42
Agents That Affect Bone Mineral Homeostasis
CASE STUDY
A 65-year-old man is referred to you from his primary care physician (PCP) for evaluation and management of possible osteoporosis. He saw his PCP for evaluation of low back pain. X-rays of the spine showed some degenerative changes in the lumbar spine plus several wedge deformities in the thoracic spine. The patient is a long-time smoker (up to two packs per day) and has two to four glasses of wine with dinner, more on the weekends. He has chronic bronchitis, presumably from smoking, and has been treated many times with oral prednisone for exacerbations of bronchitis. He is currently on 10 mg/d prednisone. Examination shows kyphosis of the thoracic spine, with some tenderness to fist percussion over the thoracic spine. The DEXA (dual-energy X-ray absorptiometry) measurement of the lumbar spine is “within the normal limits,” but the radiologist noted that the reading may be misleading because of degenerative changes. The hip measurement shows a T score (number of standard deviations by which the patient’s measured bone density differs from that of a normal young adult) in the femoral neck of –2.2. What further workup should be considered, and what therapy should be initiated?
 BASIC PHARMACOLOGY
BASIC PHARMACOLOGY
Calcium and phosphate, the major mineral constituents of bone, are also two of the most important minerals for general cellular function. Accordingly, the body has evolved complex mechanisms to carefully maintain calcium and phosphate homeostasis (Figure 42–1). Approximately 98% of the 1–2 kg of calcium and 85% of the 1 kg of phosphorus in the human adult are found in bone, the principal reservoir for these minerals. This reservoir is dynamic, with constant remodeling of bone and ready exchange of bone mineral with that in the extracellular fluid. Bone also serves as the principal structural support for the body and provides the space for hematopoiesis. This relationship is more than fortuitous as elements of the bone marrow affect skeletal processes just as skeletal elements affect hematopoeitic processes. During aging and in nutritional diseases such as anorexia nervosa and obesity, fat accumulates in the marrow, suggesting a dynamic interaction between marrow fat and bone. Abnormalities in bone mineral homeostasis can lead to a wide variety of cellular dysfunctions (eg, tetany, coma, muscle weakness), and to disturbances in structural support of the body (eg, osteoporosis with fractures) and loss of hematopoietic capacity (eg, infantile osteopetrosis).
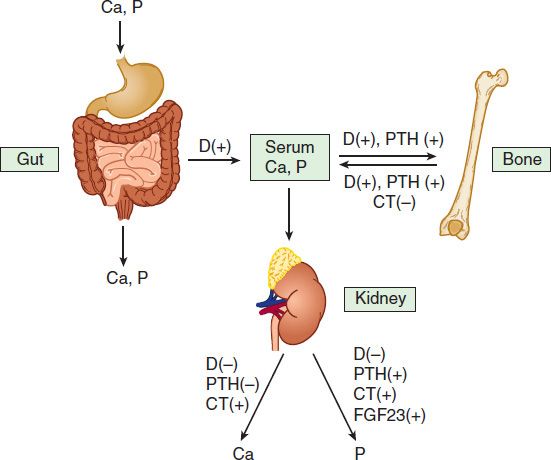
FIGURE 42–1 Mechanisms contributing to bone mineral homeostasis. Serum calcium (Ca) and phosphorus (P) concentrations are controlled principally by three hormones, 1,25-dihydroxyvitamin D (D), fibroblast growth factor 23 (FGF23), and parathyroid hormone (PTH), through their action on absorption from the gut and from bone and on renal excretion. PTH and 1,25(OH)2D increase the input of calcium and phosphorus from bone into the serum and stimulate bone formation. 1,25(OH)2D also increases calcium and phosphate absorption from the gut. In the kidney, 1,25(OH)2D decreases excretion of both calcium and phosphorus, whereas PTH reduces calcium but increases phosphorus excretion. FGF23 stimulates renal excretion of phosphate. Calcitonin (CT) is a less critical regulator of calcium homeostasis, but in pharmacologic concentrations can reduce serum calcium and phosphorus by inhibiting bone resorption and stimulating their renal excretion. Feedback may alter the effects shown; for example, 1,25(OH)2D increases urinary calcium excretion indirectly through increased calcium absorption from the gut and inhibition of PTH secretion and may increase urinary phosphate excretion because of increased phosphate absorption from the gut and stimulation of FGF23 production.
Calcium and phosphate enter the body from the intestine. The average American diet provides 600–1000 mg of calcium per day, of which approximately 100–250 mg is absorbed. This amount represents net absorption, because both absorption (principally in the duodenum and upper jejunum) and secretion (principally in the ileum) occur. The quantity of phosphorus in the American diet is about the same as that of calcium. However, the efficiency of absorption (principally in the jejunum) is greater, ranging from 70% to 90%, depending on intake. In the steady state, renal excretion of calcium and phosphate balances intestinal absorption. In general, over 98% of filtered calcium and 85% of filtered phosphate is reabsorbed by the kidney. The movement of calcium and phosphate across the intestinal and renal epithelia is closely regulated. Dysfunction of the intestine (eg, nontropical sprue) or kidney (eg, chronic renal failure) can disrupt bone mineral homeostasis.
Three hormones serve as the principal regulators of calcium and phosphate homeostasis: parathyroid hormone (PTH), fibroblast growth factor 23 (FGF23), and vitamin D via its active metabolite 1,25-dihydroxyvitamin D (1,25(OH)2D (Figure 42–2). The role of calcitonin (CT) is less critical during adult life but may play a greater role during pregnancy and lactation. The term vitamin D, when used without a subscript, refers to both vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol). This applies also to the metabolites of vitamin D2 and D3. Vitamin D2 and its metabolites differ from vitamin D3 and its metabolites only in the side chain where they contain a double bond between C-22–23 and a methyl group at C-24 (Figure 42–3). Vitamin D is considered a prohormone because it must be further metabolized to gain biologic activity (Figure 42–3). Vitamin D3 is produced in the skin under ultraviolet B (UVB) radiation (eg, in sunlight) from its precursor, 7-dehydrocholesterol. The initial product, pre-vitamin D3, undergoes a temperature-sensitive isomerization to vitamin D3. The precursor of vitamin D2 is ergosterol, found in plants and fungi (mushrooms). It undergoes a similar transformation to vitamin D2 with UVB radiation. Vitamin D2 thus comes only from the diet, whereas vitamin D3 comes from the skin or the diet, or both. The subsequent metabolism of these two forms of vitamin D is essentially the same and follows the illustration for vitamin D3 metabolism in Figure 42–3. The first step is the 25-hydroxylation of vitamin D to 25-hydroxyvitamin D (25[OH]D). A number of enzymes in the liver and other tissues perform this function, of which CYP2R1 is the most important. 25(OH)D is then metabolized to the active hormone 1,25-dihydroxyvitamin D (1,25[OH]2D) in the kidney and elsewhere. PTH stimulates the production of 1,25(OH)2D in the kidney, whereas FGF23 is inhibitory. Elevated levels of blood phosphate and calcium also inhibit 1,25(OH)2D production in part by their effects on FGF23 (high phosphate stimulates FGF23 production) and PTH (high calcium inhibits PTH production). 1,25(OH)2D inhibits its own production but, at least as important, it stimulates the enzyme 24-hydroxyase (CYP24A1), which begins the catabolism of 1,25(OH)2D, suppresses PTH production, and stimulates FGF23 production, all of which conspire to reduce 1,25(OH)2D levels. Other tissues also produce 1,25(OH)2D; the control of this production differs from that in the kidney, as will be discussed subsequently. The complex interplay among PTH, FGF23, and 1,25(OH)2D is discussed in detail later.
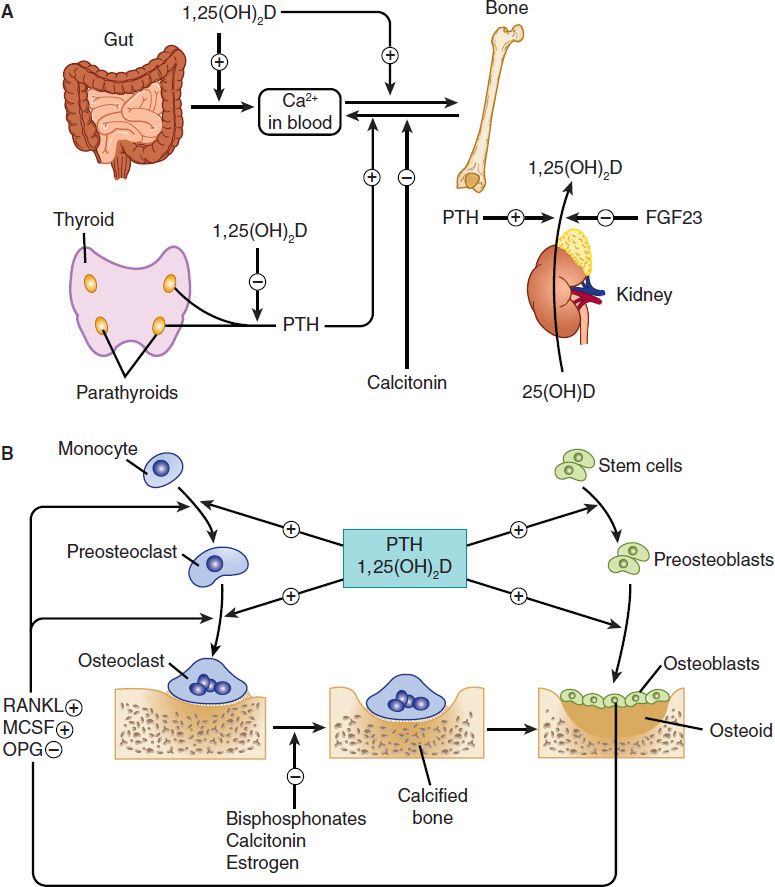
FIGURE 42–2 The hormonal interactions controlling bone mineral homeostasis. In the body (A), 1,25-dihydroxyvitamin D (1,25[OH]2D) is produced by the kidney under the control of parathyroid hormone (PTH), which stimulates its production, and fibroblast growth factor 23 (FGF23), which inhibits its production. 1,25(OH)2D in turn inhibits the production of PTH by the parathyroid glands and stimulates FGF23 release from bone. 1,25(OH)2D is the principal regulator of intestinal calcium and phosphate absorption. At the level of the bone (B), both PTH and 1,25(OH)2D regulate bone formation and resorption, with each capable of stimulating both processes. This is accomplished by their stimulation of preosteoblast proliferation and differentiation into osteoblasts, the bone-forming cell. PTH also stimulates osteoblast formation indirectly by inhibiting the osteocyte’s production of sclerostin, a protein that blocks osteoblast proliferation by inhibiting the wnt pathway (not shown). PTH and 1,25(OH)2D stimulate the expression of RANKL by the osteoblast, which, with MCSF, stimulates the differentiation and subsequent activation of osteoclasts, the bone-resorbing cell. OPG blocks RANKL action, and may be inhibited by PTH and 1,25(OH)2D. FGF23 in excess leads to osteomalacia indirectly by inhibiting 1,25(OH)2D production and lowering phosphate levels. MCSF, macrophage colony-stimulating factor; OPG, osteoprotegerin; RANKL, ligand for receptor for activation of nuclear factor-κB.
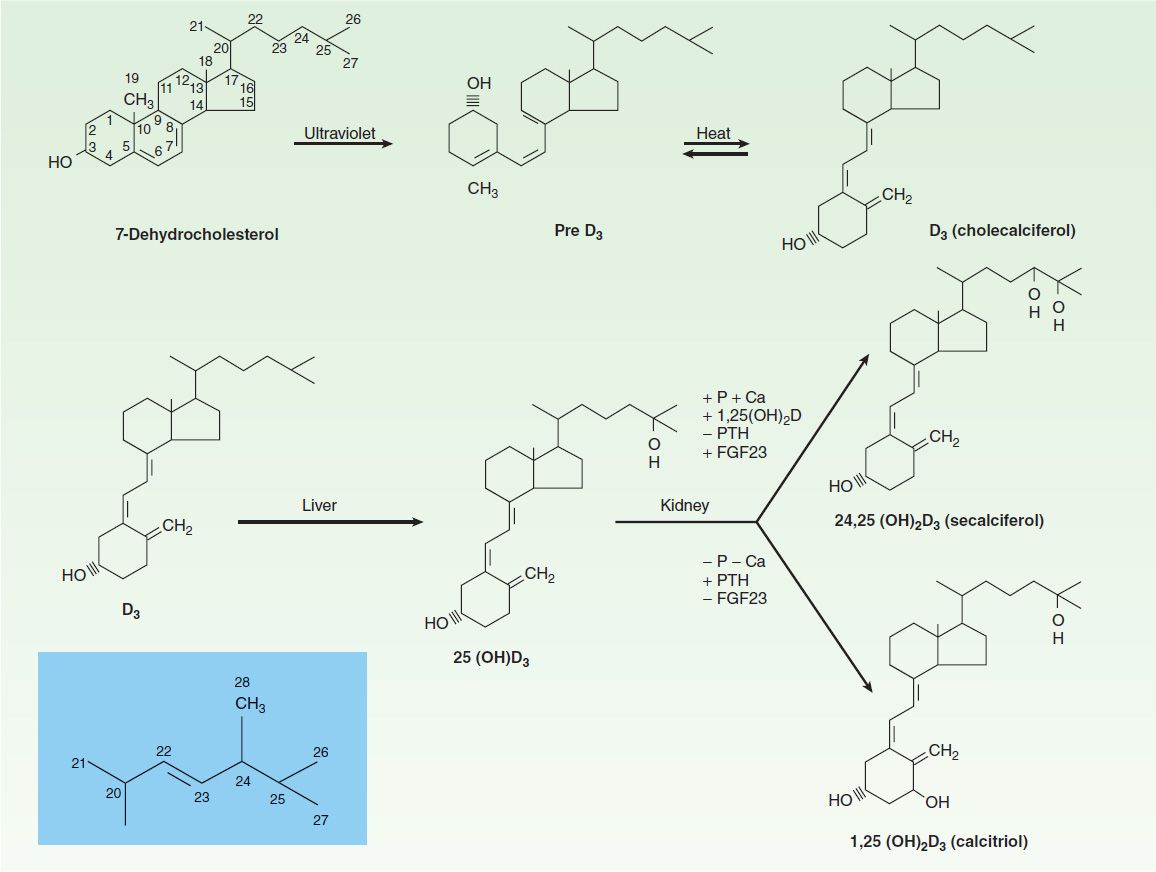
FIGURE 42–3 Conversion of 7-dehydrocholesterol to vitamin D3 in the skin and its subsequent metabolism to 25-hydroxyvitamin D3 (25[OH]D3) in the liver and to 1,25-dihydroxyvitamin D3 (1,25[OH]2D3) and 24,25-dihydroxyvitamin D3 (24,25[OH]2D3) in the kidney. Control of vitamin D metabolism is exerted primarily at the level of the kidney, where high concentrations of serum phosphorus (P) and calcium (Ca) as well as fibroblast growth factor 23 (FGF23) inhibit production of 1,25(OH)2D3 (indicated by a minus [−] sign), but promote that of 24,25(OH)2D3 (indicated by a plus [+] sign). Parathyroid hormone (PTH), on the other hand, stimulates 1,25(OH)2D3 production but inhibits 24,25(OH)2D3 production. The insert (shaded) shows the side chain for ergosterol, vitamin D2, and the active vitamin D2 metabolites. Ergosterol is converted to vitamin D2 (ergocalciferol) by UV radiation similar to the conversion of 7-dehydrocholesterol to vitamin D3. Vitamin D2, in turn, is metabolized to 25-hydroxyvitamin D2, 1,25-dihydroxyvitamin D2, and 24,25-dihydroxyvitamin D2 via the same enzymes that metabolize vitamin D3. In humans, corresponding D2 and D3 metabolites have equivalent biologic effects, although they differ in pharmacokinetics. +, facilitation; –, inhibition; P, phosphorus; Ca, calcium; PTH, parathyroid hormone; FGF23, fibroblast growth factor 23.
To summarize: 1,25(OH)2D suppresses the production of PTH as does calcium, but stimulates the production of FGF23. Phosphate stimulates both PTH and FGF23 secretion. In turn PTH stimulates 1,25(OH)2D production, whereas FGF23 is inhibitory. 1,25(OH)2D stimulates the intestinal absorption of calcium and phosphate. 1,25(OH)2D and PTH promote both bone formation and resorption in part by stimulating the proliferation and differentiation of osteoblasts and osteoclasts. Both PTH and 1,25(OH)2D enhance renal retention of calcium, but PTH promotes renal phosphate excretion as does FGF23, whereas 1,25(OH)2D promotes renal reabsorption of phosphate.
Other hormones—calcitonin, prolactin, growth hormone, insulin, thyroid hormone, glucocorticoids, and sex steroids—influence calcium and phosphate homeostasis under certain physiologic circumstances and can be considered secondary regulators. Deficiency or excess of these secondary regulators within a physiologic range does not produce the disturbance of calcium and phosphate homeostasis that is observed in situations of deficiency or excess of PTH, FGF23, and vitamin D. However, certain of these secondary regulators—especially calcitonin, glucocorticoids, and estrogens—are useful therapeutically and discussed in subsequent sections.
In addition to these hormonal regulators, calcium and phosphate themselves, other ions such as sodium and fluoride, and a variety of drugs (bisphosphonates, plicamycin, and diuretics) also alter calcium and phosphate homeostasis.
PRINCIPAL HORMONAL REGULATORS OF BONE MINERAL HOMEOSTASIS
PARATHYROID HORMONE
Parathyroid hormone (PTH) is a single-chain peptide hormone composed of 84 amino acids. It is produced in the parathyroid gland in a precursor form of 115 amino acids, the remaining 31 amino terminal amino acids being cleaved off before secretion. Within the gland is a calcium-sensitive protease capable of cleaving the intact hormone into fragments, thereby providing one mechanism by which calcium limits the production of PTH. A second mechanism involves the calcium-sensing receptor (CaSR) which, when stimulated by calcium, reduces PTH production and secretion. The parathyroid gland also contains the vitamin D receptor (VDR) and the enzyme, CYP27B1, that produces 1,25(OH)2D, thus enabling circulating or endogenously produced 1,25(OH)2D to suppress PTH production. 1,25(OH)2D also induces the CaSR, making the parathyroid gland more sensitive to suppression by calcium. Biologic activity resides in the amino terminal region of PTH such that synthetic PTH 1-34 (available as teriparatide) is fully active. Loss of the first two amino terminal amino acids eliminates most biologic activity.
The metabolic clearance of intact PTH is rapid, with a half-time of disappearance measured in minutes. Most of the clearance occurs in the liver and kidney. The inactive carboxyl terminal fragments produced by metabolism of the intact hormone have a much lower clearance, especially in renal failure. In the past, this accounted for the very high PTH values observed in patients with renal failure when the hormone was measured by radioimmunoassays directed against the carboxyl terminal region. Currently, most PTH assays differentiate between intact PTH 1-34 and large inactive fragments, so that it is possible to more accurately evaluate biologically active PTH status in patients with renal failure.
PTH regulates calcium and phosphate flux across cellular membranes in bone and kidney, resulting in increased serum calcium and decreased serum phosphate (Figure 42–1). In bone, PTH increases the activity and number of osteoclasts, the cells responsible for bone resorption (Figure 42–2). However, this stimulation of osteoclasts is not a direct effect. Rather, PTH acts on the osteoblast (the bone-forming cell) to induce membrane-bound and secreted soluble forms of a protein called RANK ligand (RANKL). RANKL acts on osteoclasts and osteoclast precursors to increase both the numbers and activity of osteoclasts. This action increases bone remodeling, a specific sequence of cellular events initiated by osteoclastic bone resorption and followed by osteoblastic bone formation. Denosumab, an antibody that inhibits the action of RANKL, has been developed for the treatment of excess bone resorption in patients with osteoporosis and certain cancers. PTH also inhibits the production and secretion of sclerostin from osteocytes. Sclerostin is one of several proteins that blocks osteoblast proliferation by inhibiting the wnt pathway. Thus, PTH indirectly increases proliferation of osteoblasts, the cells responsible for bone formation. An antibody against sclerostin is in clinical trials for the treatment of osteoporosis. Although both bone resorption and bone formation are enhanced by PTH, the net effect of excess endogenous PTH is to increase bone resorption. However, administration of exogenous PTH in low and intermittent doses increases bone formation without first stimulating bone resorption. This net anabolic action may be indirect, involving other growth factors such as insulin-like growth factor 1 (IGF-1) as well as inhibition of sclerostin as noted above. These anabolic actions have led to the approval of recombinant PTH 1-34 (teriparatide) for the treatment of osteoporosis. In the kidney, PTH increases tubular reabsorption of calcium and magnesium but reduces reabsorption of phosphate, amino acids, bicarbonate, sodium, chloride, and sulfate. As noted earlier another important action of PTH on the kidney is stimulation of 1,25(OH)2D production.
VITAMIN D
Vitamin D is a secosteroid produced in the skin from 7-dehydrocholesterol under the influence of ultraviolet radiation. Vitamin D is also found in certain foods and is used to supplement dairy products and other foods. Both the natural form (vitamin D3, cholecalciferol) and the plant-derived form (vitamin D2, ergocalciferol) are present in the diet. As discussed earlier these forms differ in that ergocalciferol contains a double bond and an additional methyl group in the side chain (Figure 42–3). Ergocalciferol and its metabolites bind less well than cholecalciferol and its metabolites to vitamin D-binding protein (DBP), the major transport protein of these compounds in blood, and have a different path of catabolism. As a result their half-lives are shorter than those of the cholecalciferol metabolites. This influences treatment strategies, as will be discussed. However, the key steps in metabolism and biologic activities of the active metabolites are comparable, so with this exception the following comments apply equally well to both forms of vitamin D.
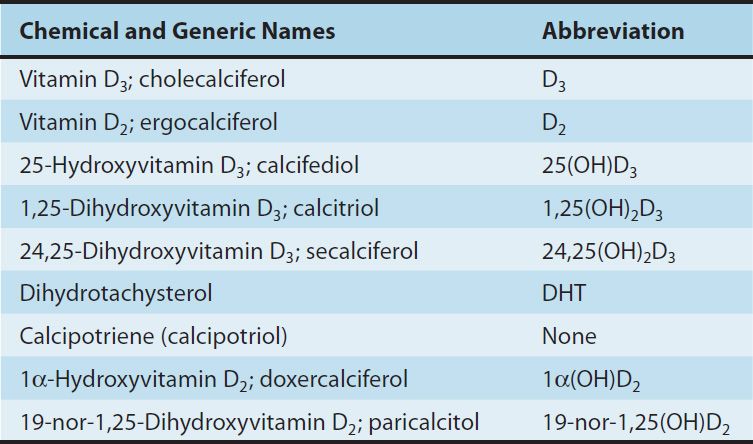
Vitamin D is a precursor to a number of biologically active metabolites (Figure 42–3). Vitamin D is first hydroxylated in the liver and other tissues to form 25(OH)D(calcifediol). As noted earlier there are a number of enzymes with 25-hydroxylase activity. This metabolite is further converted in the kidney to a number of other forms, the best studied of which are 1,25(OH)2D (calcitriol) and 24,25-dihydroxyvitamin D (24,25[OH]2D), by the enzymes CYP27B1 and CYP24A1, respectively. The regulation of vitamin D metabolism is complex, involving calcium, phosphate, and a variety of hormones, the most important of which are PTH, which stimulates, and FGF23, which inhibits the production of 1,25(OH)2D by the kidney while reciprocally inhibiting or promoting the production of 24,25(OH)2D. The importance of CYP24A1, the enzyme that 24-hydroxylates 25(OH)D and 1,25(OH)2D, is well demonstrated in children lacking this enzyme who have high levels of calcium and 1,25(OH)2D resulting in kidney damage from nephrocalcinosis and stones. Of the natural metabolites, only vitamin D and 1,25(OH)2D (as calcitriol) are available for clinical use (Table 42–1). A number of analogs of 1,25(OH)2D have been synthesized to extend the usefulness of this metabolite to a variety of nonclassic conditions. Calcipotriene (calcipotriol), for example, is being used to treat psoriasis, a hyperproliferative skin disorder (see Chapter 61). Doxercalciferol and paricalcitol are approved for the treatment of secondary hyperparathyroidism in patients with chronic kidney disease. Eldecalcitol is in phase 3 clinical trials in Japan for the treatment of osteoporosis. Other analogs are being investigated for the treatment of various malignancies.
TABLE 42–1 Vitamin D and its major metabolites and analogs.
Vitamin D and its metabolites circulate in plasma tightly bound to the DBP. This α-globulin binds 25(OH)D and 24,25(OH)2D with comparable high affinity and vitamin D and 1,25(OH)2D with lower affinity. There is increasing evidence that it is the free or unbound forms of these metabolites that have biologic activity. This is of clinical importance because there are several different forms of DBP in the population with different affinities for the vitamin D metabolites. Such individuals vary with respect to the fraction of free metabolite available. Moreover, as noted above, the affinity of DBP for the D2 metabolites is less than that for the D3 metabolites. In normal subjects, the terminal half-life of injected calcifediol (25[OH]D) is around 23 days, whereas in anephric subjects it is around 42 days. The half-life of 24,25(OH)2D is probably similar. Tracer studies with vitamin D have shown a rapid clearance from the blood. The liver appears to be the principal organ for clearance. Excess vitamin D is stored in adipose tissue. The metabolic clearance of calcitriol (1,25[OH]2D) in humans likewise indicates a rapid turnover, with a terminal half-life measured in hours. Several of the 1,25(OH)2D analogs are bound poorly by DBP. As a result, their clearance is very rapid, with a terminal half-life of minutes. Such analogs have less hypercalcemic, hypercalciuric effects than calcitriol, an important aspect of their use in the management of conditions such as psoriasis and hyperparathyroidism.
The mechanism of action of the vitamin D metabolites remains under active investigation. However, 1,25(OH)2D is well established as the most potent stimulant of intestinal calcium and phosphate transport and bone resorption. 1,25(OH)2D appears to act on the intestine both by induction of new protein synthesis (eg, calcium-binding protein and TRPV6, an intestinal calcium channel) and by modulation of calcium flux across the brush border and basolateral membranes by a process that does not require new protein synthesis. The molecular action of 1,25(OH)2D on bone has received less attention. However, like PTH, 1,25(OH)2D can induce RANKL in osteoblasts and proteins such as osteocalcin, which may regulate the mineralization process. The metabolites 25(OH)D and 24,25(OH)2D are far less potent stimulators of intestinal calcium and phosphate transport or bone resorption.
Specific receptors for 1,25(OH)2D (VDR) exist in nearly all tissues, not just intestine, bone, and kidney; as a result much effort has been made to develop analogs of 1,25(OH)2D that will target these non-classic tissues without increasing serum calcium. These non-classic actions include regulation of the secretion of PTH, insulin, and renin; dendritic cell as well as T-cell differentiation; and proliferation and differentiation of a number of cancer cells. Thus, the clinical utility of 1,25(OH)2D and its analogs is expanding.
FIBROBLAST GROWTH FACTOR 23
Fibroblast growth factor 23 (FGF23) is a single-chain protein with 251 amino acids, including a 24-amino-acid leader sequence. It inhibits 1,25(OH)2D production and phosphate reabsorption (via the sodium phosphate co-transporters NaPi 2a and 2c) in the kidney, and can lead to both hypophosphatemia and inappropriately low levels of circulating 1,25(OH)2D. Whereas FGF23 was originally identified in certain mesenchymal tumors, osteoblasts and osteocytes in bone appear to be its primary site of production. Other tissues can also produce FGF23, though at lower levels. FGF23 requires O-glycosylation for its secretion, a glycosylation mediated by the glycosyl transferase GALNT3. Mutations in GALNT3 result in abnormal deposition of calcium phosphate in periarticular tissues (tumoral calcinosis) with elevated phosphate and 1,25(OH)2D. FGF23 is normally inactivated by cleavage at an RXXR site (amino acids 176–179). Mutations in this site lead to excess FGF23, the underlying problem in autosomal dominant hypophosphatemic rickets. A similar disease, X-linked hypophosphatemic rickets, is due to mutations in PHEX, an endopeptidase, which initially was thought to cleave FGF23. However, this concept has been shown to be invalid, and the mechanism by which PHEX mutations lead to increased FGF23 levels remains obscure. FGF23 binds to FGF receptors 1 and 3c in the presence of the accessory receptor Klotho. Both Klotho and the FGF receptor must be present for signaling. Mutations in Klotho disrupt FGF23 signaling, resulting in elevated phosphate and 1,25(OH)2D levels, a phenotype quite similar to inactivating mutations in FGF23 or GALNT3. FGF23 production is stimulated by 1,25(OH)2D and phosphate and directly or indirectly inhibited by the dentin matrix protein DMP1 found in osteocytes. Mutations in DMP1 lead to increased FGF23 levels and osteomalacia.
INTERACTION OF PTH, FGF23, & VITAMIN D
A summary of the principal actions of PTH, FGF23, and vitamin D on the three main target tissues—intestine, kidney, and bone—is presented in Table 42–2. The net effect of PTH is to raise serum calcium and reduce serum phosphate; the net effect of FGF23 is to decrease serum phosphate; the net effect of vitamin D is to raise both. Regulation of calcium and phosphate homeostasis is achieved through important feedback loops. Calcium is one of two principal regulators of PTH secretion. It binds to a novel ion recognition site that is part of a Gq protein-coupled receptor called the calcium-sensing receptor (CaSR) that employs the phosphoinositide second messenger system to link changes in the extracellular calcium concentration to changes in the intracellular free calcium. As serum calcium levels rise and activate this receptor, intracellular calcium levels increase and inhibit PTH secretion. This inhibition by calcium of PTH secretion, along with inhibition of renin and atrial natriuretic peptide secretion, is the opposite of the effect in other tissues such as the beta cell of the pancreas, in which calcium stimulates secretion. Phosphate regulates PTH secretion directly and indirectly by forming complexes with calcium in the serum. Because it is the ionized free concentration of extracellular calcium that is detected by the parathyroid gland, increases in serum phosphate levels reduce the ionized calcium, leading to enhanced PTH secretion. Such feedback regulation is appropriate to the net effect of PTH to raise serum calcium and reduce serum phosphate levels. Likewise, both calcium and phosphate at high levels reduce the amount of 1,25(OH)2D produced by the kidney and increase the amount of 24,25(OH)2D produced.
TABLE 42–2 Actions of parathyroid hormone (PTH), vitamin D, and FGF23 on gut, bone, and kidney.
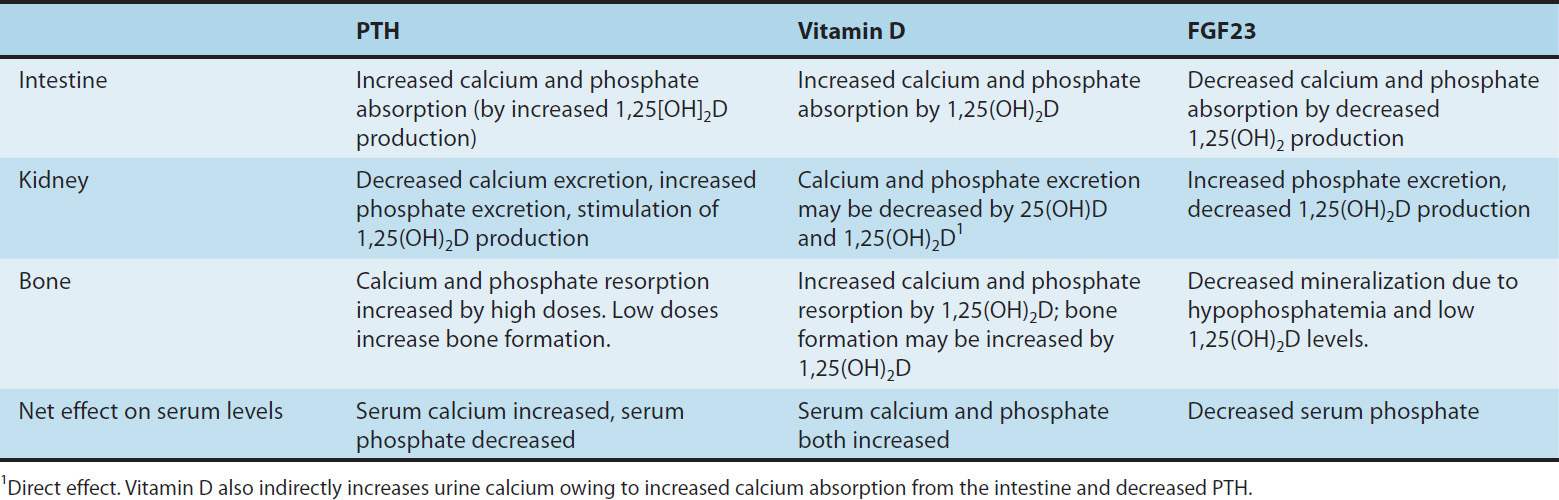
High serum calcium works directly and indirectly by reducing PTH secretion. High serum phosphate works directly and indirectly by increasing FGF23 levels. Since 1,25(OH)2D raises serum calcium and phosphate, whereas 24,25(OH)2D has less effect, such feedback regulation is again appropriate. 1,25(OH)2D directly inhibits PTH secretion (independent of its effect on serum calcium) by a direct inhibitory effect on PTH gene transcription. This provides yet another negative feedback loop. In patients with chronic renal failure who frequently are deficient in producing 1,25(OH)2D, loss of this 1,25(OH)2D-mediated feedback loop coupled with impaired phosphate excretion and intestinal calcium absorption leads to secondary hyperparathyroidism. The ability of 1,25(OH)2D to inhibit PTH secretion directly is being exploited with calcitriol analogs that have less effect on serum calcium because of their lesser effect on intestinal calcium absorption. Such drugs are proving useful in the management of secondary hyperparathyroidism accompanying chronic kidney disease and may be useful in selected cases of primary hyperparathyroidism. 1,25(OH)2D also stimulates the production of FGF23. This completes the negative feedback loop in that FGF23 inhibits 1,25(OH)2D production while promoting hypophosphatemia, which in turn inhibits FGF23 production and stimulates 1,25(OH)2D production.
SECONDARY HORMONAL REGULATORS OF BONE MINERAL HOMEOSTASIS
A number of hormones modulate the actions of PTH, FGF23, and vitamin D in regulating bone mineral homeostasis. Compared with that of PTH, FGF23, and vitamin D, the physiologic impact of such secondary regulation on bone mineral homeostasis is minor. However, in pharmacologic amounts, several of these hormones, including calcitonin, glucocorticoids, and estrogens, have actions on bone mineral homeostatic mechanisms that can be exploited therapeutically.
CALCITONIN
The calcitonin secreted by the parafollicular cells of the mammalian thyroid is a single-chain peptide hormone with 32 amino acids and a molecular weight of 3600. A disulfide bond between positions 1 and 7 is essential for biologic activity. Calcitonin is produced from a precursor with MW 15,000. The circulating forms of calcitonin are multiple, ranging in size from the monomer (MW 3600) to forms with an apparent MW of 60,000. Whether such heterogeneity includes precursor forms or covalently linked oligomers is not known. Because of its chemical heterogeneity, calcitonin preparations are standardized by bioassay in rats. Activity is compared to a standard maintained by the British Medical Research Council (MRC) and expressed as MRC units.
Human calcitonin monomer has a half-life of about 10 minutes. Salmon calcitonin has a longer half-life of 40–50 minutes, making it more attractive as a therapeutic agent. Much of the clearance occurs in the kidney by metabolism; little intact calcitonin appears in the urine.
The principal effects of calcitonin are to lower serum calcium and phosphate by actions on bone and kidney. Calcitonin inhibits osteoclastic bone resorption. Although bone formation is not impaired at first after calcitonin administration, with time both formation and resorption of bone are reduced. In the kidney, calcitonin reduces both calcium and phosphate reabsorption as well as reabsorption of other ions, including sodium, potassium, and magnesium. Tissues other than bone and kidney are also affected by calcitonin. Calcitonin in pharmacologic amounts decreases gastrin secretion and reduces gastric acid output while increasing secretion of sodium, potassium, chloride, and water in the gut. Pentagastrin is a potent stimulator of calcitonin secretion (as is hypercalcemia), suggesting a possible physiologic relationship between gastrin and calcitonin. In the adult human, no readily demonstrable problem develops in cases of calcitonin deficiency (thyroidectomy) or excess (medullary carcinoma of the thyroid). However, the ability of calcitonin to block bone resorption and lower serum calcium makes it a useful drug for the treatment of Paget’s disease, hypercalcemia, and osteoporosis, albeit a less efficacious drug than other available agents.
GLUCOCORTICOIDS
Glucocorticoid hormones alter bone mineral homeostasis by antagonizing vitamin D-stimulated intestinal calcium transport, stimulating renal calcium excretion, and blocking bone formation. Although these observations underscore the negative impact of glucocorticoids on bone mineral homeostasis, these hormones have proved useful in reversing the hypercalcemia associated with lymphomas and granulomatous diseases such as sarcoidosis (in which unregulated ectopic production of 1,25[OH]2D occurs) or in cases of vitamin D intoxication. Prolonged administration of glucocorticoids is a common cause of osteoporosis in adults and can cause stunted skeletal development in children.
ESTROGENS
Estrogens can prevent accelerated bone loss during the immediate postmenopausal period and at least transiently increase bone in postmenopausal women.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


