40
The Gonadal Hormones & Inhibitors
CASE STUDY
A 25-year-old woman with menarche at 13 years and menstrual periods until about 1 year ago complains of hot flushes, skin and vaginal dryness, weakness, poor sleep, and scanty and infrequent menstrual periods of a year’s duration. She visits her gynecologist, who obtains plasma levels of follicle-stimulating hormone and luteinizing hormone, both of which are moderately elevated. She is diagnosed with premature ovarian failure, and estrogen and progesterone replacement therapy is recommended. A dual-energy absorptiometry scan (DEXA) reveals a bone density t-score of < 2.5 SD, ie, frank osteoporosis. How should the ovarian hormones she lacks be replaced? What extra measures should she take for her osteoporosis while receiving treatment?
 THE OVARY (ESTROGENS, PROGESTINS, OTHER OVARIAN HORMONES, ORAL CONTRACEPTIVES, INHIBITORS & ANTAGONISTS, & OVULATION-INDUCING AGENTS)
THE OVARY (ESTROGENS, PROGESTINS, OTHER OVARIAN HORMONES, ORAL CONTRACEPTIVES, INHIBITORS & ANTAGONISTS, & OVULATION-INDUCING AGENTS)
The ovary has important gametogenic functions that are integrated with its hormonal activity. In the human female, the gonad is relatively quiescent during childhood, the period of rapid growth and maturation. At puberty, the ovary begins a 30- to 40-year period of cyclic function called the menstrual cycle because of the regular episodes of bleeding that are its most obvious manifestation. It then fails to respond to gonadotropins secreted by the anterior pituitary gland, and the cessation of cyclic bleeding that occurs is called menopause.
The mechanism responsible for the onset of ovarian function at the time of puberty is thought to be neural in origin, because the immature gonad can be stimulated by gonadotropins already present in the pituitary and because the pituitary is responsive to exogenous hypothalamic gonadotropin-releasing hormone. The maturation of centers in the brain may withdraw a childhood-related inhibitory effect upon hypothalamic arcuate nucleus neurons, allowing them to produce gonadotropin-releasing hormone (GnRH) in pulses with the appropriate amplitude, which stimulates the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) (see Chapter 37). At first, small amounts of the latter two hormones are released during the night, and the limited quantities of ovarian estrogen secreted in response start to cause breast development. Subsequently, FSH and LH are secreted throughout the day and night, causing secretion of higher amounts of estrogen and leading to further breast enlargement, alterations in fat distribution, and a growth spurt that culminates in epiphysial closure in the long bones. The change of ovarian function at puberty is called gonadarche.
A year or so after gonadarche, sufficient estrogen is produced to induce endometrial changes and periodic bleeding (menarche). After the first few irregular cycles, which may be anovulatory, normal cyclic function is established.
At the beginning of each cycle, a variable number of follicles (vesicular follicles), each containing an ovum, begin to enlarge in response to FSH. After 5 or 6 days, one follicle, called the dominant follicle, begins to develop more rapidly. The outer theca and inner granulosa cells of this follicle multiply and, under the influence of LH, synthesize and release estrogens at an increasing rate. The estrogens appear to inhibit FSH release and may lead to regression of the smaller, less mature follicles. The mature dominant ovarian follicle consists of an ovum surrounded by a fluid-filled antrum lined by granulosa and theca cells. The estrogen secretion reaches a peak just before midcycle, and the granulosa cells begin to secrete progesterone. These changes stimulate the brief surge in LH and FSH release that precedes and causes ovulation. When the follicle ruptures, the ovum is released into the abdominal cavity near the opening of the uterine tube.
ACRONYMS
Following the above events, the cavity of the ruptured follicle fills with blood (corpus hemorrhagicum), and the luteinized theca and granulosa cells proliferate and replace the blood to form the corpus luteum. The cells of this structure produce estrogens and progesterone for the remainder of the cycle, or longer if pregnancy occurs.
If pregnancy does not occur, the corpus luteum begins to degenerate and ceases hormone production, eventually becoming a corpus albicans. The endometrium, which proliferated during the follicular phase and developed its glandular function during the luteal phase, is shed in the process of menstruation. These events are summarized in Figure 40–1.
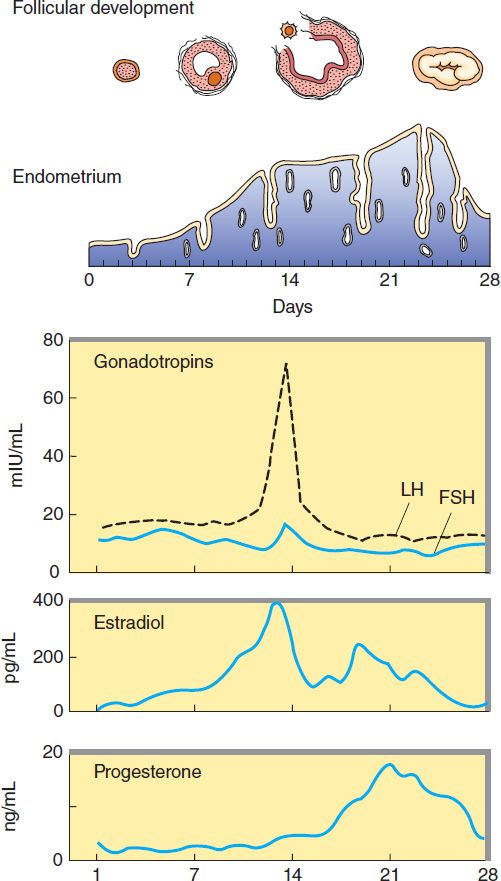
FIGURE 40–1 The menstrual cycle, showing plasma levels of pituitary and ovarian hormones and histologic changes.
The ovary normally ceases its gametogenic and endocrine function with time. This change is accompanied by a cessation in uterine bleeding (menopause) and occurs at a mean age of 52 years in the USA. Although the ovary ceases to secrete estrogen, significant levels of estrogen persist in many women as a result of conversion of adrenal and ovarian steroids such as androstenedione to estrone and estradiol in adipose and possibly other nonendocrine tissues.
Disturbances in Ovarian Function
Disturbances of cyclic function are common even during the peak years of reproduction. A minority of these result from inflammatory or neoplastic processes that influence the functions of the uterus, ovaries, or pituitary. Many of the minor disturbances leading to periods of amenorrhea or anovulatory cycles are self-limited. They are often associated with emotional or physical stress and reflect temporary alterations in the stress centers in the brain that control the secretion of GnRH. Anovulatory cycles are also associated with eating disorders (bulimia, anorexia nervosa) and with severe exercise such as distance running and swimming. Among the more common organic causes of persistent ovulatory disturbances are pituitary prolactinomas and syndromes and tumors characterized by excessive ovarian or adrenal androgen production. Normal ovarian function can be modified by androgens produced by the adrenal cortex or tumors arising from it. The ovary also gives rise to androgen-producing neoplasms such as arrhenoblastomas, as well as to estrogen-producing granulosa cell tumors.
THE ESTROGENS
Estrogenic activity is shared by a large number of chemical substances. In addition to the variety of steroidal estrogens derived from animal sources, numerous nonsteroidal estrogens have been synthesized. Many phenols are estrogenic, and estrogenic activity has been identified in such diverse forms of life as those found in ocean sediments. Estrogen-mimetic compounds (flavonoids) are found in many plants, including saw palmetto, and soybeans and other foods. Studies have shown that a diet rich in these plant products may cause slight estrogenic effects. Additionally, some compounds used in the manufacture of plastics (bisphenols, alkylphenols, phthalate phenols) have been found to be estrogenic. It has been proposed that these agents are associated with an increased breast cancer incidence in both women and men in the industrialized world.
Natural Estrogens
The major estrogens produced by women are estradiol (estradiol-17β, E2), estrone (E1), and estriol (E3) (Figure 40–2). Estradiol is the major secretory product of the ovary. Although some estrone is produced in the ovary, most estrone and estriol are formed in the liver from estradiol or in peripheral tissues from androstenedione and other androgens (see Figure 39–1). As noted above, during the first part of the menstrual cycle estrogens are produced in the ovarian follicle by the theca and granulosa cells. After ovulation, the estrogens as well as progesterone are synthesized by the luteinized granulosa and theca cells of the corpus luteum, and the pathways of biosynthesis are slightly different.
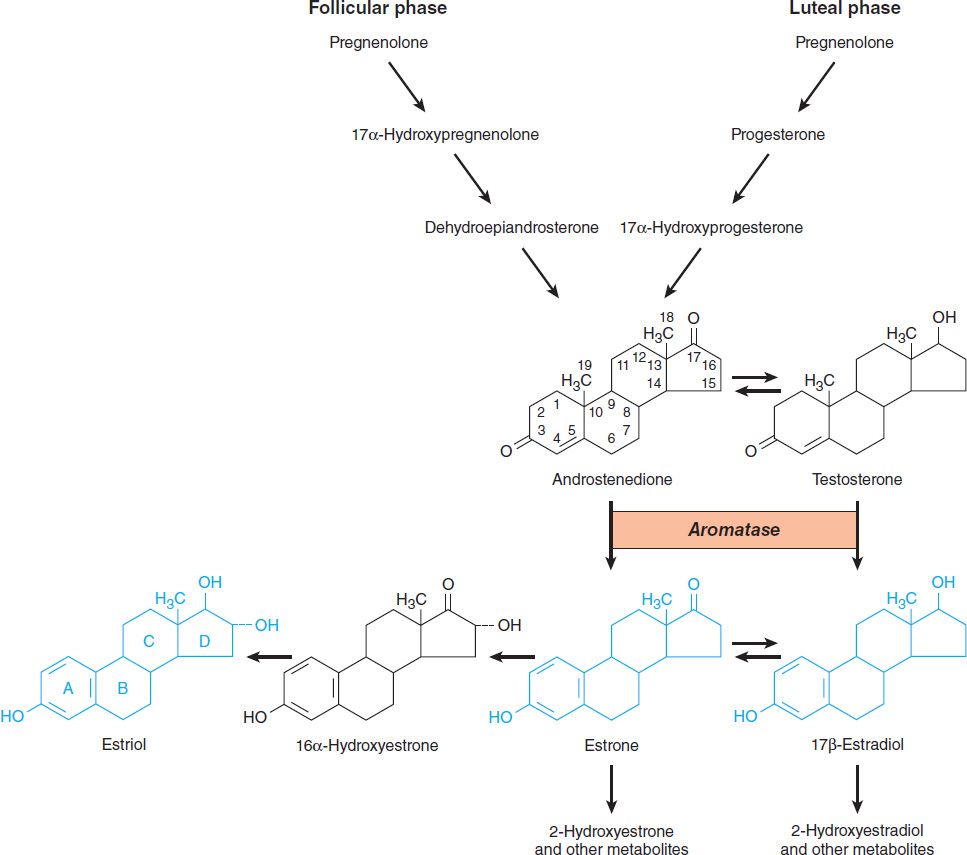
FIGURE 40–2 Biosynthesis and metabolism of estrogens and testosterone.
During pregnancy, a large amount of estrogen is synthesized by the fetoplacental unit—consisting of the fetal adrenal zone, secreting androgen precursor, and the placenta, which aromatizes it into estrogen. The estriol synthesized by the fetoplacental unit is released into the maternal circulation and excreted into the urine. Repeated assay of maternal urinary estriol excretion has been used in the assessment of fetal well-being.
One of the most prolific natural sources of estrogenic substances is the stallion, which liberates more of these hormones than the pregnant mare or pregnant woman. The equine estrogens—equilenin and equilin—and their congeners are unsaturated in the B as well as the A ring and are excreted in large quantities in urine, from which they can be recovered and used for medicinal purposes.
In normal women, estradiol is produced at a rate that varies during the menstrual cycle, resulting in plasma levels as low as 50 pg/mL in the early follicular phase to as high as 350–850 pg/mL at the time of the preovulatory peak (Figure 40–1).
Synthetic Estrogens
A variety of chemical alterations have been applied to the natural estrogens. The most important effect of these alterations has been to increase their oral effectiveness. Some structures are shown in Figure 40–3. Those with therapeutic use are listed in Table 40–1.
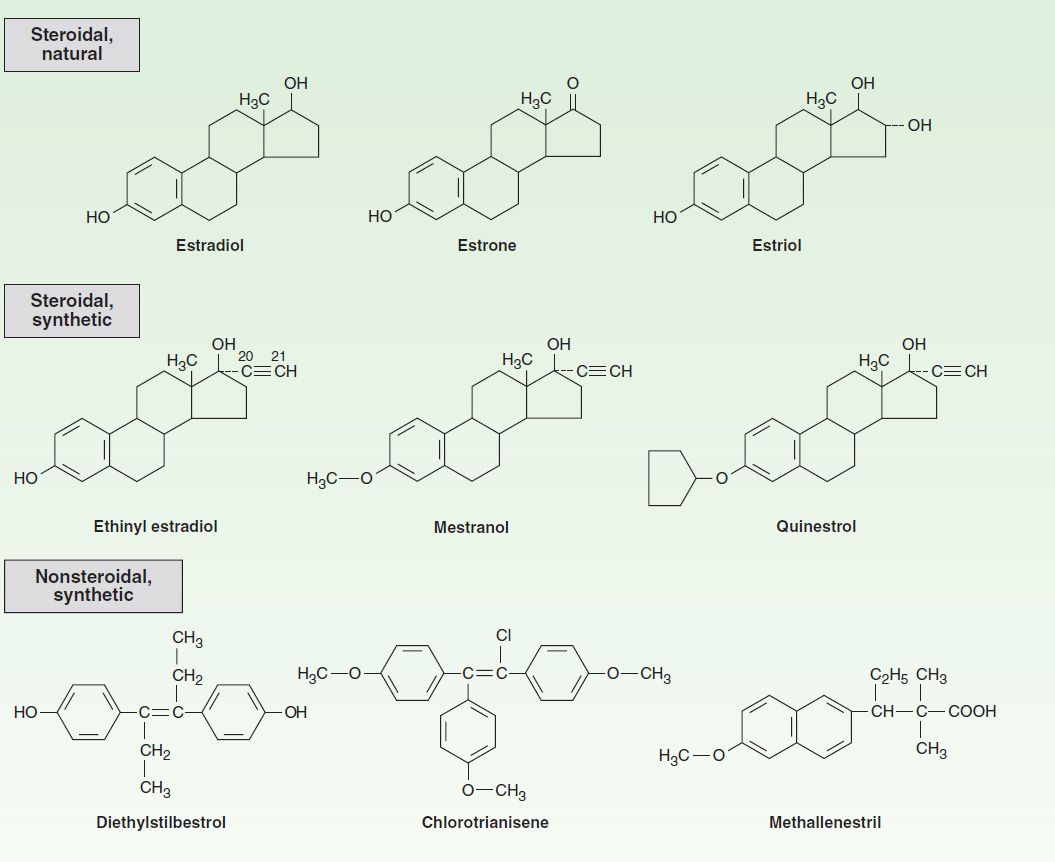
FIGURE 40–3 Compounds with estrogenic activity.
TABLE 40–1 Commonly used estrogens.
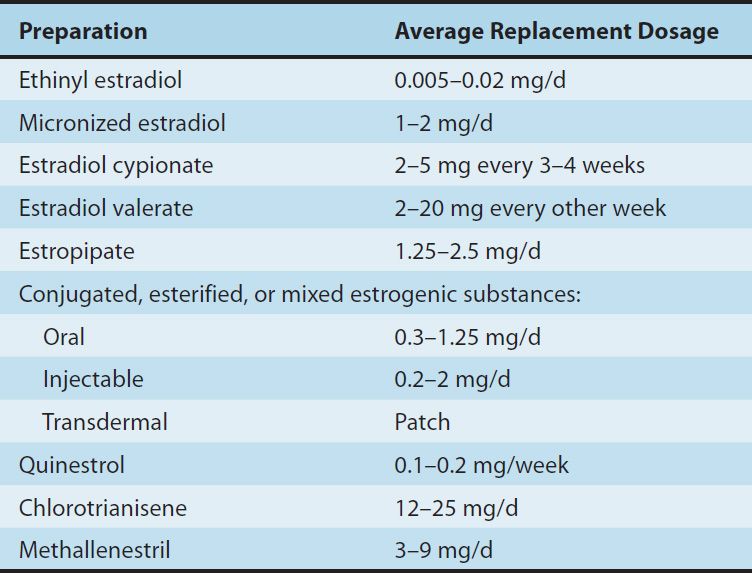
In addition to the steroidal estrogens, a variety of nonsteroidal compounds with estrogenic activity have been synthesized and used clinically. These include dienestrol, diethylstilbestrol, benzestrol, hexestrol, methestrol, methallenestril, and chlorotrianisene.
Pharmacokinetics
When released into the circulation, estradiol binds strongly to an α2 globulin (sex hormone-binding globulin [SHBG]) and with lower affinity to albumin. Bound estrogen is relatively unavailable for diffusion into cells, and it is the free fraction that is physiologically active. Estradiol is converted by the liver and other tissues to estrone and estriol (Figure 40–2) and their 2-hydroxylated derivatives and conjugated metabolites (which are too insoluble in lipid to cross the cell membrane readily) and excreted in the bile. Estrone and estriol have low affinity for the estrogen receptor. However, the conjugates may be hydrolyzed in the intestine to active, reabsorbable compounds. Estrogens are also excreted in small amounts in the breast milk of nursing mothers.
Because significant amounts of estrogens and their active metabolites are excreted in the bile and reabsorbed from the intestine, the resulting enterohepatic circulation ensures that orally administered estrogens will have a high ratio of hepatic to peripheral effects. As noted below, the hepatic effects are thought to be responsible for some undesirable actions such as synthesis of increased clotting factors and plasma renin substrate. The hepatic effects of estrogen can be minimized by routes that avoid first-pass liver exposure, ie, vaginal, transdermal, or by injection.
Physiologic Effects
A. Mechanism
Estrogens in the blood and interstitial fluid are bound to SHBG, from which they dissociate to cross the cell membrane, enter the nucleus, and bind to their receptor. Two genes code for two estrogen receptor isoforms, α and β, which are members of the superfamily of steroid, sterol, retinoic acid, and thyroid receptors. Unlike glucocorticoid receptors, estrogen receptors are found predominantly in the nucleus bound to heat shock proteins that stabilize them (see Figure 39–4).
Binding of the hormone to its receptor alters its conformation and releases it from the stabilizing proteins (predominantly Hsp90). The receptor-hormone complex forms dimers (usually ERα-ERα, ERβ-ERβ, or ERα-ERβ) that bind to a specific sequence of nucleotides, called estrogen response elements (EREs), in the regulatory regions of various genes and regulate their transcription. The ERE is composed of two half-sites arranged as a palindrome separated by a small group of nucleotides called the spacer. The interaction of a receptor dimer with the ERE also involves a number of nuclear proteins, the coregulators, as well as components of the transcription machinery. Complex interactions with various coregulators appear to be responsible for some of the tissue-specific effects that govern the actions of selective estrogen receptor modulators (SERMs, see below). The receptor may also bind to other transcription factors to influence the effects of these factors on their responsive genes. Interestingly, although ERβ has its own separate actions from ERα, it also acts as a dominant negative inhibitor of ERα. Thus, while ERα has many growth-promoting properties, ERβ has antigrowth effects. Many phytoestrogens act via the ERβ protecting cells from the pro-growth effects of ERα.
The relative concentrations and types of receptors, receptor coregulators, and transcription factors confer the cell specificity of the hormone’s actions. The genomic effects of estrogens are mainly due to proteins synthesized by translation of RNA transcribed from a responsive gene. Some of the effects of estrogens are indirect, mediated by the autocrine and paracrine actions of autacoids such as growth factors, lipids, glycolipids, and cytokines produced by the target cells in response to estrogen.
Rapid estrogen-induced effects such as granulosa cell Ca2+ uptake and increased uterine blood flow do not require gene activation. These appear to be mediated by nongenomic effects of the classic estrogen receptor-estrogen complex, influencing several intracellular signaling pathways.
Recently, all steroid receptors except the mineralocorticoid receptors were shown to have palmitoylation motifs that allow enzymatic addition of palmitate and increased localization of the receptors in the vicinity of plasma membranes. Such receptors are available for direct interactions with, and effects on, various membrane-associated or cytoplasmic proteins without the need for entry into the nucleus and induction of transcriptional actions.
B. Female Maturation
Estrogens are required for the normal sexual maturation and growth of the female. They stimulate the development of the vagina, uterus, and uterine tubes as well as the secondary sex characteristics. They stimulate stromal development and ductal growth in the breast and are responsible for the accelerated growth phase and the closing of the epiphyses of the long bones that occur at puberty. They contribute to the growth of axillary and pubic hair and alter the distribution of body fat to produce typical female body contours. Larger quantities also stimulate development of pigmentation in the skin, most prominent in the region of the nipples and areolae and in the genital region.
C. Endometrial Effects
In addition to its growth effects on uterine muscle, estrogen plays an important role in the development of the endometrial lining. When estrogen production is properly coordinated with the production of progesterone during the normal human menstrual cycle, regular periodic bleeding and shedding of the endometrial lining occur. Continuous exposure to estrogens for prolonged periods leads to hyperplasia of the endometrium that is usually associated with abnormal bleeding patterns.
D. Metabolic and Cardiovascular Effects
Estrogens have a number of important metabolic and cardiovascular effects. They seem to be partially responsible for maintenance of the normal structure and function of the skin and blood vessels in women. Estrogens also decrease the rate of resorption of bone by promoting the apoptosis of osteoclasts and by antagonizing the osteoclastogenic and pro-osteoclastic effects of parathyroid hormone and interleukin-6. Estrogens also stimulate adipose tissue production of leptin and are in part responsible for the higher levels of this hormone in women than in men.
In addition to stimulating the synthesis of enzymes and growth factors leading to uterine and breast growth and differentiation, estrogens alter the production and activity of many other proteins in the body. Metabolic alterations in the liver are especially important, so that there is a higher circulating level of proteins such as transcortin (corticosteroid-binding globulin [CBG]), thyroxine-binding globulin (TBG), SHBG, transferrin, renin substrate, and fibrinogen. This leads to increased circulating levels of thyroxine, estrogen, testosterone, iron, copper, and other substances.
Alterations in the composition of the plasma lipids caused by estrogens are characterized by an increase in the high-density lipoproteins (HDL), a slight reduction in the low-density lipoproteins (LDL), and a reduction in total plasma cholesterol levels. Plasma triglyceride levels are increased. Estrogens decrease hepatic oxidation of adipose tissue lipid to ketones and increase synthesis of triglycerides.
E. Effects on Blood Coagulation
Estrogens enhance the coagulability of blood. Many changes in factors influencing coagulation have been reported, including increased circulating levels of factors II, VII, IX, and X and decreased antithrombin III, partially as a result of the hepatic effects mentioned above. Increased plasminogen levels and decreased platelet adhesiveness have also been found (see Hormonal Contraception, below).
F. Other Effects
Estrogens induce the synthesis of progesterone receptors. They are responsible for estrous behavior in animals and may influence behavior and libido in humans. Administration of estrogens stimulates central components of the stress system, including the production of corticotropin-releasing hormone and the activity of the sympathetic system, and promotes a sense of well-being when given to women who are estrogen-deficient. They also facilitate the loss of intravascular fluid into the extracellular space, producing edema. The resulting decrease in plasma volume causes a compensatory retention of sodium and water by the kidney. Estrogens also modulate sympathetic nervous system control of smooth muscle function.
Clinical Uses*
A. Primary Hypogonadism
Estrogens have been used extensively for replacement therapy in estrogen-deficient patients. The estrogen deficiency may be due to primary failure of development of the ovaries, premature menopause, castration, or menopause.
Treatment of primary hypogonadism is usually begun at 11–13 years of age in order to stimulate the development of secondary sex characteristics and menses, to stimulate optimal growth, to prevent osteoporosis, and to avoid the psychological consequences of delayed puberty and estrogen deficiency. Treatment attempts to mimic the physiology of puberty. It is initiated with small doses of estrogen (0.3 mg conjugated estrogens or 5–10 mcg ethinyl estradiol) on days 1–21 each month and is slowly increased to adult doses and then maintained until the age of menopause (approximately 51 years of age). A progestin is added after the first uterine bleeding. When growth is completed, chronic therapy consists mainly of the administration of adult doses of both estrogens and progestins, as described below.
B. Postmenopausal Hormonal Therapy
In addition to the signs and symptoms that follow closely upon the cessation of normal ovarian function—such as loss of menstrual periods, vasomotor symptoms, sleep disturbances, and genital atrophy—there are longer-lasting changes that influence the health and well-being of postmenopausal women. These include an acceleration of bone loss, which in susceptible women may lead to vertebral, hip, and wrist fractures; and lipid changes, which may contribute to the acceleration of atherosclerotic cardiovascular disease noted in postmenopausal women. The effects of estrogens on bone have been extensively studied, and the effects of hormone withdrawal have been well-characterized. However, the role of estrogens and progestins in the cause and prevention of cardiovascular disease, which is responsible for 350,000 deaths per year, and breast cancer, which causes 35,000 deaths per year, is less well understood.
When normal ovulatory function ceases and the estrogen levels fall after menopause, oophorectomy, or premature ovarian failure, there is an accelerated rise in plasma cholesterol and LDL concentrations, while LDL receptors decline. HDL is not much affected, and levels remain higher than in men. Very-low-density lipoprotein and triglyceride levels are also relatively unaffected. Since cardiovascular disorders account for most deaths in this age group, the risk for these disorders constitutes a major consideration in deciding whether or not hormonal “replacement” therapy (HRT, also correctly called HT) is indicated and influences the selection of hormones to be administered. Estrogen replacement therapy has a beneficial effect on circulating lipids and lipoproteins, and this was earlier thought to be accompanied by a reduction in myocardial infarction by about 50% and of fatal strokes by as much as 40%. These findings, however, have been disputed by the results of a large study from the Women’s Health Initiative (WHI) project showing no cardiovascular benefit from estrogen plus progestin replacement therapy in perimenopausal or older postmenopausal patients. In fact, there may be a small increase in cardiovascular problems as well as breast cancer in women who received the replacement therapy. Interestingly, a small protective effect against colon cancer was observed. Although current clinical guidelines do not recommend routine hormone therapy in postmenopausal women, the validity of the WHI report has been questioned. In any case, there is no increased risk for breast cancer if therapy is given immediately after menopause and for the first 7 years, while the cardiovascular risk depends on the degree of atherosclerosis at the onset of therapy. Transdermal or vaginal administration of estrogen may be associated with decreased cardiovascular risk because it bypasses the liver circulation. Women with premature menopause should definitely receive hormone therapy.
In some studies, a protective effect of estrogen replacement therapy against Alzheimer’s disease was observed. However, several other studies have not supported these results.
Progestins antagonize estrogen’s effects on LDL and HDL to a variable extent. However, one large study has shown that the addition of a progestin to estrogen replacement therapy does not influence the cardiovascular risk.
Optimal management of the postmenopausal patient requires careful assessment of her symptoms as well as consideration of her age and the presence of (or risks for) cardiovascular disease, osteoporosis, breast cancer, and endometrial cancer. Bearing in mind the effects of the gonadal hormones on each of these disorders, the goals of therapy can then be defined and the risks of therapy assessed and discussed with the patient.
If the main indication for therapy is hot flushes and sleep disturbances, therapy with the lowest dose of estrogen required for symptomatic relief is recommended. Treatment may be required for only a limited period of time and the possible increased risk for breast cancer avoided. In women who have undergone hysterectomy, estrogens alone can be given 5 days per week or continuously, since progestins are not required to reduce the risk for endometrial hyperplasia and cancer. Hot flushes, sweating, insomnia, and atrophic vaginitis are generally relieved by estrogens; many patients experience some increased sense of well-being; and climacteric depression and other psychopathologic states are improved.
The role of estrogens in the prevention and treatment of osteoporosis has been carefully studied (see Chapter 42). The amount of bone present in the body is maximal in the young active adult in the third decade of life and begins to decline more rapidly in middle age in both men and women. The development of osteoporosis also depends on the amount of bone present at the start of this process, on vitamin D and calcium intake, and on the degree of physical activity. The risk of osteoporosis is highest in smokers who are thin, Caucasian, and inactive and have a low calcium intake and a strong family history of osteoporosis. Depression also is a major risk factor for development of osteoporosis in women.
Estrogens should be used in the smallest dosage consistent with relief of symptoms. In women who have not undergone hysterectomy, it is most convenient to prescribe estrogen on the first 21–25 days of each month. The recommended dosages of estrogen are 0.3–1.25 mg/d of conjugated estrogen or 0.01–0.02 mg/d of ethinyl estradiol. Dosages in the middle of these ranges have been shown to be maximally effective in preventing the decrease in bone density occurring at menopause. From this point of view, it is important to begin therapy as soon as possible after the menopause for maximum effect. In these patients and others not taking estrogen, calcium supplements that bring the total daily calcium intake up to 1500 mg are useful.
Patients at low risk of developing osteoporosis who manifest only mild atrophic vaginitis can be treated with topical preparations. The vaginal route of application is also useful in the treatment of urinary tract symptoms in these patients. It is important to realize, however, that although locally administered estrogens escape the first-pass effect (so that some undesirable hepatic effects are reduced), they are almost completely absorbed into the circulation, and these preparations should be given cyclically.
As noted below, the administration of estrogen is associated with an increased risk of endometrial carcinoma. The administration of a progestational agent with the estrogen prevents endometrial hyperplasia and markedly reduces the risk of this cancer. When estrogen is given for the first 25 days of the month and the progestin medroxyprogesterone (10 mg/d) is added during the last 10–14 days, the risk is only half of that in women not receiving hormone replacement therapy. On this regimen, some women will experience a return of symptoms during the period off estrogen administration. In these patients, the estrogen can be given continuously. If the progestin produces sedation or other undesirable effects, its dose can be reduced to 2.5–5 mg for the last 10 days of the cycle with a slight increase in the risk for endometrial hyperplasia. These regimens are usually accompanied by bleeding at the end of each cycle. Some women experience migraine headaches during the last few days of the cycle. The use of a continuous estrogen regimen will often prevent their occurrence. Women who object to the cyclic bleeding associated with sequential therapy can also consider continuous therapy. Daily therapy with 0.625 mg of conjugated equine estrogens and 2.5–5 mg of medroxyprogesterone will eliminate cyclic bleeding, control vasomotor symptoms, prevent genital atrophy, maintain bone density, and show a favorable lipid profile with a small decrease in LDL and an increase in HDL concentrations. These women have endometrial atrophy on biopsy. About half of these patients experience breakthrough bleeding during the first few months of therapy. Seventy to 80 percent become amenorrheic after the first 4 months, and most remain so. The main disadvantage of continuous therapy is the need for uterine biopsy if bleeding occurs after the first few months.
As noted above, estrogens may also be administered vaginally or transdermally. When estrogens are given by these routes, the liver is bypassed on the first circulation, and the ratio of the liver effects to peripheral effects is reduced.
In patients in whom estrogen replacement therapy is contraindicated, such as those with estrogen-sensitive tumors, relief of vasomotor symptoms may be obtained by the use of clonidine.
C. Other Uses
Estrogens combined with progestins can be used to suppress ovulation in patients with intractable dysmenorrhea or when suppression of ovarian function is used in the treatment of hirsutism and amenorrhea due to excessive secretion of androgens by the ovary. Under these circumstances, greater suppression may be needed, and oral contraceptives containing 50 mcg of estrogen or a combination of a low estrogen pill with GnRH suppression may be required.
Adverse Effects
Adverse effects of variable severity have been reported with the therapeutic use of estrogens. Many other effects reported in conjunction with hormonal contraceptives may be related to their estrogen content. These are discussed below.
A. Uterine Bleeding
Estrogen therapy is a major cause of postmenopausal uterine bleeding. Unfortunately, vaginal bleeding at this time of life may also be due to carcinoma of the endometrium. To avoid confusion, patients should be treated with the smallest amount of estrogen possible. It should be given cyclically so that bleeding, if it occurs, will be more likely to occur during the withdrawal period. As noted above, endometrial hyperplasia can be prevented by administration of a progestational agent with estrogen in each cycle.
B. Cancer
The relation of estrogen therapy to cancer continues to be the subject of active investigation. Although no adverse effect of short-term estrogen therapy on the incidence of breast cancer has been demonstrated, a small increase in the incidence of this tumor may occur with prolonged therapy. Although the risk factor is small (1.25), the impact may be great since this tumor occurs in 10% of women, and addition of progesterone does not confer a protective effect. Studies indicate that following unilateral excision of breast cancer, women receiving tamoxifen (an estrogen partial agonist, see below) show a 35% decrease in contralateral breast cancer compared with controls. These studies also demonstrate that tamoxifen is well tolerated by most patients, produces estrogen-like alterations in plasma lipid levels, and stabilizes bone mineral loss. Studies bearing on the possible efficacy of tamoxifen and raloxifene in postmenopausal women at high risk for breast cancer show decreases of risk for at least 5 years, but of unknown further duration. A recent study shows that postmenopausal hormone replacement therapy with estrogens plus progestins was associated with greater breast epithelial cell proliferation and breast epithelial cell density than estrogens alone or no replacement therapy. Furthermore, with estrogens plus progestins, breast proliferation was localized to the terminal duct-lobular unit of the breast, which is the main site of development of breast cancer. Thus, further studies are needed to conclusively assess the possible association between progestins and breast cancer risk.
Many studies show an increased risk of endometrial carcinoma in patients taking estrogens alone. The risk seems to vary with the dose and duration of treatment: 15 times greater in patients taking large doses of estrogen for 5 or more years, in contrast with two to four times greater in patients receiving lower doses for short periods. However, as noted above, the concomitant use of a progestin prevents this increased risk and may in fact reduce the incidence of endometrial cancer to less than that in the general population.
There have been a number of reports of adenocarcinoma of the vagina in young women whose mothers were treated with large doses of diethylstilbestrol early in pregnancy. These cancers are most common in young women (ages 14–44). The incidence is less than 1 per 1000 women exposed—too low to establish a cause-and-effect relationship with certainty. However, the risks for infertility, ectopic pregnancy, and premature delivery are also increased. It is now recognized that there is no indication for the use of diethylstilbestrol during pregnancy, and it should be avoided. It is not known whether other estrogens have a similar effect or whether the observed phenomena are peculiar to diethylstilbestrol. This agent should be used only in the treatment of cancer (eg, of the prostate) or as a “morning after” contraceptive (see page 712).
C. Other Effects
Nausea and breast tenderness are common and can be minimized by using the smallest effective dose of estrogen. Hyperpigmentation also occurs. Estrogen therapy is associated with an increase in frequency of migraine headaches as well as cholestasis, gallbladder disease, and hypertension.
Contraindications
Estrogens should not be used in patients with estrogen-dependent neoplasms such as carcinoma of the endometrium or in those with—or at high risk for—carcinoma of the breast. They should be avoided in patients with undiagnosed genital bleeding, liver disease, or a history of thromboembolic disorder. In addition, the use of estrogens should be avoided by heavy smokers.
Preparations & Dosages
The dosages of commonly used natural and synthetic preparations are listed in Table 40–1. Although all of the estrogens produce almost the same hormonal effects, their potencies vary both between agents and depending on the route of administration. As noted above, estradiol is the most active endogenous estrogen, and it has the highest affinity for the estrogen receptor. However, its metabolites estrone and estriol have weak uterine effects.
For a given level of gonadotropin suppression, oral estrogen preparations have more effect on the circulating levels of CBG, SHBG, and a host of other liver proteins, including angiotensinogen, than do transdermal preparations. The oral route of administration allows greater concentrations of hormone to reach the liver, thus increasing the synthesis of these proteins. Transdermal preparations were developed to avoid this effect. When administered transdermally, 50–100 mcg of estradiol has effects similar to those of 0.625–1.25 mg of conjugated oral estrogens on gonadotropin concentrations, endometrium, and vaginal epithelium. Furthermore, the transdermal estrogen preparations do not significantly increase the concentrations of renin substrate, CBG, and TBG and do not produce the characteristic changes in serum lipids. Combined oral preparations containing 0.625 mg of conjugated estrogens and 2.5 mg of medroxyprogesterone acetate are available for menopausal replacement therapy. Tablets containing 0.625 mg of conjugated estrogens and 5 mg of medroxyprogesterone acetate are available to be used in conjunction with conjugated estrogens in a sequential fashion. Estrogens alone are taken on days 1–14 and the combination on days 15–28.
THE PROGESTINS
Natural Progestins: Progesterone
Progesterone is the most important progestin in humans. In addition to having important hormonal effects, it serves as a precursor to the estrogens, androgens, and adrenocortical steroids. It is synthesized in the ovary, testis, and adrenal cortex from circulating cholesterol. Large amounts are also synthesized and released by the placenta during pregnancy.
In the ovary, progesterone is produced primarily by the corpus luteum. Normal males appear to secrete 1–5 mg of progesterone daily, resulting in plasma levels of about 0.03 mcg/dL. The level is only slightly higher in the female during the follicular phase of the cycle, when only a few milligrams per day of progesterone are secreted. During the luteal phase, plasma levels range from 0.5 mcg/dL to more than 2 mcg/dL (Figure 40–1). Plasma levels of progesterone are further elevated and reach their peak levels in the third trimester of pregnancy.
Synthetic Progestins
A variety of progestational compounds have been synthesized. Some are active when given by mouth. They are not a uniform group of compounds, and all of them differ from progesterone in one or more respects. Table 40–2 lists some of these compounds and their effects. In general, the 21-carbon compounds (hydroxyprogesterone, medroxyprogesterone, megestrol, and dimethisterone) are the most closely related, pharmacologically as well as chemically, to progesterone. A new group of third-generation synthetic progestins has been introduced, principally as components of oral contraceptives. These “19-nor, 13-ethyl” steroid compounds include desogestrel (Figure 40–4), gestodene, and norgestimate. They are claimed to have lower androgenic activity than older synthetic progestins.
TABLE 40–2 Properties of some progestational agents.
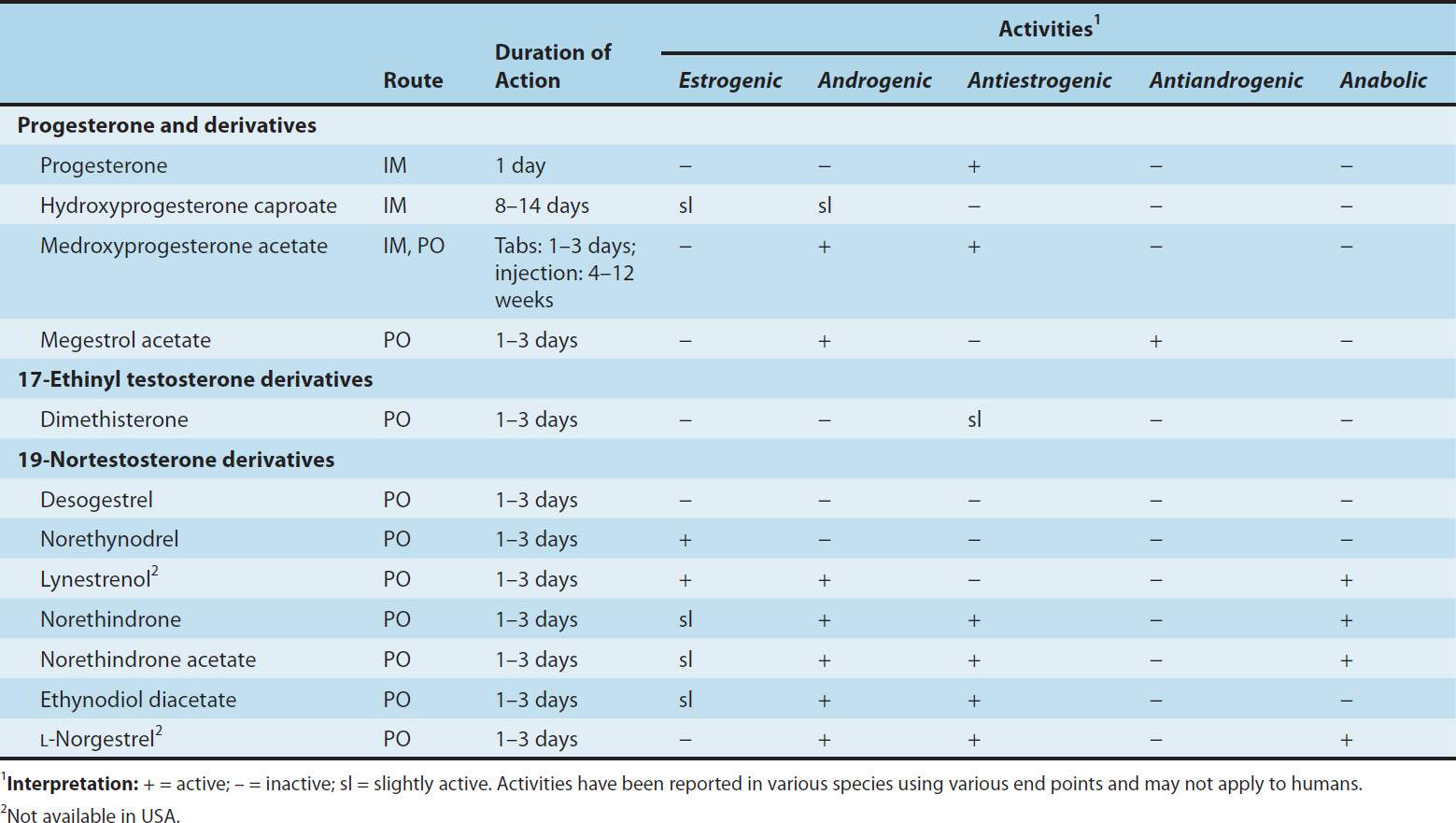
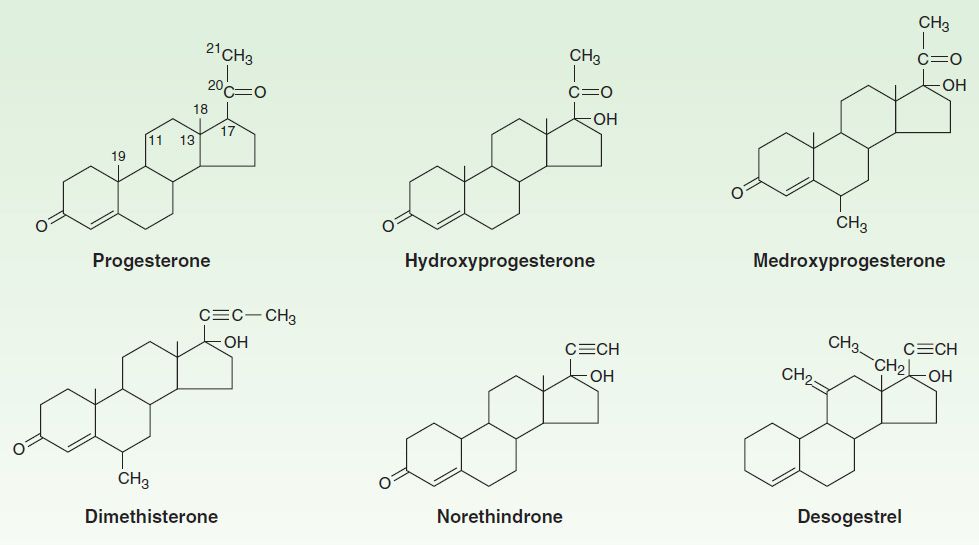
FIGURE 40–4 Progesterone and some progestational agents in clinical use.
Pharmacokinetics
Progesterone is rapidly absorbed following administration by any route. Its half-life in the plasma is approximately 5 minutes, and small amounts are stored temporarily in body fat. It is almost completely metabolized in one passage through the liver, and for that reason it is quite ineffective when the usual formulation is administered orally. However, high-dose oral micronized progesterone preparations have been developed that provide adequate progestational effect.
In the liver, progesterone is metabolized to pregnanediol and conjugated with glucuronic acid. It is excreted into the urine as pregnanediol glucuronide. The amount of pregnanediol in the urine has been used as an index of progesterone secretion. This measure has been very useful despite the fact that the proportion of secreted progesterone converted to this compound varies from day to day and from individual to individual. In addition to progesterone, 20α- and 20β-hydroxyprogesterone (20α- and 20β-hydroxy-4-pregnene-3-one) are also found. These compounds have about one fifth the progestational activity of progesterone in humans and other species. Little is known of their physiologic role, but 20α-hydroxyprogesterone is produced in large amounts in some species and may be of some importance biologically.
The usual routes of administration and durations of action of the synthetic progestins are listed in Table 40–2. Most of these agents are extensively metabolized to inactive products that are excreted mainly in the urine.
Physiologic Effects
A. Mechanism
The mechanism of action of progesterone—described in more detail above—is similar to that of other steroid hormones. Progestins enter the cell and bind to progesterone receptors that are distributed between the nucleus and the cytoplasm. The ligand-receptor complex binds to a progesterone response element (PRE) to activate gene transcription. The response element for progesterone appears to be similar to the corticosteroid response element, and the specificity of the response depends upon which receptor is present in the cell as well as upon other cell-specific receptor coregulators and interacting transcription factors. The progesterone-receptor complex forms a dimer before binding to DNA. Like the estrogen receptor, it can form heterodimers as well as homodimers between two isoforms, A and B. These isoforms are produced by alternative splicing of the same gene.
B. Effects of Progesterone
Progesterone has little effect on protein metabolism. It stimulates lipoprotein lipase activity and seems to favor fat deposition. The effects on carbohydrate metabolism are more marked. Progesterone increases basal insulin levels and the insulin response to glucose. There is usually no manifest change in carbohydrate tolerance. In the liver, progesterone promotes glycogen storage, possibly by facilitating the effect of insulin. Progesterone also promotes ketogenesis.
Progesterone can compete with aldosterone for the mineralocorticoid receptor of the renal tubule, causing a decrease in Na+ reabsorption. This leads to an increased secretion of aldosterone by the adrenal cortex (eg, in pregnancy). Progesterone increases body temperature in humans. The mechanism of this effect is not known, but an alteration of the temperature-regulating centers in the hypothalamus has been suggested. Progesterone also alters the function of the respiratory centers. The ventilatory response to CO2 is increased by progesterone but synthetic progestins with an ethinyl group do not have respiratory effects. This leads to a measurable reduction in arterial and alveolar PCO2 during pregnancy and in the luteal phase of the menstrual cycle. Progesterone and related steroids also have depressant and hypnotic effects on the brain.
Progesterone is responsible for the alveolobular development of the secretory apparatus in the breast. It also participates in the preovulatory LH surge and causes the maturation and secretory changes in the endometrium that are seen following ovulation (Figure 40–1).
Progesterone decreases the plasma levels of many amino acids and leads to increased urinary nitrogen excretion. It induces changes in the structure and function of smooth endoplasmic reticulum in experimental animals.
Other effects of progesterone and its analogs are noted below in the section, Hormonal Contraception.
C. Synthetic Progestins
The 21-carbon progesterone analogs antagonize aldosterone-induced sodium retention (see above). The remaining compounds (“19-nortestosterone” third-generation agents) produce a decidual change in the endometrial stroma, do not support pregnancy in test animals, are more effective gonadotropin inhibitors, and may have minimal estrogenic and androgenic or anabolic activity (Table 40–2; Figure 40–4). They are sometimes referred to as “impeded androgens.” Progestins without androgenic activity include desogestrel, norgestimate, and gestodene. The first two of these compounds are dispensed in combination with ethinyl estradiol for oral contraception (Table 40–3) in the USA. Oral contraceptives containing the progestins cyproterone acetate (also an antiandrogen) in combination with ethinyl estradiol are investigational in the USA.
TABLE 40–3 Some oral and implantable contraceptive agents in use.1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


