INTRODUCTION
Asthma is a chronic disease characterized by inflammation of the airways and exaggerated airway smooth muscle constriction. The symptoms of asthma include dyspnea and wheezing as well as mucus production and cough, particularly at night. Asthma is both an obstructive lung disease and an inflammatory disease; the obstructive component is characterized by bronchoconstriction, whereas the inflammatory component is marked by airway edema, goblet cell hyperplasia, mucus secretion, and infiltration and cytokine release by immune and inflammatory cells. Although the airway obstruction is generally reversible during acute asthma attacks, over time, the disease may cause airway remodeling and permanent deterioration in pulmonary function.
Medications used to treat asthma act in one of two ways: by relaxing bronchial smooth muscle or by preventing and reducing inflammation. This chapter approaches asthma as both a bronchoconstrictive and an inflammatory disease. After discussing the physiologic control of bronchial tone and the function of immune pathways in the airways, the chapter turns to the pathophysiology of asthma. Current therapies are then discussed, including the pharmacology of both bronchodilators and airway anti-inflammatory agents.
 WY is a 51-year-old man with a long-standing history of asthma and allergies, first diagnosed at the age of 6. His asthma had been managed successfully for many years on a regimen of twice-daily inhaled fluticasone (an inhaled corticosteroid) and albuterol (a β-adrenergic agonist) as needed whenever he developed shortness of breath or wheezing. Over the past year, Mr. Y has noticed worsening symptoms and more frequent asthma attacks. He has had shortness of breath, wheezing, and chest tightness when running to catch the bus. He has also developed a significant amount of coughing, especially at night, and has found himself using albuterol several times a day.
WY is a 51-year-old man with a long-standing history of asthma and allergies, first diagnosed at the age of 6. His asthma had been managed successfully for many years on a regimen of twice-daily inhaled fluticasone (an inhaled corticosteroid) and albuterol (a β-adrenergic agonist) as needed whenever he developed shortness of breath or wheezing. Over the past year, Mr. Y has noticed worsening symptoms and more frequent asthma attacks. He has had shortness of breath, wheezing, and chest tightness when running to catch the bus. He has also developed a significant amount of coughing, especially at night, and has found himself using albuterol several times a day.
One hazy, hot summer day, Mr. Y develops substantial coughing, wheezing, and shortness of breath at rest. He takes two puffs of his albuterol inhaler but finds that it provides only minimal relief. He calls his doctor but has trouble even speaking in full sentences. His doctor advises him to go to the emergency department (ED) immediately.
On presentation to the ED, Mr. Y is immediately given albuterol via nebulizer and a large dose of intravenous methylprednisolone (a corticosteroid). Although he is now more comfortable, he continues to “feel tight” and he has quiet breath sounds on exam. Fortunately, with continued administrations of nebulized albuterol and a treatment with inhaled ipratropium (an anticholinergic agent), Mr. Y begins to feel better over the next few hours. He is hospitalized for 2 days and sent home on a tapering dose of prednisone, an oral corticosteroid.
At a follow-up appointment with his pulmonologist, Mr. Y is concerned that his asthma has significantly worsened. Even though the acute event has passed, he continues to have frequent asthma symptoms, and his examination and pulmonary function tests suggest that his pulmonary function is significantly reduced. His pulmonologist discusses medication adherence and the proper way to use inhalers, including use of a spacer with the fluticasone inhaler and the need for Mr. Y to rinse his mouth after its use. Mr. Y’s pulmonologist also increases the intensity of his drug regimen by adding salmeterol, a long-acting β-agonist, as well as montelukast, a cysteinyl leukotriene receptor antagonist.
Three months later, Mr. Y reports that his baseline symptoms have improved but that he has had an interval asthma exacerbation requiring treatment with prednisone. Because his asthma is still not under adequate control and his laboratory tests show an elevated IgE level, Mr. Y’s physician recommends that he start omalizumab, an anti-IgE monoclonal antibody. Mr. Y now receives twice-monthly injections of omalizumab and he has had only one mild asthma exacerbation in the 6 months he has been on this therapy.
Questions
1. Why did Mr. Y develop asthma?
2. Why was Mr. Y initially managed with both an inhaled corticosteroid (fluticasone), taken twice daily, and a β-adrenergic agonist (albuterol), taken only as needed?
3. Why was it preferable to keep Mr. Y on a maintenance regimen of an inhaled corticosteroid (fluticasone) instead of a systemic corticosteroid? Why was it necessary to administer systemic corticosteroids (methylprednisolone intravenously and prednisone orally) to treat his asthma exacerbations?
4. How does omalizumab, an anti-IgE monoclonal antibody, prevent exacerbations of asthma?
 PHYSIOLOGY OF AIRWAY SMOOTH MUSCLE TONE AND IMMUNE FUNCTION
PHYSIOLOGY OF AIRWAY SMOOTH MUSCLE TONE AND IMMUNE FUNCTION
Asthma involves dysfunction in the pathways that regulate both smooth muscle tone and immune function in the airways. It is therefore important to review the normal physiology of these systems before discussing the pathophysiology of asthma.
Physiology of Airway Smooth Muscle Contraction
As discussed in Chapter 9, Principles of Nervous System Physiology and Pharmacology, involuntary responses of smooth muscle are regulated by the autonomic nervous system. In the airways, sympathetic (adrenergic) tone causes bronchodilation and parasympathetic (cholinergic) tone causes bronchoconstriction. Bronchial smooth muscle tone is also regulated by nonadrenergic, noncholinergic (NANC) fibers that innervate the respiratory tree.
Sympathetic innervation of the lung is concentrated primarily on pulmonary blood vessels and the submucosal glands. There is little direct sympathetic innervation of the bronchial smooth muscle. However, airway smooth muscle cells express β2-adrenergic receptors (and, to a lesser extent, β1-adrenergic receptors) that are responsive to circulating catecholamines. β2-Adrenergic receptors are activated by epinephrine, which is secreted by the adrenal medulla and causes bronchodilation. Exogenous epinephrine was one of the first pharmacotherapies for asthma and was available until recently in some over-the-counter formulations. Newer, β2-selective adrenergic agonists, such as the albuterol used by Mr. Y, are now considered the first-line bronchodilators for treatment of acute asthmatic symptoms.
The vagus nerve provides parasympathetic innervation to the lungs. Airway smooth muscle cells express muscarinic receptors, especially the excitatory M3 subtype of muscarinic receptor. Parasympathetic postganglionic neurons release acetylcholine, which stimulates these muscarinic receptors and induces bronchoconstriction. Parasympathetic neurons are dominant in maintaining airway smooth muscle tone, and anticholinergic agents can cause bronchorelaxation. These agents are used primarily in the treatment of chronic obstructive pulmonary disease (see Box 48-1) but can also be used in acute asthma exacerbations (as was the case with Mr. Y) or when β-adrenergic agonists are contraindicated.
BOX 48-1 Pharmacology of Chronic Obstructive Pulmonary Disease |
Chronic obstructive pulmonary disease (COPD) describes a spectrum of disorders that result in obstructive lung disease. Unlike asthma, COPD is generally not reversible. COPD is caused by an abnormal inflammatory response to an inhaled environmental insult. In 90% of cases, this insult to the lungs is tobacco smoke. Clinically, COPD is divided into two frequently overlapping diseases: emphysema and chronic bronchitis. Pulmonary emphysema refers to alveolar enlargement caused by destruction of alveolar walls and loss of pulmonary elastic recoil, whereas chronic bronchitis is a clinical diagnosis made on the basis of a chronic cough for 3 or more months during 2 consecutive years that cannot be attributed to another cause. As noted above, COPD is caused by an abnormal response to inhalation of tobacco smoke or other toxic agents. In contrast to asthma, where CD4+ T lymphocytes, B lymphocytes, mast cells, and eosinophils are the primary inflammatory cells, the inflammatory response to tobacco smoke is primarily neutrophilic and monocytic. Tobacco smoke stimulates resident alveolar macrophages to produce chemokines that attract neutrophils. These neutrophils and resident macrophages release proteinases, particularly matrix metalloproteinases. The proteinases degrade elastin, which provides elastic recoil to the alveoli, as well as other proteins that compose the matrix supporting the lung parenchyma. Cell death follows, due to impaired attachment of alveolar cells to the degraded matrix and to the toxic actions of inflammatory cells and the environmental insult. The result is that alveoli degrade and coalesce, forming the characteristic enlargement of air spaces typical of emphysema. There is also enhanced mucus production and fibrosis, although the mechanisms underlying these pathologic phenomena have not been well characterized. Although it is tempting to think that inflammation in COPD could be held in check by inhaled corticosteroids, steroids are unfortunately of limited benefit in this disease. The lack of steroid efficacy likely results from the fact that the inflammatory cells responsible for COPD are macrophages and neutrophils, which are less responsive than lymphocytes and eosinophils to the actions of corticosteroids. Moreover, the activity of histone deacetylase is impaired in COPD, so the inhibition of proinflammatory transcription factors is limited. A number of studies have examined the effects of inhaled corticosteroids on lung function in COPD, but none have found a statistically significant benefit. However, inhaled corticosteroids have been found to reduce the frequency and severity of acute exacerbations of COPD. Therefore, while corticosteroids are not routinely recommended for the treatment of COPD, they may be indicated in patients who develop frequent, severe exacerbations. Because cysteinyl leukotrienes, mast cells, and IgE have no role in the pathophysiology of COPD, specific treatments for asthma that target these pathways are not useful in COPD. Interestingly, although leukotriene B4 (LTB4) is a potent chemotactic factor for neutrophils, clinical studies of LTB4 antagonism have not shown a benefit to date. Bronchodilators produce only a modest improvement in airflow in patients with COPD. However, even a small improvement in airflow can significantly improve symptoms in patients with COPD, especially in those whose lungs have become hyperinflated. Asthma is punctuated by acute attacks, while most patients with COPD have chronic breathlessness that is worsened with exertion. Therefore, short-acting “reliever” medications are less beneficial than long-acting drugs in COPD. Both β-adrenergic agonists and inhaled anticholinergic agents cause bronchodilation in COPD. However, many patients with COPD have concomitant coronary artery disease, so anticholinergic agents may be preferred in this subset of patients. There is evidence that the bronchodilatory effects of β-agonists and anticholinergic agents (and theophylline) are additive; therefore, patients with severe COPD may benefit from combination therapies such as formoterol and tiotropium. |

Airway NANC fibers are primarily under parasympathetic control. These fibers can be either stimulatory (causing bronchoconstriction) or inhibitory (causing bronchodilation). NANC fibers do not release either norepinephrine or acetylcholine but instead release neuropeptides. Bronchoconstricting peptides released by NANC fibers include neurokinin A, calcitonin gene-related peptide, substance P, bradykinin, tachykinin, and neuropeptide Y; the bronchodilating peptide vasoactive intestinal polypeptide (VIP) is also released by NANC fibers, as is the bronchodilating gasotransmitter nitric oxide (NO). Although no pharmacologic agents have yet been developed to take advantage of the NANC system, nitric oxide is a marker of the intensity of airway inflammation, and NO measurements have been used to assess the severity of asthma and titrate therapy accordingly.
As described in Chapter 42, Principles of Inflammation and the Immune System, T lymphocytes play a key role in controlling the immune response. T lymphocytes are classified as CD8+ TC (cytotoxic) cells, which are mediators of cellular adaptive immunity, and CD4+ TH (helper) cells, which regulate adaptive immune responses. TH cells are subclassified as TH1 and TH2 cells based on the cytokines they produce. TH1 cells, which produce predominantly interferon-γ, IL-2, and TNF-α, favor a cellular immune response involving T lymphocytes. TH2 cells, on the other hand, produce IL-4, IL-5, IL-6, IL-9, IL-10, and IL-13 and favor a humoral immune response involving antibody production by B cells. Because cytokines produced by activated TH1 and TH2 cells are mutually inhibitory, any given immune stimulus elicits predominantly one or the other response (Fig. 48-1).
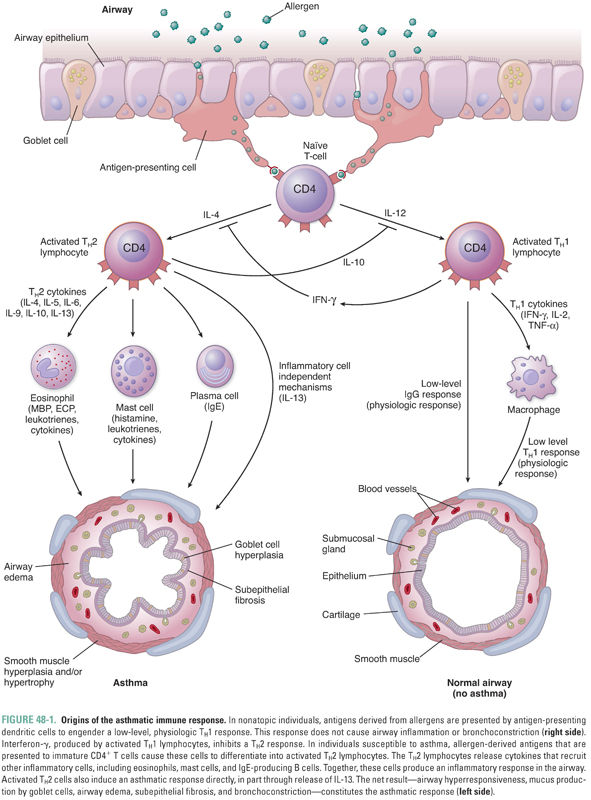
All individuals continually inhale environmental aeroallergens such as pollens, cat dander, dust mites, and a host of other antigens. These allergens are phagocytosed by antigen-presenting cells lining the airways. The antigens are recognized as foreign by TH cells and generate a low-level TH1 response mediated primarily by interferon-γ, as well as a low-level IgG antibody response. However, in asthma, an exaggerated TH2 response often predominates, generating airway inflammation and bronchial hyperresponsiveness (Fig. 48-1).

Asthma is a complex disease characterized by airway inflammation, airway smooth muscle hyperresponsiveness, and symptomatic bronchoconstriction. Because the most prominent clinical feature of asthma is bronchoconstriction, a simplistic approach to understanding the disease focuses on airway smooth muscle contraction. At its most fundamental level, however, asthma is an inflammatory disease of the airways, and treatment of the underlying inflammation is crucial to maintain normal airway function. Therefore, as detailed below, the treatment of asthma employs both bronchodilators and anti-inflammatory agents.
Asthma as a Bronchoconstrictive Disease
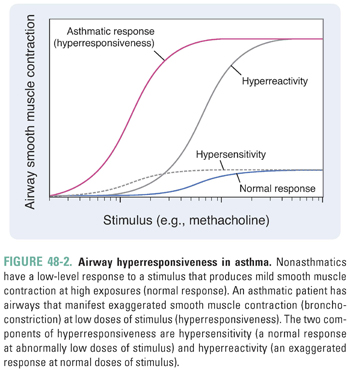
The propensity for the airways of asthmatic patients to constrict in response to a wide variety of stimuli, including allergens, environmental irritants, exercise, cold air, and infections, is termed hyperresponsiveness. Two features of airway hyperresponsiveness separate the response to stimuli in asthmatic patients from the nonasthmatic response: hypersensitivity and hyperreactivity. Hypersensitivity describes a normal response at abnormally low levels of stimuli (i.e., the airways of asthmatic patients constrict to stimuli that do not elicit a response in healthy individuals). Hyperreactivity describes an exaggerated response at normal levels of stimuli (i.e., the airways of asthmatic patients respond too vigorously). In Figure 48-2, hypersensitivity describes a shift of the stimulus–response curve to the left, while hyperreactivity describes an upward shift. The overall response to stimuli in asthmatic patients represents the combination of hypersensitivity and hyperreactivity.
The causes of airway hyperresponsiveness in asthma have not been completely elucidated. The hyperreactive response may be explained by alterations in airway smooth muscle mass due to the increase in size (hypertrophy) and number (hyperplasia) of myocytes that occurs in response to inflammation (Fig. 48-1). The hypersensitivity response is due to alterations in smooth muscle excitation–contraction coupling. Possible mechanisms of altered coupling in asthma include greater responsiveness of intracellular calcium release channels, increased calcium sensitization, and changes in the expression of ion channels, receptors, and second messengers.
Asthma as an Inflammatory Disease
Although the primary symptoms (wheezing and shortness of breath) of most asthmatic patients are due to bronchoconstriction, the underlying cause of asthma is an allergic inflammation of the airways. The inflammatory process is visible histologically as airway edema, goblet cell hyperplasia, subepithelial fibrosis, mucus hypersecretion, and infiltration by a variety of inflammatory cells, including TH2 lymphocytes, antigen-presenting cells, plasma cells, mast cells, neutrophils, and eosinophils (Fig. 48-1). Airway inflammation can lead to a chronic cough in asthmatic individuals, even in those who do not develop symptomatic bronchoconstriction (this diagnosis is known as cough variant asthma). Many inflammatory mediators and cytokines govern the interplay among the various immune cells. Anti-inflammatory medications, particularly corticosteroids, are mainstays in the pharmacologic treatment of asthma. As the complex pathophysiology of asthma has been further elucidated, more targeted therapies that block specific inflammatory pathways are being developed.
TH2 Cells and the Origin of Asthma
Although the exact causes of asthma are not fully understood, one theory suggests that asthma, like other allergic diseases, is the result of an immune imbalance favoring TH2 lymphocytes over TH1 lymphocytes. TH2 lymphocytes contribute to asthma through three mechanisms. First, in patients with a hereditary predisposition to atopy (from the Greek, meaning “out of place”), an allergen can trigger a type I hypersensitivity response. In normal (non-atopic) individuals, an allergen is phagocytosed by antigen-presenting cells, stimulating a low-level TH1 response and the production of appropriate amounts of IgG antibodies directed against the allergen. In atopic individuals, however, the same allergen induces a strong TH2 response mediated through the release of IL-4, which induces B cells to produce exaggerated amounts of IgE antibodies directed against the allergen (Fig. 48-1). The IgE antibodies bind to high-affinity IgE receptors on mast cells, and subsequent cross-linking of the IgE receptors upon reexposure to the allergen causes mast cell degranulation and triggers an allergic reaction (Fig. 48-2, and see below). Second, TH2 cells can directly induce a type IV hypersensitivity reaction through the production of IL-13 (and, to a lesser degree, IL-4). In the airway, IL-13 causes goblet cell hyperplasia, increased mucus production, and smooth muscle hyperplasia and/or hypertrophy. IL-13 also stimulates B cells to produce IgE (Fig. 48-1). Third, TH2 lymphocytes recruit eosinophils by producing IL-5 as well as GM-CSF and IL-4. These cytokines (especially IL-5) induce eosinophil proliferation and release from the bone marrow and promote eosinophil survival in the circulation and tissues. As in many patients with asthma, Mr. Y had a high level of circulating eosinophils and elevated levels of serum IgE.
What causes the imbalance between TH1 and TH2 lymphocytes in patients with asthma? Although the exact reasons remain to be fully elucidated, they likely involve environmental effects on genetically susceptible individuals. Epidemiologic studies have found that exposures to tuberculosis and viruses such as measles and hepatitis A are protective against the development of asthma. Having older siblings and/or encountering other children through attendance at a day care facility (both of which are associated with increased exposure to infectious agents) are also associated with a decreased incidence of asthma. Living in a rural environment (where there is substantial contact with bacterial endotoxins) is also protective. One leading theory suggests that a “Western lifestyle,” including decreased exposure early in life to microbes that engender TH1-lymphocyte responses, contributes to the development of asthma and other allergic diseases in susceptible individuals. Although this “hygiene hypothesis” is probably too simplistic to explain the origins of a complex disease such as asthma, it forms a useful model for thinking about the disease and is a possible explanation for the dramatic rise in the incidence of asthma in the Western hemisphere. It is impossible to know exactly what caused Mr. Y’s asthma; however, the fact that he had allergic rhinitis and elevated levels of IgE suggests that he had an atopic predisposition triggered by environmental allergens.
Plasma Cells, IgE, Mast Cells, and Leukotrienes
As noted above, an IgE-mediated type I hypersensitivity response is one mechanism by which allergens cause the pathologic and clinical manifestations of asthma (Fig. 48-3). The allergic response is initiated when a dendritic cell phagocytoses an inhaled allergen. The dendritic cell presents the processed allergen to TH2 cells and activates them. The activated TH2 cells bind to and activate B lymphocytes via CD40 on the B-cell surface. Activated TH2 cells also generate IL-4 and IL-13, which induce B-cell transformation into IgE-producing plasma cells.
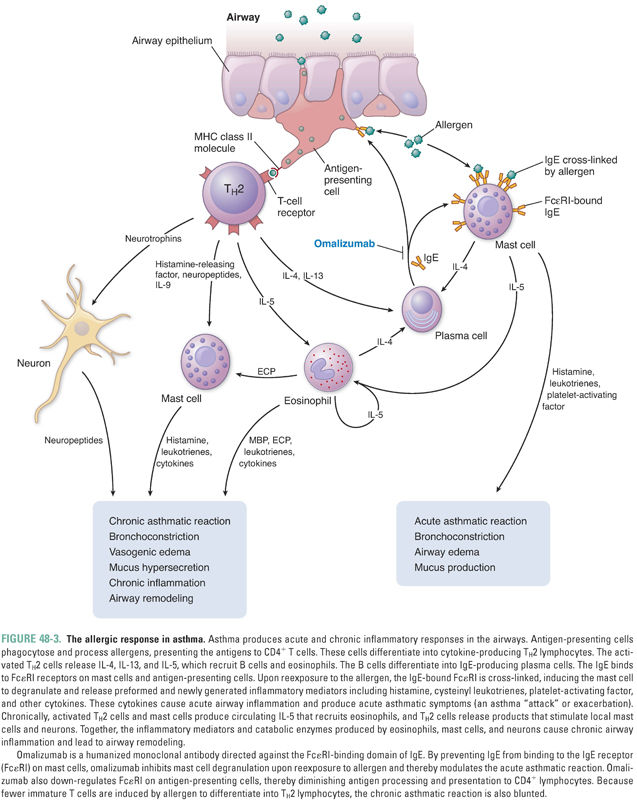
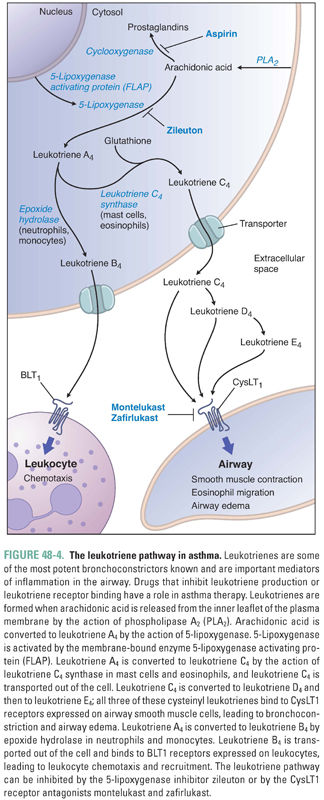
IgE circulates briefly in the bloodstream before binding to high-affinity IgE receptors (FcεRI) on mast cells. Upon reexposure, the allergen binds to and cross-links the IgE–FcεRI complexes, thereby activating the mast cell. The activated mast cell degranulates, releasing its preformed inflammatory mediators. These molecules include histamine, proteolytic enzymes, and certain cytokines (such as platelet-activating factor). The activated mast cell also releases arachidonic acid from its plasma membrane and produces leukotrienes and prostaglandin D2 (Fig. 48-4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


