Jerry Avorn

Because medications act by interfering with one or more aspects of molecular and cellular function, it is difficult to do so without also causing an undesirable effect either by that perturbation or by another (perhaps unexpected) drug action. Because all drugs have risks, the goal of pharmacotherapy cannot be to prescribe a risk-free regimen. Instead, it is to ensure that the risks of drug therapy are as low as possible and are acceptable in the context of a medication’s clinical benefit.
Some adverse effects of a drug are apparent during its early development and often result from the same on-target mechanism responsible for its therapeutic effect (e.g., cytotoxic cancer chemotherapy). Even in such situations, however, it is necessary to know how those expected adverse effects will be manifested when the drug is in routine use—in terms of both their frequency and their severity. After a drug has been approved for clinical use, the goal becomes detecting and quantifying the risks as quickly and rigorously as possible.
Serious or even life-threatening adverse effects have led to the withdrawal of widely used drugs. This has heightened the sensitivity of clinicians and patients to the growing field of pharmacoepidemiology—the measurement of drug effects in large, “real-world” populations of patients. Advances in informatics and analytic techniques in this field hold promise for enhancing our understanding of drug risks so that they can be better understood and managed, with the goal of putting a drug’s benefits into context and guiding clinical decision making and regulatory action.
 Mr. Keeley is a 67-year-old man with severe degenerative joint disease of both hips. He brings to his doctor several magazine ads and newspaper clippings that describe the nonsteroidal anti-inflammatory drug rofecoxib. These ads claim that rofecoxib provides excellent relief of arthritic pain with a lower risk of gastrointestinal toxicity. Rofecoxib selectively inhibits cyclooxygenase-2, which mediates pain and inflammation, rather than cyclooxygenase-1, which maintains gastrointestinal mucosal integrity and whose inhibition can cause gastrointestinal bleeding. This drug is widely hailed as a “super-aspirin” with minimal gastrointestinal toxicity and is heavily promoted. Mr. Keeley’s physician decides to prescribe rofecoxib, and the patient reports that it works better than the acetaminophen he had previously been using.
Mr. Keeley is a 67-year-old man with severe degenerative joint disease of both hips. He brings to his doctor several magazine ads and newspaper clippings that describe the nonsteroidal anti-inflammatory drug rofecoxib. These ads claim that rofecoxib provides excellent relief of arthritic pain with a lower risk of gastrointestinal toxicity. Rofecoxib selectively inhibits cyclooxygenase-2, which mediates pain and inflammation, rather than cyclooxygenase-1, which maintains gastrointestinal mucosal integrity and whose inhibition can cause gastrointestinal bleeding. This drug is widely hailed as a “super-aspirin” with minimal gastrointestinal toxicity and is heavily promoted. Mr. Keeley’s physician decides to prescribe rofecoxib, and the patient reports that it works better than the acetaminophen he had previously been using.
The patient continues to experience good relief of his arthritic pain. In the ensuing months, he develops mild hypertension that is easily managed with a thiazide medication but is otherwise well. Nine months after Mr. Keeley begins taking rofecoxib, his wife calls to report that her husband has been hospitalized with a myocardial infarction. He survives several episodes of arrhythmia and cardiogenic shock and is discharged to home. His physician is not surprised at this report, since the patient was an active smoker, had elevated serum cholesterol, and has a recent diagnosis of hypertension.
Two years after Mr. Keeley’s myocardial infarction, rofecoxib is withdrawn from the market when a randomized controlled trial reveals that rofecoxib nearly doubles the risk of myocardial infarction and stroke.
Questions
1. How do regulatory agencies such as the US Food and Drug Administration assess the safety of medications before they are approved?
2. How do physicians, patients, and the FDA learn about the adverse effects of drugs once they are in widespread use?
3. How can observational studies be used to determine the adverse effects of drugs that are in widespread use?
4. What issues must be considered in interpreting and acting on the results of such analyses?
 CHALLENGES IN THE ASCERTAINMENT OF DRUG SAFETY
CHALLENGES IN THE ASCERTAINMENT OF DRUG SAFETY
The randomized controlled trial (RCT) is the gold standard for determining the efficacy of a drug and is the main criterion used by regulatory agencies, such as the US Food and Drug Administration (FDA), in deciding whether to approve a new medication for use. But this valuable tool also has limits, and it is important to understand those limits when assessing the benefits and risks of a given agent.
Study Size and Generalizability
Compared to the number of patients who eventually use a drug, the number of subjects in clinical trials supporting the approval of that drug is relatively modest. Approval decisions are generally made on the basis of trials that include 2,000–4,000 participants, or fewer for rare conditions. If a particular adverse event occurs just once in every 1,000 patients, it may not occur at all during clinical trials, or if it does occur, it may be difficult or impossible to determine whether its rate of occurrence is meaningfully higher among study subjects compared to controls. One in 1,000 may seem like a rare event, but if 10 million people take a drug each year, that rate would result in 10,000 occurrences of the adverse event annually. For a life-threatening adverse effect such as fulminant hepatotoxicity, this could have important clinical and public health consequences.
Subjects in clinical trials of new drugs are nearly always volunteers—people who have come forward to participate in medical research and have given their informed consent to take part in the study. There is ample evidence that such people tend to be different from typical patients who will receive the drug when it is in routine use; study subjects tend to be younger, healthier, better educated, and of higher socioeconomic status. This problem is often exacerbated by strict exclusion criteria in preapproval study protocols. Some of these exclusions prohibit participation of patients over a given age cutoff (such as 65 or 70), even if the drug is expected to be used disproportionately by the elderly. Other entry criteria may exclude patients who have important comorbidities in addition to the disease being studied (thereby also excluding those who are taking multiple other medications). While this may be the “cleanest” way to test the efficacy of a new agent, there is growing concern that the data thus generated have limited generalizability to the populations who ultimately use these medications. Other kinds of patients may be excluded for unassailable ethical reasons, such as not allowing pregnant women or children into most preapproval drug trials. However, when such patients then take these drugs in routine care, there is little information to guide their use.
By definition, clinical trials are conducted by physicians and support staff who have experience in clinical research and who work in settings accustomed to such activities. Their actions are guided by study protocols that often require close monitoring for adverse effects as well as efficacy. Such protocols also ensure that patients are taking the prescribed product as directed. This, too, is far different from routine care in typical settings, in which both patient adherence and the intensity of surveillance for early detection of adverse events are generally lower.
Surrogate Outcomes and Comparators
It would be difficult to postpone the approval of every new antihypertensive drug until it had been shown to reduce stroke rates, or not to allow a new statin lipid-lowering drug on the market until it had been shown to prevent myocardial infarctions. Such a requirement could delay the availability of potentially useful new therapies, as well as further increase their cost. As a consequence, new products may be approved on the basis of their effect on “surrogate outcomes,” such as blood pressure for antihypertensives, hemoglobin A1c level for drugs used to manage diabetes, serum LDL cholesterol level for statins, intraocular pressure for drugs used to treat glaucoma, or biomarkers of tumor growth for oncology therapies. While such a metric can be useful in making drug approval quicker and more efficient, its utility depends on the association between the surrogate marker and the clinical outcome of concern. These may correlate well, but not always. For example, the antiarrhythmics encainide and flecainide reduced the surrogate outcome of ventricular ectopy after myocardial infarction, but a larger study (the CAST trial) demonstrated that they actually increased mortality in such patients, despite their success in “treating” the surrogate marker. Similarly, rosiglitazone (Avandia®) was approved based on its capacity to reduce hemoglobin A1c levels in preapproval trials. However, once the drug was in widespread use, meta-analysis of those trials found that it increased the risk of myocardial infarction.
When feasible, placebos are the comparison treatment preferred by manufacturers and the FDA for premarket trials used to determine approval. Such comparisons provide the clearest contrasts and the most straightforward statistical analysis, and there is no possibility of confusion resulting from therapeutic or adverse events caused by an active agent used in the control group. Placebo controls also facilitate the approval of new products whose efficacy is similar to that of existing drugs; performing “equivalency” or “noninferiority” studies against active therapies requires larger numbers of patients and is more demanding statistically. If it is ethically or pragmatically impossible to conduct placebo-controlled trials (e.g., with a new AIDS drug or an antibiotic for a serious bacterial infection), then an active comparator is used.
However, while the “better than placebo” comparison may be sufficient for a manufacturer to meet the FDA’s legal requirements for drug approval, the data it yields often fall short of what the clinician, patient, or payor needs to know about a new drug’s safety or comparative effectiveness. A new drug may work better than placebo, but is it better than an existing treatment the physician may choose instead? Or is it even as good? The new drug may produce a serious adverse effect (e.g., rhabdomyolysis with a statin), but is the rate of occurrence of the effect higher or lower than that seen with older therapies? And even if it poses a higher risk of a given adverse effect, does the new drug also provide greater efficacy (in this case, prevention of ischemic cardiac events)? If so, the trade-off might possibly be acceptable; if not, it would not be. But if no such comparative data exist, the question cannot even be considered.
Duration and Post-Approval Studies
The duration of efficacy trials for certain new drugs can be as short as 8–16 weeks, if the comparator is placebo and surrogate endpoints are used to meet a legal definition of efficacy. However, such short-term trials may yield little useful information about benefits and risks that occur beyond this time frame. The FDA requires a minimum of 6 months of safety testing for a new drug that is designed for chronic use (where chronic is defined as any period longer than 6 months), although even this duration of safety testing may be too short for a chronically administered medication that may be taken for many years.
In approving a new drug for widespread use, the FDA may ask the manufacturer to conduct additional postmarketing studies (phase IV studies) to address questions that were not resolved by the evidence submitted prior to approval. Sometimes, useful new data about a drug’s benefits and risks are obtained in this way. But until 2007, the agency had little authority to oblige a drug’s sponsor to complete these studies, since its main regulatory power, once a drug had been approved, had been confined to the “nuclear option” of threatening to take it off the market—an action that was often not possible in the absence of additional data. Each year, the agency reports how well such “postmarketing commitments” are being met by manufacturers. A report by the Government Accountability Office noted that up to half of the “mandated” postmarketing safety studies requested by the agency had not been initiated, even years after the drugs had entered widespread use. Concern about these problems was intensified by public concern over several prominent drug safety problems, particularly rofecoxib (Vioxx®). The drug had been used widely for 5 years before it was withdrawn from the market following a study demonstrating that it nearly doubled the risk of myocardial infarction or stroke. A 2006 report by the Institute of Medicine recommended sweeping changes in the way the FDA addresses drug safety (see below).

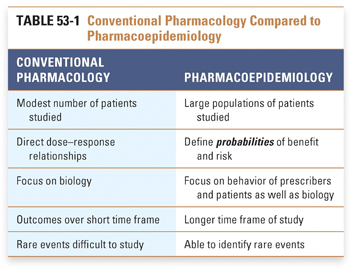
Pharmacoepidemiology is the study of drug outcomes as documented in observations of clinical data from large populations of typical patients receiving routine care. To understand this approach, it is necessary to think about drug effects in ways that are different from those of conventional pharmacology (Table 53-1). This perspective considers the population as the experimental system being studied. Medications can be considered to be variables introduced into this system much as they might be studied in an individual patient, in tissue culture, or in an isolated single-cell preparation. The differences are that, in populations, true randomization does not occur, the intervening decision making and behavior of doctors and patients can alter the drug’s effect, outcomes are measured in terms of probabilities (or rates) of events, and the magnitude of drug experience in the analysis is much larger than that of conventional pharmacology, ranging to millions of patients and millions of person-years of exposure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


