Elizabeth A. Brezinski, Lloyd B. Klickstein, and April W. Armstrong

Patients with autoimmune disease and patients who have received transplanted tissues or organs typically require therapy with immunosuppressive drugs. Immunosuppressive agents have been in use for more than 50 years, beginning with corticosteroids, antimetabolites, and alkylating agents. These early agents assisted in the treatment of previously incurable conditions, but their lack of specificity led to many serious adverse effects. Over the past 20 years, the field of immunosuppression has shifted to specific inhibitors of immunity that affect distinct immune pathways. This shift is important both because of the greater efficacy and reduced toxicity of these agents and because, as the mechanisms of these agents are discovered, insights are gained into the operation of the immune system.
 Mr. W is 59 years old when he undergoes right kidney transplantation in the spring of 2000 for chronic kidney failure resulting from long-standing uncontrolled hypertension. His induction immunosuppressant regimen consists of antithymocyte globulin. The antithymocyte globulin is co-administered with a glucocorticoid, an antihistamine, and an antipyretic. On postoperative day 1, he is transitioned to a maintenance immunosuppressant regimen of tacrolimus, mycophenolate mofetil, and prednisone. Progress during the first 2 months post-transplant is excellent, but then Mr. W develops headaches and laboratory testing shows an increase in his creatinine. The glucocorticoid dose is increased and his creatinine improves.
Mr. W is 59 years old when he undergoes right kidney transplantation in the spring of 2000 for chronic kidney failure resulting from long-standing uncontrolled hypertension. His induction immunosuppressant regimen consists of antithymocyte globulin. The antithymocyte globulin is co-administered with a glucocorticoid, an antihistamine, and an antipyretic. On postoperative day 1, he is transitioned to a maintenance immunosuppressant regimen of tacrolimus, mycophenolate mofetil, and prednisone. Progress during the first 2 months post-transplant is excellent, but then Mr. W develops headaches and laboratory testing shows an increase in his creatinine. The glucocorticoid dose is increased and his creatinine improves.
Three months after surgery, Mr. W is admitted to the hospital with fever, decreased urine output, and graft tenderness. Laboratory testing again shows increased serum creatinine and the tacrolimus serum level is within therapeutic limits. Infectious workup is negative and renal ultrasound shows no evidence of obstruction. A renal allograft biopsy demonstrates tubulitis and intimal arteritis, evidence of acute cellular rejection. He is treated with high-dose pulsed glucocorticoid therapy, which produces adverse effects of a metallic taste and abdominal discomfort. Mr. W’s renal function improves and he is discharged. Over the next few weeks, his steroid dose is slowly tapered down to his maintenance dose.
In December 2010, Mr. W arrives at clinic for his regular annual examination. He is in good health on a maintenance immunosuppressant regimen of tacrolimus, mycophenolate mofetil, and prednisone, and his monthly creatinine values have remained normal. There has been no evidence of rejection since 2000. Renal ultrasound shows remarkably normal graft structure, perhaps as a result of the aggressive hypertension management by his physicians. However, his glucose levels are consistently elevated on routine laboratory testing, an indication that he may have developed new-onset diabetes after transplantation. Because of his new-onset diabetes, Mr. W’s tacrolimus and prednisone doses are decreased and he is started on oral therapy to manage his diabetes. Over the next 2 years, his glucose and hemoglobin A1c levels remain at goal and he is able to enjoy time with his family.
Questions
1. How does each of the drugs prescribed for Mr. W reduce the likelihood of rejection?
2. Why is antithymocyte globulin administered with glucocorticoids, an antihistamine, and an antipyretic?
3. Why does Mr. W’s pulsed steroid dose need to be slowly tapered down to his maintenance dose level?
4. What is the likely cause of Mr. W’s new-onset diabetes? Why were his tacrolimus and prednisone doses decreased?

The first transplant performed successfully in humans was a kidney transplant between identical twins. No immunosuppression was used, and the individuals did well. Currently, most organ transplantation occurs between unrelated individuals, termed an allograft. Donor and recipient tissues express different major histocompatibility complex (MHC) class I molecules, one class of alloantigens, and recipient immune cells therefore recognize the transplanted tissues as foreign. This is termed alloimmunity, and it occurs when the recipient’s immune system attacks a transplanted organ. In the case of a bone marrow or stem cell transplant, graft-versus-host disease (GVHD) can result when donor lymphocytes mount an assault on recipient tissues.
Transplant rejection of solid organs can be divided into three major phases according to the time to onset. These phases, hyperacute, acute, and chronic rejection, are caused by different mechanisms and are therefore treated differently. The following three sections examine each of these processes, and Table 46-1 summarizes their differences.
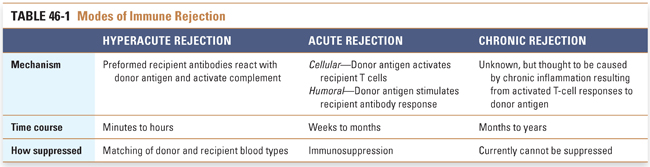
Hyperacute Rejection
Hyperacute rejection is mediated by preformed recipient antibodies against donor antigen. Because these antibodies are present at the time of organ implantation, hyperacute rejection occurs almost immediately after reperfusion of the transplanted organ. In fact, the surgeon can observe the changes in the organ minutes after restoration of blood flow. The normal, healthy, pink appearance of the transplanted organ rapidly becomes cyanotic, mottled, and flaccid. This rapid change is the result of complement activation by antibody binding to endothelial cells of the transplanted organ, resulting in thrombosis and ischemia. Most commonly, hyperacute rejection is mediated by recipient antibodies that react with blood group antigens in donor organs (e.g., type AB donor in a type O recipient). Matching of blood types between donor and recipient prevents hyperacute rejection; therefore, drug therapy for hyperacute rejection is typically not necessary. Hyperacute rejection also occurs in xenotransplantation (i.e., organ transplantation between species, such as a pig heart transplanted into a human recipient), due to the presence of preformed human antibodies that react against antigenic proteins and carbohydrates expressed by the donor species.
Acute Rejection
Acute rejection has cellular and humoral components. Acute cellular rejection is mediated by cytotoxic T cells and causes interstitial as well as vascular damage. This cellular response is most commonly seen in the initial months after transplantation. Immunosuppression of T cells is highly effective at preventing or limiting activation of the recipient immune system by the transplanted organ, thereby preventing acute cellular rejection. In acute humoral rejection, recipient B cells become sensitized to donor antigens in the transplanted organ and produce antibodies against these alloantigens after a period of 7–10 days. The antibody response is typically directed against endothelial cells and is thus also known as acute vascular rejection. Like acute cellular rejection, acute humoral rejection can usually be prevented by immunosuppression of the recipient after transplantation. Even with immunosuppression, however, episodes of acute rejection can occur months or even years after transplantation. Transplant recipients experiencing acute rejection are usually asymptomatic, and symptoms of fever or malaise are usually nonspecific.
Chronic Rejection
Chronic rejection is believed to be both cellular and humoral in nature and does not occur until months or years after transplantation. Because hyperacute and acute rejection are generally well controlled by donor/recipient matching and immunosuppressive therapy, chronic rejection is now the most common life-threatening pathology associated with organ transplantation.
Chronic rejection is thought to result from chronic inflammation caused by the response of activated recipient T cells to donor antigen. Activated T cells release cytokines that recruit macrophages into the graft. The macrophages induce chronic inflammation that leads to intimal proliferation of the vasculature and scarring of the graft tissue. The chronic changes eventually lead to irreversible organ failure. Other contributing nonimmune factors can include ischemia-reperfusion injury and infection.
No effective treatment regimens are currently available to eliminate chronic rejection. It is believed, however, that several experimental therapies have a reasonable chance of reducing chronic rejection. Especially promising is the possibility of developing tolerance through elimination of costimulation (see below).
Graft-Versus-Host Disease (GVHD)
Leukemia, primary immunodeficiency, and other conditions can be treated with bone marrow or peripheral stem cell transplantation. In this procedure, hematopoietic and immune function is restored after the patient’s bone marrow has been eradicated by aggressive chemotherapy and/or radiation therapy. GVHD is a major complication of allogeneic bone marrow or stem cell transplantation. GVHD is an alloimmune inflammatory reaction that occurs when transplanted immune cells attack the cells of the recipient. The severity of GVHD ranges from mild to life-threatening and typically involves the skin (rash), gastrointestinal tract (diarrhea), lungs (pneumonitis), and liver (veno-occlusive disease). GVHD can often be ameliorated by removing T cells from the donor bone marrow before transplantation. Mild to moderate GVHD can also be beneficial when donor immune cells attack recipient tumor cells that have survived the aggressive chemotherapy and radiation therapy. (In the case of leukemia, this is called the graft-versus-leukemia effect, or GVL.) Therefore, although removing donor T cells from the “graft” reduces the risk of GVHD, this may not be the best approach for marrow transplants used in antineoplastic therapy.
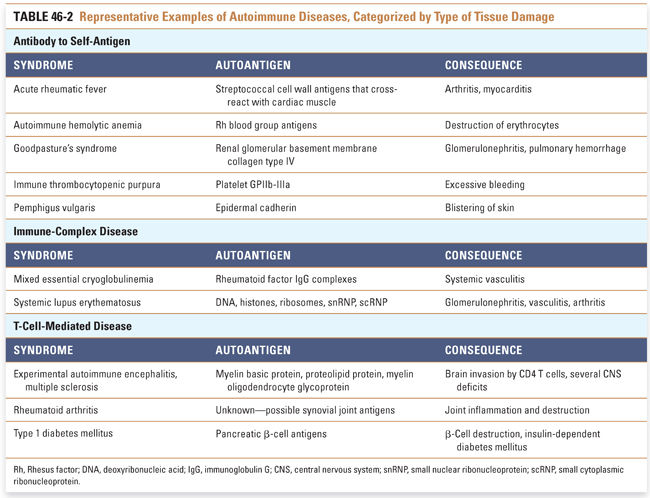
Autoimmune diseases occur when the host immune system attacks its own tissues, mistaking self-antigen for foreign. The typical result is chronic inflammation in the tissue(s) expressing the antigen.
Autoimmune diseases are most commonly due to a breakdown of self-tolerance, both central and peripheral. Central tolerance refers to the specific clonal deletion of autoreactive T and B cells during their development from precursor cells in the thymus (T cells) and bone marrow (B cells). Central tolerance ensures that the majority of immature autoreactive T and B cells do not develop into self-reactive clones. The thymus and bone marrow do not express every antigen in the body, however; a number of proteins are expressed only in specific tissues. For this reason, peripheral tolerance is also important. Peripheral tolerance results from deletion of autoreactive T cells by Fas-Fas ligand-mediated apoptosis, activation of T suppressor cells, or induction of T-cell anergy due to antigen presentation in the absence of costimulation.
Although breakdown in tolerance lies at the center of virtually all autoimmune diseases, the inciting stimulus leading to loss of tolerance is often unknown. Genetic factors may play a role, in that the presence of certain MHC subtypes may predispose T cells to the loss of self-tolerance. For example, human leukocyte antigen (HLA)-B27 is causally related to many forms of autoimmune spondylitis. Several other autoimmune diseases are linked to specific HLA loci, supporting an association, if not a causal role, for genetic predisposition to autoimmunity. Molecular mimicry, in which epitopes from infectious agents are similar to self-antigens, can also lead to a breakdown of tolerance and may be the mechanism underlying poststreptococcal glomerulonephritis. Several other processes, including failure of T-cell apoptosis, polyclonal lymphocyte activation, and exposure of cryptic self-antigens, have also been hypothesized to lead to autoimmunity. The details of these mechanisms are beyond the scope of this book; however, the result of each is a loss of tolerance.
Once self-tolerance has been compromised, the specific expression of autoimmunity can take one of three general forms (Table 46-2). In some diseases, production of autoantibodies against a specific antigen causes antibody-dependent opsonization of cells in the target organ, with subsequent cytotoxicity. One example is Goodpasture’s syndrome, which results from autoantibodies against collagen type IV in the renal glomerular basement membrane. In some autoimmune vasculitis syndromes, circulating antibody–antigen complexes deposit in blood vessels, causing inflammation and injury to the vessels. Two examples of immune complex disease are mixed essential cryoglobulinemia and systemic lupus erythematosus. Finally, T-cell-mediated diseases are caused by cytotoxic T cells that react with a specific self-antigen, resulting in destruction of the tissue(s) expressing that antigen. One example is type 1 diabetes mellitus, in which the cytotoxic T cells react against self-antigens in pancreatic β-cells.
The pharmacologic therapy for autoimmune diseases does not yet match the exquisite specificity of the offending biological process. Most currently available pharmacologic agents cause generalized immunosuppression and do not target the specific pathophysiology. Better understanding of the molecular pathways leading to autoimmune diseases should reveal new pharmacologic targets that can be used to suppress the specific autoimmune response before disease arises.
 PHARMACOLOGIC CLASSES AND AGENTS
PHARMACOLOGIC CLASSES AND AGENTS
Pharmacologic suppression of the immune system utilizes eight mechanistic approaches (Fig. 46-1):
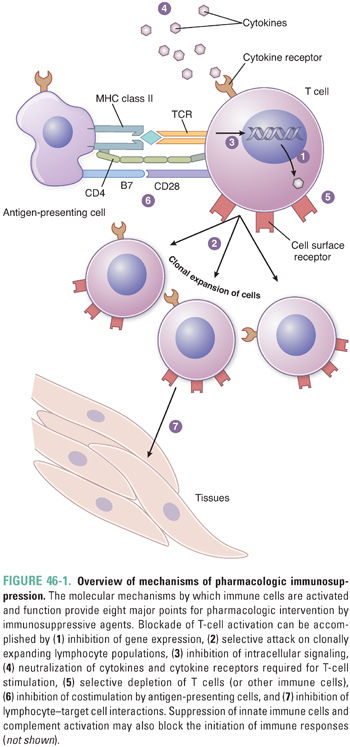
1. Inhibition of gene expression to modulate inflammatory responses
2. Depletion of expanding lymphocyte populations with cytotoxic agents
3. Inhibition of lymphocyte signaling to block activation of lymphocytes and expansion of lymphocyte populations
4. Neutralization of cytokines and cytokine receptors essential for mediating the immune response
5. Depletion of specific immune cells, usually via cell-specific antibodies
6. Blockade of costimulation to induce anergy
7. Blockade of cell adhesion to prevent migration and homing of inflammatory cells
8. Inhibition of innate immunity, including complement activation
Glucocorticoids have broad anti-inflammatory effects. The intimate relationship between cortisol and the immune system is discussed in Chapter 29, Pharmacology of the Adrenal Cortex. Briefly, glucocorticoids are steroid hormones that exert their physiologic actions by binding to the cytosolic glucocorticoid receptor. The glucocorticoid–glucocorticoid receptor complex translocates to the nucleus and binds to glucocorticoid response elements (GREs) in the promoter region of specific genes, either up-regulating or down-regulating gene expression.
Glucocorticoids have important metabolic effects on essentially all cells of the body and, in pharmacologic doses, suppress the activation and function of innate and adaptive immune cells. Glucocorticoids down-regulate the expression of many inflammatory mediators, including key cytokines such as tumor necrosis factor (TNF)-α, interleukin-1 (IL-1), and IL-4. The role of glucocorticoids in suppressing eicosanoid biosynthesis and signaling is discussed in Chapter 43, Pharmacology of Eicosanoids. The overall effect of glucocorticoid administration is profoundly anti-inflammatory and immunosuppressive, explaining the use of glucocorticoids for the treatment of numerous inflammatory diseases such as rheumatoid arthritis and transplant rejection.
Long-term glucocorticoid administration has important adverse effects. Diabetes, reduced resistance to infection, osteoporosis, cataracts, increased appetite leading to weight gain, hypertension and its sequelae, and the masking of inflammation must all be closely monitored in patients receiving glucocorticoids. Abrupt cessation of glucocorticoid therapy can result in acute adrenal insufficiency because the hypothalamus and pituitary gland require weeks to months to reestablish adequate ACTH production. During this time, the underlying disease can worsen because of disinhibition of the immune system. To prevent the latter complications, glucocorticoid dosage should be tapered slowly as therapy is terminated. This is why Mr. W’s dose of glucocorticoids was tapered slowly after his pulsed steroid therapy for treatment of acute cellular rejection.
Cytotoxic agents are used both for immunosuppression and for antineoplastic chemotherapy. The therapeutic goal in both cases is the elimination of pathogenic cells. Two classes of cytotoxic agents, antimetabolites and alkylating agents, are commonly used as immunosuppressants. Antimetabolites are structural analogues of natural metabolites that inhibit essential pathways involving these metabolites. Alkylating agents interfere with DNA replication and gene expression by alkylation of DNA.
Antimetabolites have been a mainstay of immunosuppressive treatment for many years. Their powerful suppressive effect on immune cells is accompanied by many adverse effects related to their lack of selectivity. The older antimetabolites, such as azathioprine and methotrexate, affect all rapidly dividing cells and can have toxic effects on the gastrointestinal mucosa and bone marrow. Newer antimetabolites, such as mycophenolate mofetil and leflunomide, cause fewer adverse effects. Mycophenolate mofetil may also be relatively selective for immune cells, further reducing its toxicity. Antimetabolites typically affect both cell-mediated and humoral immunity, rendering patients more susceptible to infection than would occur if only one of these immune systems were affected.
Antimetabolites are widely used in the treatment of cancer, and their mechanisms of action are described in detail in Chapter 39, Pharmacology of Cancer: Genome Synthesis, Stability, and Maintenance. Here, we focus on the antimetabolites used for immunosuppression and briefly discuss the anti-inflammatory aspects of their mechanisms of action.
Azathioprine
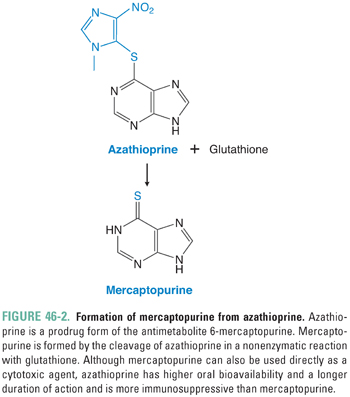
Azathioprine (AZA) was the first drug to be used for suppression of the immune system after organ transplantation, and it remains a mainstay for this indication. AZA is a prodrug of the purine analogue 6-mercaptopurine (6-MP), which is slowly released as AZA reacts nonenzymatically with sulfhydryl compounds such as glutathione (Fig. 46-2). The slow release of 6-MP from AZA favors immunosuppression. Although AZA prolongs organ graft survival, this drug is less efficacious than mycophenolate mofetil in improving the long-term survival of kidney allografts. AZA and 6-MP are also used as immunosuppressants in the treatment of inflammatory bowel disease, acute lymphoblastic leukemia, and autoimmune skin disorders.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


