflammation with NSAIDs often results in relief of pain for significant periods. Furthermore, most of the nonopioid analgesics (aspirin, etc) have anti-inflammatory effects, so they are appropriate for the treatment of both acute and chronic inflammatory conditions.
The glucocorticoids also have powerful anti-inflammatory effects and when first introduced were considered to be the ultimate answer to the treatment of inflammatory arthritis. Although there are data indicating that low-dose corticosteroids have disease-modifying properties, their toxicity makes them less favored than other medications, when it is possible to use the others. However, the glucocorticoids continue to have a significant role in the long-term treatment of arthritis.
Another important group of agents is characterized as DMARDs including biologics (a subset of the DMARDs). They decrease inflammation, improve symptoms, and slow the bone damage associated with RA. They affect more basic inflammatory mechanisms than do glucocorticoids or the NSAIDs. They may also be more toxic than those alternative medications.
 NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
Salicylates and other similar agents used to treat rheumatic disease share the capacity to suppress the signs and symptoms of inflammation including pain. These drugs also exert antipyretic effects.
Since aspirin, the original NSAID, has a number of adverse effects, many other NSAIDs have been developed in attempts to improve upon aspirin’s efficacy and decrease its toxicity.
Chemistry & Pharmacokinetics
The NSAIDs are grouped in several chemical classes, as shown in Figure 36–1. This chemical diversity yields a broad range of pharmacokinetic characteristics (Table 36–1). Although there are many differences in the kinetics of NSAIDs, they have some general properties in common. All but one of the NSAIDs are weak organic acids as given; the exception, nabumetone, is a ketone prodrug that is metabolized to the acidic active drug.
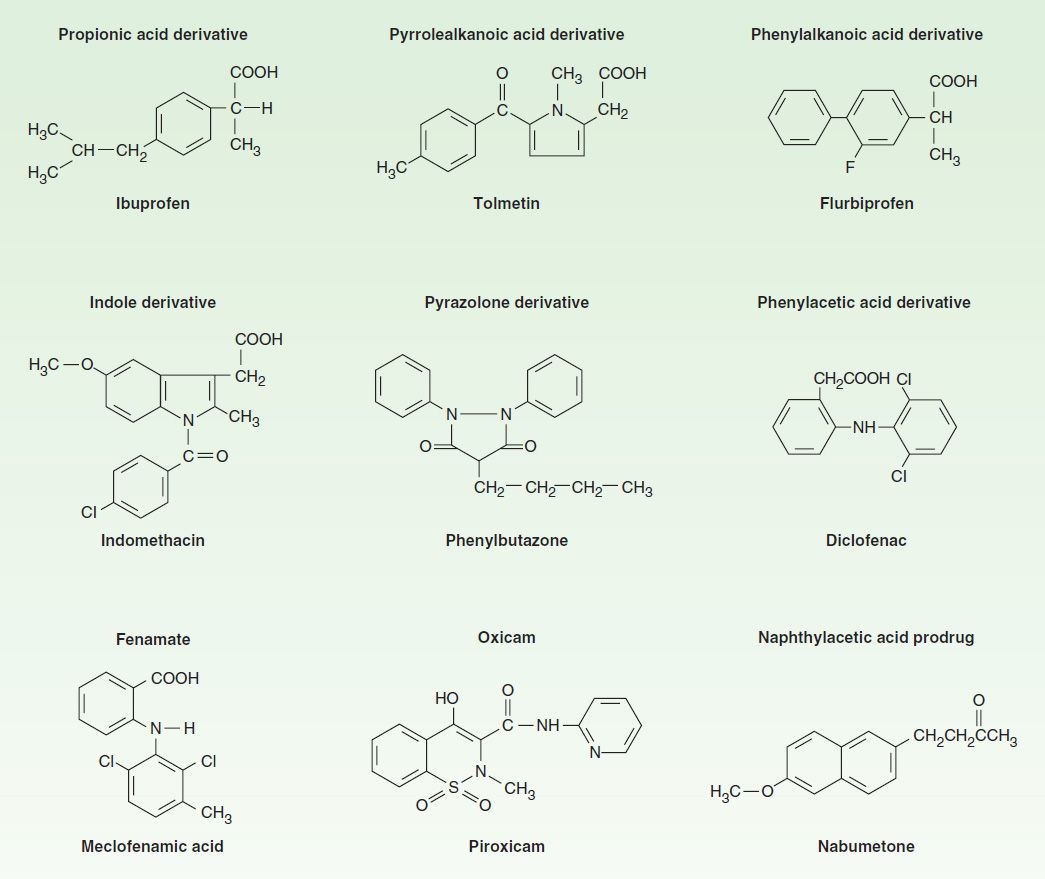
FIGURE 36–1 Chemical structures of some NSAIDs.
TABLE 36–1 PROPERTIES OF ASPIRIN AND SOME OTHER NONSTEROIDAL ANTI-INFLAMMATORY DRUGS.
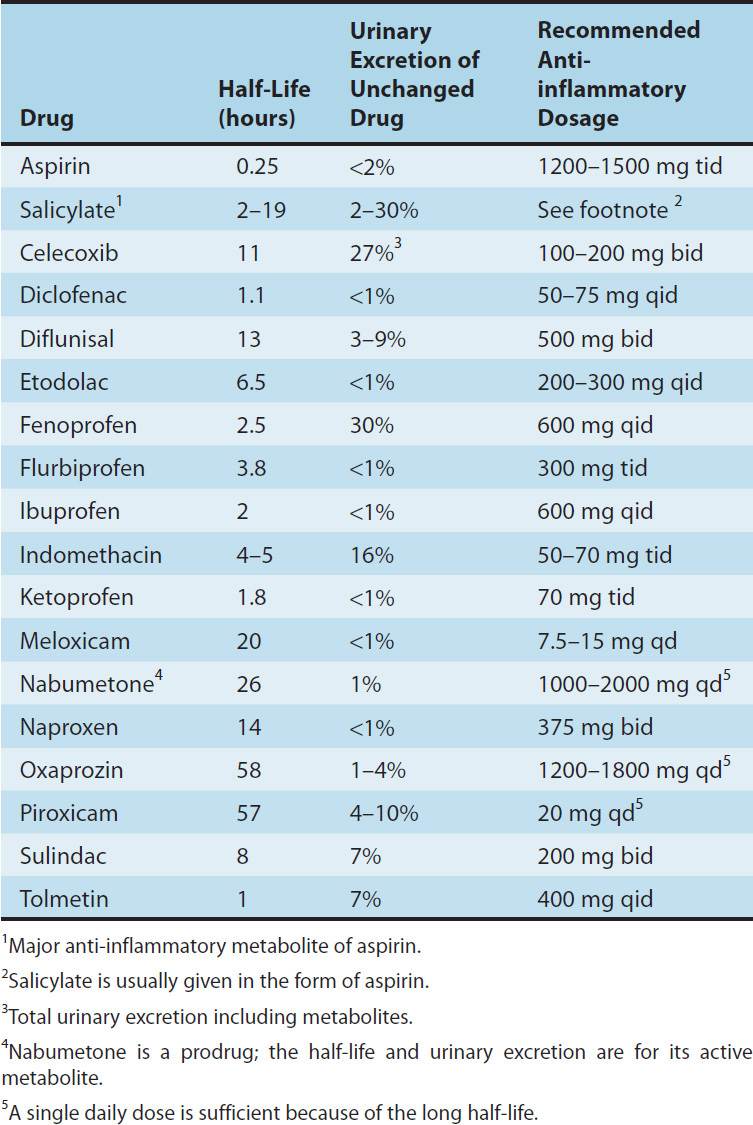
Most of these drugs are well absorbed, and food does not substantially change their bioavailability. Most of the NSAIDs are highly metabolized, some by phase I followed by phase II mechanisms and others by direct glucuronidation (phase II) alone. NSAID metabolism proceeds, in large part, by way of the CYP3A or CYP2C families of P450 enzymes in the liver (see Chapter 4). While renal excretion is the most important route for final elimination, nearly all undergo varying degrees of biliary excretion and reabsorption (enterohepatic circulation). In fact, the degree of lower gastrointestinal (GI) tract irritation correlates with the amount of enterohepatic circulation. Most of the NSAIDs are highly protein-bound (~ 98%), usually to albumin. Most of the NSAIDs (eg, ibuprofen, ketoprofen) are racemic mixtures, while one, naproxen, is provided as a single enantiomer and a few have no chiral center (eg, diclofenac).
All NSAIDs can be found in synovial fluid after repeated dosing. Drugs with short half-lives remain in the joints longer than would be predicted from their half-lives, while drugs with longer half-lives disappear from the synovial fluid at a rate proportionate to their half-lives.
Pharmacodynamics
NSAID anti-inflammatory activity is mediated chiefly through inhibition of prostaglandin biosynthesis (Figure 36–2). Various NSAIDs have additional possible mechanisms of action, including inhibition of chemotaxis, down-regulation of IL-1 production, decreased production of free radicals and superoxide, and interference with calcium-mediated intracellular events. Aspirin irreversibly acetylates and blocks platelet COX, while the non-COX-selective NSAIDs are reversible inhibitors.
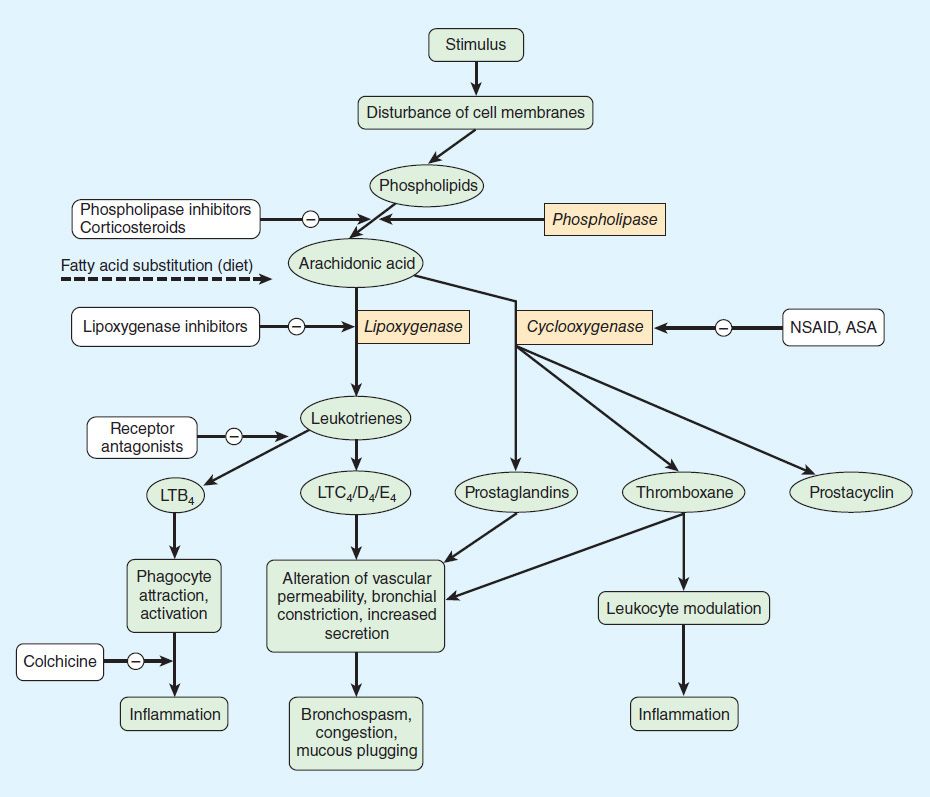
FIGURE 36–2 Prostanoid mediators derived from arachidonic acid and sites of drug action. ASA, acetylsalicylic acid (aspirin); LT, leukotriene; NSAID, nonsteroidal anti-inflammatory drug.
Selectivity for COX-1 versus COX-2 is variable and incomplete for the older NSAIDs, but selective COX-2 inhibitors have been synthesized. The selective COX-2 inhibitors do not affect platelet function at their usual doses. The efficacy of COX-2-selective drugs equals that of the older NSAIDs, while GI safety may be improved. On the other hand, selective COX-2 inhibitors increase the incidence of edema, hypertension, and possibly, myocardial infarction. As of August 2011, celecoxib and the less selective meloxicam were the only COX-2 inhibitors marketed in the USA. Celecoxib has an FDA-initiated “black box” warning concerning cardiovascular risks. It has been recommended that all NSAID product labels be revised to mention cardiovascular risks.
The NSAIDs decrease the sensitivity of vessels to bradykinin and histamine, affect lymphokine production from T lymphocytes, and reverse the vasodilation of inflammation. To varying degrees, all newer NSAIDs are analgesic, anti-inflammatory, and antipyretic, and all (except the COX-2-selective agents and the nonacetylated salicylates) inhibit platelet aggregation. NSAIDs are all gastric irritants and can be associated with GI ulcers and bleeds as well, although as a group the newer agents tend to cause less GI irritation than aspirin. Nephrotoxicity, reported for all NSAIDs, is due, in part, to interference with the autoregulation of renal blood flow, which is modulated by prostaglandins. Hepatotoxicity can also occur with any NSAID.
Although these drugs effectively inhibit inflammation, there is no evidence that—in contrast to drugs such as methotrexate, biologics, and other DMARDs—they alter the course of any arthritic disorder.
Several NSAIDs (including aspirin) reduce the incidence of colon cancer when taken chronically. Several large epidemiologic studies have shown a 50% reduction in relative risk for this neoplasm when the drugs are taken for 5 years or longer. The mechanism for this protective effect is unclear.
Although not all NSAIDs are approved by the FDA for the whole range of rheumatic diseases, most are probably effective in RA, sero-negative spondyloarthropathies (eg, PA and arthritis associated with inflammatory bowel disease), OA, localized musculoskeletal syndromes (eg, sprains and strains, low back pain), and gout (except tolmetin, which appears to be ineffective in gout).
Adverse effects are generally quite similar for all of the NSAIDs:
1. Central nervous system: Headaches, tinnitus, dizziness, and rarely, aseptic meningitis.
2. Cardiovascular: Fluid retention, hypertension, edema, and rarely, myocardial infarction and congestive heart failure (CHF).
3. Gastrointestinal: Abdominal pain, dysplasia, nausea, vomiting, and rarely, ulcers or bleeding.
4. Hematologic: Rare thrombocytopenia, neutropenia, or even aplastic anemia.
5. Hepatic: Abnormal liver function test results and rare liver failure.
6. Pulmonary: Asthma.
7. Skin: Rashes, all types, pruritus.
8. Renal: Renal insufficiency, renal failure, hyperkalemia, and proteinuria.
ASPIRIN
Aspirin’s long use and availability without prescription diminishes its glamour compared with that of the newer NSAIDs. Aspirin is now rarely used as an anti-inflammatory medication and will be reviewed only in terms of its antiplatelet effects (ie, doses of 81–325 mg once daily).
1. Pharmacokinetics: Salicylic acid is a simple organic acid with a pKa of 3.0. Aspirin (acetylsalicylic acid; ASA) has a pKa of 3.5 (see Table 1–3). Aspirin is absorbed as such and is rapidly hydrolyzed (serum half-life 15 minutes) to acetic acid and salicylate by esterases in tissue and blood (Figure 36–3). Salicylate is nonlinearly bound to albumin. Alkalinization of the urine increases the rate of excretion of free salicylate and its water-soluble conjugates.
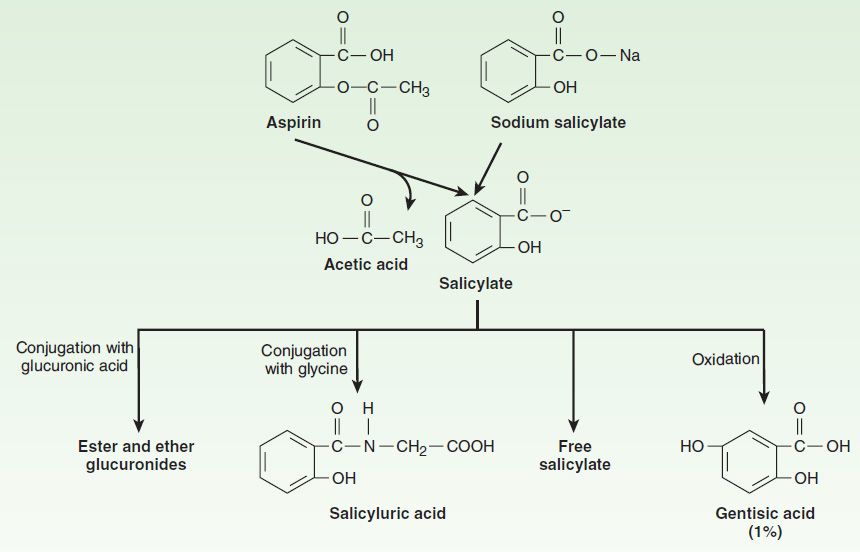
FIGURE 36–3 Structure and metabolism of the salicylates. (Reproduced, with permission, from Meyers FH, Jawetz E, Goldfien A: Review of Medical Pharmacology, 7th ed. McGraw-Hill, 1980. Copyright © The McGraw-Hill Companies, Inc.)
2. Mechanisms of Action: Aspirin irreversibly inhibits platelet COX so that aspirin’s antiplatelet effect lasts 8–10 days (the life of the platelet). In other tissues, synthesis of new COX replaces the inactivated enzyme so that ordinary doses have a duration of action of 6–12 hours.
3. Clinical Uses: Aspirin decreases the incidence of transient ischemic attacks, unstable angina, coronary artery thrombosis with myocardial infarction, and thrombosis after coronary artery bypass grafting (see Chapter 34).
4. Epidemiologic studies suggest that long-term use of aspirin at low dosage is associated with a lower incidence of colon cancer, possibly related to its COX-inhibiting effects.
5. Adverse Effects: In addition to the common side effects listed above, aspirin’s main adverse effects at antithrombotic doses are gastric upset (intolerance) and gastric and duodenal ulcers. Hepatotoxicity, asthma, rashes, GI bleeding, and renal toxicity rarely if ever occur at antithrombotic doses.
6. The antiplatelet action of aspirin contraindicates its use by patients with hemophilia. Although previously not recommended during pregnancy, aspirin may be valuable in treating preeclampsia-eclampsia.
NONACETYLATED SALICYLATES
These drugs include magnesium choline salicylate, sodium salicylate, and salicyl salicylate. All nonacetylated salicylates are effective anti-inflammatory drugs, although they may be less effective analgesics than aspirin. Because they are much less effective than aspirin as COX inhibitors and they do not inhibit platelet aggregation, they may be preferable when COX inhibition is undesirable such as in patients with asthma, those with bleeding tendencies, and even (under close supervision) those with renal dysfunction.
The nonacetylated salicylates are administered in doses up to 3–4 g of salicylate a day and can be monitored using serum salicylate measurements.
COX-2 SELECTIVE INHIBITORS
COX-2 selective inhibitors, or coxibs, were developed in an attempt to inhibit prostaglandin synthesis by the COX-2 isozyme induced at sites of inflammation without affecting the action of the constitutively active “housekeeping” COX-1 isozyme found in the GI tract, kidneys, and platelets. COX-2 inhibitors at usual doses have no impact on platelet aggregation, which is mediated by thromboxane produced by the COX-1 isozyme. In contrast, they do inhibit COX-2-mediated prostacyclin synthesis in the vascular endothelium. As a result, COX-2 inhibitors do not offer the cardioprotective effects of traditional nonselective NSAIDs. Recommended doses of COX-2 inhibitors cause renal toxicities similar to those associated with traditional NSAIDs. Clinical data suggested a higher incidence of cardiovascular thrombotic events associated with COX-2 inhibitors such as rofecoxib and valdecoxib, resulting in their withdrawal from the market.
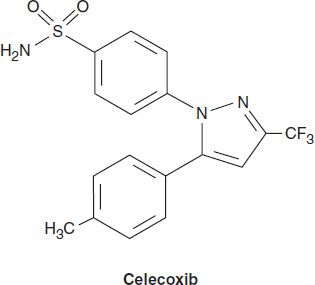
Celecoxib
Celecoxib is a selective COX-2 inhibitor—about 10–20 times more selective for COX-2 than for COX-1. Pharmacokinetic and dosage considerations are given in Table 36–1.
Celecoxib is associated with fewer endoscopic ulcers than most other NSAIDs. Probably because it is a sulfonamide, celecoxib may cause rashes. It does not affect platelet aggregation at usual doses. It interacts occasionally with warfarin—as would be expected of a drug metabolized via CYP2C9. Adverse effects are the common toxicities listed above.
Meloxicam
Meloxicam is an enolcarboxamide related to piroxicam that preferentially inhibits COX-2 over COX-1, particularly at its lowest therapeutic dose of 7.5 mg/d. It is not as selective as celecoxib and may be considered “preferentially” selective rather than “highly” selective. It is associated with fewer clinical GI symptoms and complications than piroxicam, diclofenac, and naproxen. Similarly, while meloxicam is known to inhibit synthesis of thromboxane A2, even at supratherapeutic doses, its blockade of thromboxane A2 does not reach levels that result in decreased in vivo platelet function (see common adverse effects above).
NONSELECTIVE COX INHIBITORS*
Diclofenac
Diclofenac is a phenylacetic acid derivative that is relatively nonselective as a COX inhibitor. Pharmacokinetic and dosage characteristics are set forth in Table 36–1.
Gastrointestinal ulceration may occur less frequently than with some other NSAIDs. A preparation combining diclofenac and misoprostol decreases upper gastrointestinal ulceration but may result in diarrhea. Another combination of diclofenac and omeprazole was also effective with respect to the prevention of recurrent bleeding, but renal adverse effects were common in high-risk patients. Diclofenac, 150 mg/d, appears to impair renal blood flow and glomerular filtration rate. Elevation of serum aminotransferases occurs more commonly with this drug than with other NSAIDs.
A 0.1% ophthalmic preparation is promoted for prevention of postoperative ophthalmic inflammation and can be used after intraocular lens implantation and strabismus surgery. A topical gel containing 3% diclofenac is effective for solar keratoses. Diclofenac in rectal suppository form can be considered for preemptive analgesia and postoperative nausea. In Europe, diclofenac is also available as an oral mouthwash and for intramuscular administration.
Diflunisal
Although diflunisal is derived from salicylic acid, it is not metabolized to salicylic acid or salicylate. It undergoes an enterohepatic cycle with reabsorption of its glucuronide metabolite followed by cleavage of the glucuronide to again release the active moiety. Diflunisal is subject to capacity-limited metabolism, with serum half-lives at various dosages approximating that of salicylates (Table 36–1). In RA the recommended dose is 500–1000 mg daily in two divided doses. It is claimed to be particularly effective for cancer pain with bone metastases and for pain control in dental (third molar) surgery. A 2% diflunisal oral ointment is a clinically useful analgesic for painful oral lesions.
Because its clearance depends on renal function as well as hepatic metabolism, diflunisal’s dosage should be limited in patients with significant renal impairment.
Etodolac
Etodolac is a racemic acetic acid derivative with an intermediate half-life (Table 36–1). The analgesic dosage of etodolac is 200–400 mg three to four times daily. The recommended dose in OA and RA is 300 mg twice or three times a day up to 500 mg twice a day initially followed by a maintenance of 600 mg/d.
Flurbiprofen
Flurbiprofen is a propionic acid derivative with a possibly more complex mechanism of action than other NSAIDs. Its (S)(−) enantiomer inhibits COX nonselectively, but it has been shown in rat tissue to also affect tumor necrosis factor-α (TNF-α) and nitric oxide synthesis. Hepatic metabolism is extensive; its (R)(+) and (S)(−) enantiomers are metabolized differently, and it does not undergo chiral conversion. It does demonstrate enterohepatic circulation.
Flurbiprofen is also available in a topical ophthalmic formulation for inhibition of intraoperative miosis. Flurbiprofen intravenously is effective for perioperative analgesia in minor ear, neck, and nose surgery and in lozenge form for sore throat.
Although its adverse effect profile is similar to that of other NSAIDs in most ways, flurbiprofen is also rarely associated with cogwheel rigidity, ataxia, tremor, and myoclonus.
Ibuprofen
Ibuprofen is a simple derivative of phenylpropionic acid (Figure 36–1). In doses of about 2400 mg daily, ibuprofen is equivalent to 4 g of aspirin in anti-inflammatory effect. Pharmacokinetic characteristics are given in Table 36–1.
Oral ibuprofen is often prescribed in lower doses (<2400 mg/d), at which it has analgesic but not anti-inflammatory efficacy. It is available over the counter in low-dose forms under several trade names.
Ibuprofen oral and IV is effective in closing patent ductus arteriosus in preterm infants, with much the same efficacy and safety as indomethacin. A topical cream preparation appears to be absorbed into fascia and muscle; ibuprofen cream was more effective than placebo cream in the treatment of primary knee OA. A liquid gel preparation of ibuprofen, 400 mg, provides prompt relief and good overall efficacy in postsurgical dental pain.
In comparison with indomethacin, ibuprofen decreases urine output less and also causes less fluid retention. The drug is relatively contraindicated in individuals with nasal polyps, angio-edema, and bronchospastic reactivity to aspirin. Aseptic meningitis (particularly in patients with SLE), and fluid retention have been reported. The concomitant administration of ibuprofen and aspirin antagonizes the irreversible platelet inhibition induced by aspirin. Thus, treatment with ibuprofen in patients with increased cardiovascular risk may limit the cardioprotective effects of aspirin. Furthermore, the use of ibuprofen concomitantly with aspirin may decrease the total anti-inflammatory effect. Common adverse effects are listed on pages 620-621; rare hematologic effects include agranulocytosis and aplastic anemia.
Indomethacin
Indomethacin, introduced in 1963, is an indole derivative (Figure 36–1). It is a potent nonselective COX inhibitor and may also inhibit phospholipase A and C, reduce neutrophil migration, and decrease T-cell and B-cell proliferation.
Indomethacin differs somewhat from other NSAIDs in its indications and toxicities. It has been used to accelerate closure of patent ductus arteriosus. Indomethacin has been tried in numerous small or uncontrolled trials for many other conditions, including Sweet’s syndrome, juvenile RA, pleurisy, nephrotic syndrome, diabetes insipidus, urticarial vasculitis, postepisiotomy pain, and prophylaxis of heterotopic ossification in arthroplasty.
An ophthalmic preparation is efficacious for conjunctival inflammation and to reduce pain after traumatic corneal abrasion. Gingival inflammation is reduced after administration of indomethacin oral rinse. Epidural injections produce a degree of pain relief similar to that achieved with methylprednisolone in postlaminectomy syndrome.
At usual doses, indomethacin has the common side effects listed above. The GI effects may include pancreatitis. Headache is experienced by 15–25% of patients and may be associated with dizziness, confusion, and depression. Renal papillary necrosis has also been observed. A number of interactions with other drugs have been reported (see Chapter 66).
Ketoprofen
Ketoprofen is a propionic acid derivative that inhibits both COX (nonselectively) and lipoxygenase. Its pharmacokinetic characteristics are given in Table 36–1. Concurrent administration of probenecid elevates ketoprofen levels and prolongs its plasma half-life.
The effectiveness of ketoprofen at dosages of 100–300 mg/d is equivalent to that of other NSAIDs. Its major adverse effects are on the GI tract and the central nervous system (see common adverse effects above).
Nabumetone
Nabumetone is the only nonacid NSAID in current use; it is given as a ketone prodrug (Figure 36–1) and resembles naproxen in structure. Its half-life of more than 24 hours (Table 36–1) permits once-daily dosing, and the drug does not appear to undergo enterohepatic circulation. Renal impairment results in a doubling of its half-life and a 30% increase in the area under the curve.
Its properties are very similar to those of other NSAIDs, though it may be less damaging to the stomach. Unfortunately, higher dosages (eg, 1500–2000 mg/d) are often needed, and this is a very expensive NSAID. Like naproxen, nabumetone has been associated with pseudoporphyria and photosensitivity in some patients.
Naproxen
Naproxen is a naphthylpropionic acid derivative. It is the only NSAID presently marketed as a single enantiomer. Naproxen’s free fraction is significantly higher in women than in men, but half-life is similar in both sexes (Table 36–1). Naproxen is effective for the usual rheumatologic indications and is available in a slow-release formulation, as an oral suspension, and over the counter. A topical preparation and an ophthalmic solution are also available.
The incidence of upper GI bleeding in over-the-counter use is low but still double that of over-the-counter ibuprofen (perhaps due to a dose effect). Rare cases of allergic pneumonitis, leukocytoclastic vasculitis, and pseudoporphyria as well as the common NSAID-associated adverse effects have been noted.
Oxaprozin
Oxaprozin is another propionic acid derivative NSAID. As noted in Table 36–1, its major difference from the other members of this subgroup is a very long half-life (50–60 hours), although oxaprozin does not undergo enterohepatic circulation. It is mildly uricosuric. Otherwise, the drug has the same benefits and risks that are associated with other NSAIDs.
Piroxicam
Piroxicam, an oxicam (Figure 36–1), is a nonselective COX inhibitor that at high concentrations also inhibits polymorphonuclear leukocyte migration, decreases oxygen radical production, and inhibits lymphocyte function. Its long half-life (Table 36–1) permits once-daily dosing.
Piroxicam can be used for the usual rheumatic indications. When piroxicam is used in dosages higher than 20 mg/d, an increased incidence of peptic ulcer and bleeding is encountered—as much as 9.5 times higher than with other NSAIDs (see common adverse effects above).
Sulindac
Sulindac is a sulfoxide prodrug. It is reversibly metabolized to the active sulfide metabolite, which is excreted in bile and then reabsorbed from the intestine. The enterohepatic cycling prolongs the duration of action to 12–16 hours.
In addition to its rheumatic disease indications, sulindac suppresses familial intestinal polyposis and it may inhibit the development of colon, breast, and prostate cancer in humans. Among the more severe adverse reactions, Stevens-Johnson epidermal necrolysis syndrome, thrombocytopenia, agranulocytosis, and nephrotic syndrome have all been observed. It is sometimes associated with cholestatic liver damage.
Tolmetin
Tolmetin is a nonselective COX inhibitor with a short half-life (1–2 hours) and is not often used. It is ineffective (for unknown reasons) in the treatment of gout.
Other NSAIDs
Azapropazone, carprofen, meclofenamate, and tenoxicam are rarely used and are not reviewed here.
CHOICE OF NSAID
All NSAIDs, including aspirin, are about equally efficacious with a few exceptions—tolmetin seems not to be effective for gout, and aspirin is less effective than other NSAIDs (eg, indomethacin) for AS.
Thus, NSAIDs tend to be differentiated on the basis of toxicity and cost-effectiveness. For example, the GI and renal side effects of ketorolac limit its use. Some surveys suggest that indomethacin and tolmetin are the NSAIDs associated with the greatest toxicity, while salsalate, aspirin, and ibuprofen are least toxic. The selective COX-2 inhibitors were not included in these analyses.
For patients with renal insufficiency, nonacetylated salicylates may be best. Diclofenac and sulindac are associated with more liver function test abnormalities than other NSAIDs. The relatively expensive, selective COX-2 inhibitor celecoxib is probably safest for patients at high risk for GI bleeding but may have a higher risk of cardiovascular toxicity. Celecoxib or a nonselective NSAID plus omeprazole or misoprostol may be appropriate in patients at highest risk for GI bleeding; in this subpopulation of patients, they are cost-effective despite their high acquisition costs.
The choice of an NSAID thus requires a balance of efficacy, cost-effectiveness, safety, and numerous personal factors (eg, other drugs also being used, concurrent illness, compliance, medical insurance coverage), so that there is no best NSAID for all patients. There may, however, be one or two best NSAIDs for a specific person.
 DISEASE-MODIFYING ANTIRHEUMATIC DRUGS
DISEASE-MODIFYING ANTIRHEUMATIC DRUGS
RA is a progressive immunologic disease that causes significant systemic effects, shortens life, and reduces mobility and quality of life. Interest has centered on finding treatments that might arrest—or at least slow—this progression by modifying the disease itself. The effects of disease-modifying therapies may take 2 weeks to 6 months to become clinically evident.
These therapies include nonbiologic and biologic disease-modifying antirheumatic drugs (usually designated “DMARDs”). The nonbiologic agents include small molecule drugs such as methotrexate, azathioprine, chloroquine and hydroxychloroquine, cyclophosphamide, cyclosporine, leflunomide, mycophenolate mofetil, and sulfasalazine. Tofacitinib, though marketed as a biologic, is actually a well-tolerated nonbiologic DMARD. Gold salts, which were once extensively used, are no longer recommended because of their significant toxicities and questionable efficacy. Biologics are large-molecule therapeutic agents, usually proteins, that are often produced by recombinant DNA technology. The biologic DMARDs approved for RA include: a T-cell-modulating biologic (abatacept), a B-cell cytotoxic agent (rituximab), an anti-IL-6 receptor antibody (tocilizumab), IL-1-inhibiting agents (anakinra, rilonacept, canakinumab), and the TNF-α-blocking agents (five drugs).
The small-molecule DMARDs and biologics are discussed alphabetically, independent of origin.
ABATACEPT
1. Mechanism of action: Abatacept is a co-stimulation modulator biologic that inhibits the activation of T cells (see also Chapter 55). After a T cell has engaged an antigen-presenting cell (APC), a second signal is produced by CD28 on the T cell that interacts with CD80 or CD86 on the APC, leading to T-cell activation. Abatacept (which contains the endogenous ligand CTLA-4) binds to CD80 and 86, thereby inhibiting the binding to CD28 and preventing the activation of T cells.
2. Pharmacokinetics: The recommended dose of abatacept for the treatment of adult patients with RA is three intravenous infusion “induction” doses (day 0, week 2, and week 4), followed by monthly infusions. The dose is based on body weight; patients weighing less than 60 kg receiving 500 mg, those 60–100 kg receiving 750 mg, and those more than 100 kg receiving 1000 mg. Abatacept is also available as a subcutaneous formulation and is given as 125 mg subcutaneously once weekly.
JIA can also be treated with abatacept with an induction schedule at day 0, week 2, and week 4, followed by intravenous infusion every 4 weeks. The recommended dose for patients 6–17 years of age and weighing less than 75 kg is 10 mg/kg, while those weighing 75 kg or more follow the adult intravenous doses to a maximum not to exceed 1000 mg. The terminal serum half-life is 13–16 days. Co-administration with methotrexate, NSAIDs, and corticosteroids does not influence abatacept clearance.
Most patients respond to abatacept within 12–16 weeks after the initiation of the treatment; however, some patients can respond in as few as 2–4 weeks. A study showed equivalence between adalimumab and abatacept.
3. Indications: Abatacept can be used as monotherapy or in combination with methotrexate or other DMARDs in patients with moderate to severe RA or severe PJIA. It is being tested in early RA and methotrexate-naïve patients.
4. Adverse Effects: There is a slightly increased risk of infection (as with other biologic DMARDs), predominantly of the upper respiratory tract. Concomitant use with TNF-α antagonists or other biologics is not recommended due to the increased incidence of serious infection. All patients should be screened for latent tuberculosis and viral hepatitis before starting this medication. Live vaccines should be avoided in patients while taking abatacept and up to 3 months after discontinuation. Infusion-related reactions and hypersensitivity reactions, including anaphylaxis, have been reported but are rare. Anti-abatacept antibody formation is infrequent (<5%) and has no effect on clinical outcomes. There is a possible increase in lymphomas but not other malignancies when using abatacept.
AZATHIOPRINE
1. Mechanism of Action: Azathioprine is a synthetic nonbiologic DMARD that acts through its major metabolite, 6-thioguanine. 6-Thioguanine suppresses inosinic acid synthesis, B-cell and T-cell function, immunoglobulin production, and IL-2 secretion (see Chapter 55).
2. Pharmacokinetics: Azathioprine can be given orally or parenterally. Its metabolism is bimodal in humans, with rapid metabolizers clearing the drug four times faster than slow metabolizers. Production of 6-thioguanine is dependent on thiopurine methyltransferase (TPMT), and patients with low or absent TPMT activity (0.3% of the population) are at particularly high risk of myelosuppression by excess concentrations of the parent drug, if dosage is not adjusted.
3. Indications: Azathioprine is approved for use in RA at a dosage of 2 mg/kg/d. It is also used for the prevention of kidney transplant rejection in combination with other immune suppressants. Controlled trials show efficacy in PA, reactive arthritis, polymyositis, SLE, maintenance of remission in vasculitis, and Behçet’s disease. Azathioprine is also used in scleroderma; however, in one study, it was found to be less effective than cyclophosphamide in controlling the progression of scleroderma lung disease.
4. Adverse Effects: Azathioprine’s toxicity includes bone marrow suppression, GI disturbances, and some increase in infection risk. As noted in Chapter 55, lymphomas may be increased with azathioprine use. Rarely, fever, rash, and hepatotoxicity signal acute allergic reactions.
CHLOROQUINE & HYDROXYCHLOROQUINE
1. Mechanism of Action: Chloroquine and hydroxychloroquine are nonbiologic drugs mainly used for malaria (see Chapter 52) and in the rheumatic diseases. The following mechanisms have been proposed: suppression of T-lymphocyte responses to mitogens, inhibition of leukocyte chemotaxis, stabilization of lysosomal enzymes, processing through the Fc-receptor, inhibition of DNA and RNA synthesis, and the trapping of free radicals.
2. Pharmacokinetics: Antimalarials are rapidly absorbed and 50% protein-bound in the plasma. They are very extensively tissue-bound, particularly in melanin-containing tissues such as the eyes. The drugs are deaminated in the liver and have blood elimination half-lives of up to 45 days.
3. Indications: Antimalarials are approved for RA, but they are not considered very effective DMARDs. Dose-loading may increase rate of response. There is no evidence that these compounds alter bony damage in RA at their usual dosages (up to 6.4 mg/kg/d for hydroxychloroquine or 200 mg/d for chloroquine). It usually takes 3–6 months to obtain a response. Antimalarials are used very commonly in SLE because they decrease mortality and the skin manifestations, serositis, and joint pains of this disease. They have also been used in Sjögren’s syndrome.
4. Adverse Effects: Although ocular toxicity (see Chapter 52) may occur at dosages greater than 250 mg/d for chloroquine and greater than 6.4 mg/kg/d for hydroxychloroquine, it rarely occurs at lower doses. Nevertheless, ophthalmologic monitoring every 12 months is advised. Other toxicities include dyspepsia, nausea, vomiting, abdominal pain, rashes, and nightmares. These drugs appear to be relatively safe in pregnancy.
CYCLOPHOSPHAMIDE
1. Mechanism of Action: Cyclophosphamide is a synthetic nonbiologic DMARD. Its major active metabolite is phosphoramide mustard, which cross-links DNA to prevent cell replication. It suppresses T-cell and B-cell function by 30–40%; T-cell suppression correlates with clinical response in the rheumatic diseases. Its pharmacokinetics and toxicities are discussed in Chapter 54.
2. Indications: Cyclophosphamide is used regularly at 2 mg/kg/d to treat SLE, vasculitis, Wegener’s granulomatosis, and other severe rheumatic diseases.
CYCLOSPORINE
1. Mechanism of Action:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


