INTRODUCTION
Cancer therapy has traditionally been based on the principle that tumor cells are traversing the cell cycle frequently and are thus more sensitive than normal cells to interference with DNA synthesis and mitosis. Indeed, the antimetabolites, a class of agents that are analogues of endogenous folates, purines, and pyrimidines, and that function as inhibitors of the enzymes of nucleotide synthesis, were some of the first drugs to be tested as chemotherapeutic agents. In the late 1940s, Sidney Farber and colleagues administered the antifolate compound aminopterin to patients with acute leukemia and observed temporary remissions in more than half of the patients.
Because of their rapid growth and division, cancer cells are also thought to be more sensitive than normal cells to the effect of DNA-damaging agents. Also in the late 1940s, nitrogen mustards—derivatives of agents that had been found to cause bone marrow suppression through wartime exposures—were tested in patients with lymphoma and leukemia and shown to induce remissions.
These and other findings have since led to the development of multiple classes of antineoplastic drugs designed to interfere with the building blocks of DNA synthesis and mitosis, or to produce DNA damage and chromosomal instability, and thereby to promote cytotoxicity and programmed cell death (apoptosis). Unfortunately, the therapeutic window of these drugs is narrow because they also affect normal cells that routinely undergo cell division in tissues such as the gastrointestinal tract and bone marrow. That being said, cancer cells can often be induced to undergo apoptosis more readily than normal cells, providing some degree of selectivity. Use of combination chemotherapy with agents from different classes has helped to enhance efficacy while minimizing overlapping dose-limiting toxicities, but the ability to cure patients with most types of advanced cancer remains limited. In part, this limited efficacy is due to the development of multiple resistance mechanisms, including the failure of tumor cells to undergo apoptosis in response to DNA damage or stress. In addition, it is increasingly apparent that populations of cancer stem cells may have low proliferation rates and other properties that render them resistant to cytotoxic chemotherapy.
 One day, JL, a 23-year-old graduate student who has heretofore been in good health, notices while showering that he has developed a hard lump in his left testis. Concerned by the finding, JL’s physician orders an ultrasound examination, which shows a solid mass suggestive of cancer. The testis is removed surgically; pathologic review confirms the diagnosis of testicular cancer. A chest x-ray reveals several lung nodules, which are thought to represent metastatic spread of the cancer. JL is treated with several cycles of combination chemotherapy, including bleomycin, etoposide, and cisplatin. The lung nodules disappear completely. One year later, JL is able to resume his studies, and there are no signs of recurrence of the cancer. Nonetheless, at every subsequent follow-up visit, JL’s physician asks him whether he is developing shortness of breath.
One day, JL, a 23-year-old graduate student who has heretofore been in good health, notices while showering that he has developed a hard lump in his left testis. Concerned by the finding, JL’s physician orders an ultrasound examination, which shows a solid mass suggestive of cancer. The testis is removed surgically; pathologic review confirms the diagnosis of testicular cancer. A chest x-ray reveals several lung nodules, which are thought to represent metastatic spread of the cancer. JL is treated with several cycles of combination chemotherapy, including bleomycin, etoposide, and cisplatin. The lung nodules disappear completely. One year later, JL is able to resume his studies, and there are no signs of recurrence of the cancer. Nonetheless, at every subsequent follow-up visit, JL’s physician asks him whether he is developing shortness of breath.
Questions
1. How did serendipity lead to the discovery of cisplatin, the most efficacious drug against testicular cancer?
2. What is the molecular target of each of the drugs in JL’s combination chemotherapy regimen?
3. Why does JL’s physician inquire about shortness of breath at each follow-up visit?
4. By what mechanisms could bleomycin, etoposide, and cisplatin act synergistically against JL’s testicular cancer?
 BIOCHEMISTRY OF GENOME SYNTHESIS, STABILITY, AND MAINTENANCE
BIOCHEMISTRY OF GENOME SYNTHESIS, STABILITY, AND MAINTENANCE
The central dogma of molecular biology states that DNA contains all the information necessary to encode cellular macromolecules—specifically, that DNA is transcribed into RNA, and RNA is then translated into proteins. Antimetabolites inhibit the synthesis of nucleotides, which are the building blocks of both DNA and RNA. Figure 39-1A provides an overview of nucleotide synthesis, and Figure 39-1B shows the steps at which some of the drugs discussed in this chapter inhibit nucleotide metabolism.
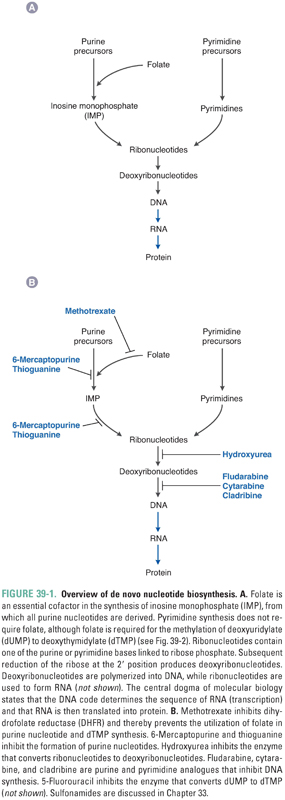
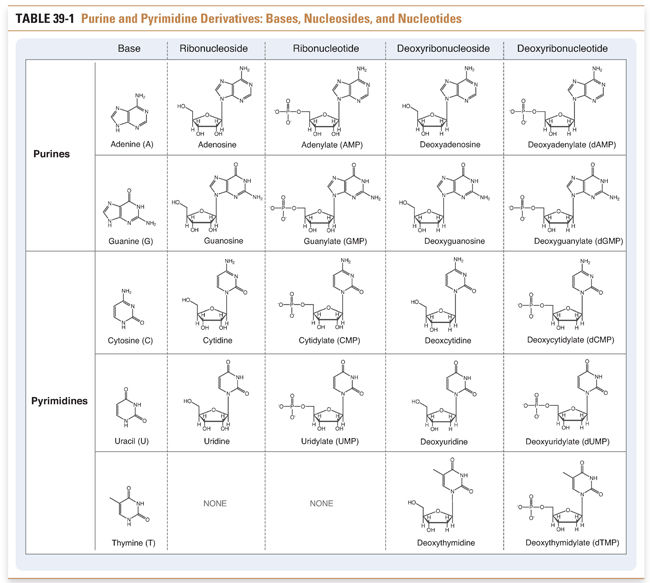
Nucleotides, the precursors of DNA and RNA, include the purine nucleotides and the pyrimidine nucleotides. Purines and pyrimidines are the bases that are used to determine the chemical code within DNA and RNA. Adenine and guanine are purines; cytosine, thymine, and uracil are pyrimidines. Nucleosides are derivatives of purines and pyrimidines that are conjugated to ribose or deoxyribose. Nucleotides are monophosphate, diphosphate, and triphosphate esters of the corresponding nucleosides. For example, an adenine base covalently linked to a ribose sugar and a diphosphate ester is called adenosine diphosphate (ADP). The various purine and pyrimidine bases, nucleosides, and nucleotides are shown in Table 39-1.
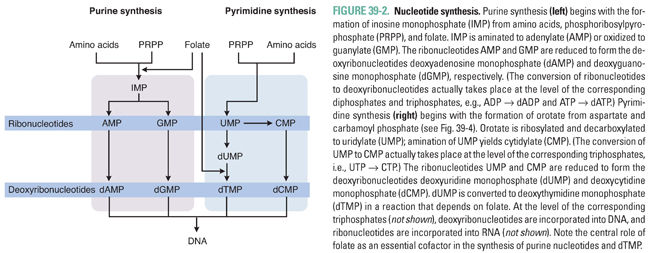
Nucleotide synthesis involves three general sets of sequential reactions: (1) synthesis of ribonucleotides, (2) reduction of ribonucleotides to deoxyribonucleotides, and (3) conversion of deoxyuridylate (dUMP) to deoxythymidylate (dTMP) (Fig. 39-2). Ribonucleotide synthesis differs for purines and pyrimidines; therefore, the synthesis of each class of molecules is discussed individually. All ribonucleotides are reduced to deoxyribonucleotides by a single enzyme, ribonucleotide reductase. Deoxyribonucleotides generated from ribonucleotides and from dUMP are used for DNA synthesis. Because folate is an essential cofactor for the synthesis of purine ribonucleotides and dTMP, folate metabolism is discussed separately (see Chapter 33, Principles of Antimicrobial and Antineoplastic Pharmacology).
Purine Ribonucleotide Synthesis
Adenine and guanine, the purine bases shown in Table 39-1, are synthesized as components of ribonucleotides (for RNA synthesis) and deoxyribonucleotides (for DNA synthesis). Derivatives of adenine and guanine, which include ATP, GTP, cAMP, and cGMP, are also used for energy storage and cell signaling. Purine synthesis begins with the assembly of inosinate (IMP) from a ribose phosphate, moieties derived from the amino acids glycine, aspartate, and glutamine, and one-carbon transfers catalyzed by tetrahydrofolate (THF), as shown in Figure 39-2. Because of the central role of THF in purine synthesis, one important chemotherapeutic strategy is to reduce the amount of THF available to the cell and thereby to inhibit purine synthesis.
Figure 39-3 shows the central role of IMP in purine synthesis. IMP can be aminated to AMP or oxidized to GMP. In turn, AMP and GMP can be converted to ATP and GTP, respectively, and then incorporated into RNA, or reduced to dAMP and dGMP, respectively, as described below.
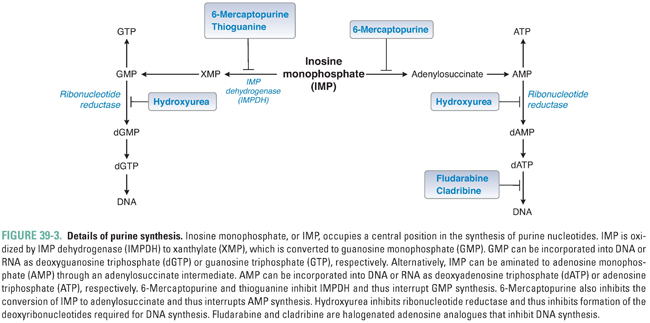
Purine bases, nucleosides, and nucleotides are readily interconverted by multiple enzymes within the cell. In one such reaction, the enzyme adenosine deaminase (ADA) catalyzes the irreversible conversion of adenosine or 2′-deoxyadenosine to inosine or 2′-deoxyinosine, respectively. Inhibition of ADA causes the intracellular stores of adenosine and 2′-deoxyadenosine to exceed those of the other purines, ultimately resulting in metabolic effects that are toxic to the cell (see discussion of pentostatin below).
Pyrimidine Ribonucleotide Synthesis
Pyrimidine ribonucleotides are synthesized according to the metabolic pathway shown in Figure 39-4. The basic pyrimidine ring, orotate, is assembled from carbamoyl phosphate and aspartate. Orotate then reacts with a ribose phosphate; the decarboxylation product of this reaction yields uridylate (UMP). As with IMP in purine synthesis, UMP has a central role in pyrimidine synthesis. UMP is itself a nucleotide component of RNA, as well as the common precursor of the RNA and DNA components cytidylate (CMP), deoxycytidylate (dCMP), and deoxythymidylate (dTMP). CTP is formed by the amination of UTP.
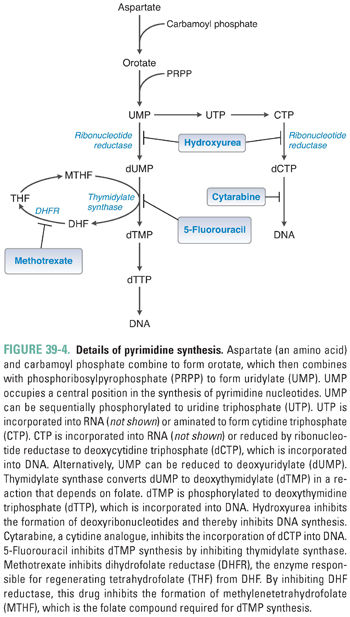
Ribonucleotide Reduction and Thymidylate Synthesis
The ribonucleotides ATP, GTP, UTP, and CTP, which are required for RNA synthesis, are assembled on a DNA template and linked to form RNA. Alternatively, ribonucleotides can be reduced at the 2′ position on ribose to form the deoxyribonucleotides dATP, dGTP, dUTP, and dCTP. The conversion of ribonucleotides to deoxyribonucleotides is catalyzed by the enzyme ribonucleotide reductase. (In actuality, ribonucleotide reductase uses as substrates the diphosphate forms of the four ribonucleotides to produce dADP, dGDP, dUDP, and dCDP; nucleotides can, however, be readily interconverted among their monophosphate, diphosphate, and triphosphate forms.)
Note, in Figures 39-2 through 39-4, that ribonucleotide reductase catalyzes the formation of the DNA precursors dATP, dGTP, and dCTP. The DNA precursor dTTP is not synthesized directly by ribonucleotide reductase, however. Rather, dUMP must be modified to form dTMP. As demonstrated in Table 39-1, dTMP is the product of dUMP methylation. The methylation of dUMP to dTMP is catalyzed by thymidylate synthase, with methylenetetrahydrofolate (MTHF) serving as the donor of the methyl group (Fig. 39-4). As MTHF donates its methyl group, it is oxidized to dihydrofolate (DHF). DHF must be reduced to THF by dihydrofolate reductase (DHFR) and then converted to MTHF in order to serve as the cofactor for another cycle of dTMP synthesis. Inhibition of DHFR prevents the regeneration of tetrahydrofolate and thereby inhibits the conversion of dUMP to dTMP, eventually resulting in an insufficient cellular level of dTMP for DNA replication.
Provided that sufficient levels of nucleotides are available, DNA and RNA can be synthesized, and protein synthesis, cell growth, and cell division can occur. Many drugs, including the antimetabolites discussed in this chapter, inhibit both DNA and RNA synthesis. To avoid repetition, a detailed discussion of DNA and RNA synthesis is provided in Chapter 34, Pharmacology of Bacterial Infections: DNA Replication, Transcription, and Translation. For the purposes of this chapter, the reader should be aware that RNA and DNA are formed by polymerization of ribonucleotides and deoxyribonucleotides, respectively. RNA polymers are elongated by the enzyme RNA polymerase, and DNA is elongated by DNA polymerase. Although antimetabolites primarily inhibit the enzymes that mediate nucleotide synthesis, some antimetabolites also inhibit DNA and RNA polymerases (see below).
DNA Repair and Chromosome Maintenance
Mutations and other DNA lesions can arise spontaneously or as a result of exposure to DNA-damaging chemical agents or radiation. Several general pathways exist for repair of these lesions, including mismatch repair (MMR) for DNA replication errors, base excision repair (BER) for small base modifications and single-strand breaks, nucleotide excision repair (NER) for removal of bulky adducts, and homologous recombination or nonhomologous end-joining for double-strand breaks (Fig. 39-5). DNA repair pathways are important not only because they can alter the efficacy of chemotherapy but also because loss of these pathways frequently contributes to tumor development via impairment of genomic integrity and facilitation of mutations in oncogenes and tumor suppressor genes. Telomeres, the repeat sequences that cap the ends of chromosomes, also play an important role in genome stability and prevention of chromosome fusions. The enzyme telomerase, which prevents telomere shortening in cancer cells, represents a key component in the process of immortalization and oncogenic transformation.
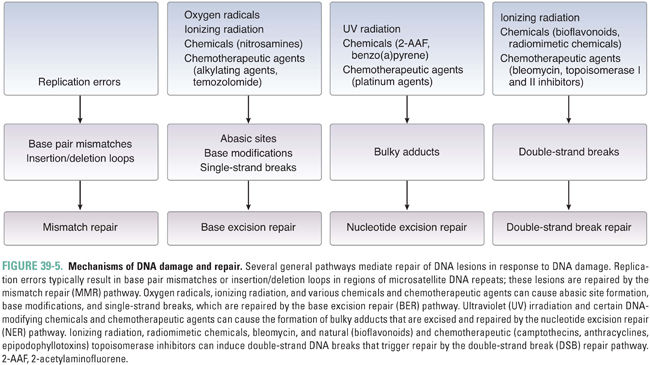
During DNA replication, errors such as single-base mismatches and insertions or deletions of microsatellite repeat sequences (microsatellite instability) are recognized and repaired by proteins of the mismatch repair (MMR) system. For single-base mismatches, recognition involves a heterodimer between the MSH2 protein and MSH6, while for insertion/deletion loops, MSH2 can also partner with MSH3 (Fig. 39-6). These complexes recruit the proteins MLH1 and PMS2 (as well as MLH3 for insertion/deletion loops), which, in turn, recruit exonucleases and components of the DNA replication machinery for excision and repair of the lesion. Germline mutations in MLH1, PMS2, MSH2, or MSH6 are associated with 70–80% of cases of hereditary nonpolyposis colon cancer. In addition, microsatellite instability, a hallmark of defective MMR, is observed in 15–25% of sporadic colorectal cancers.
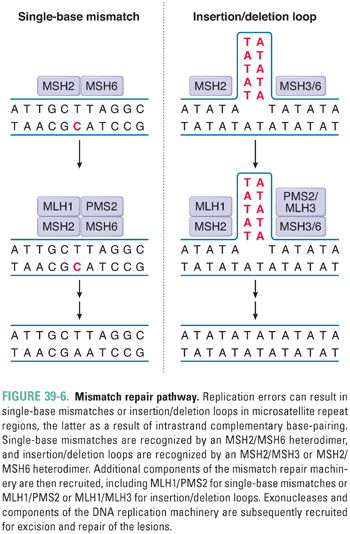
DNA single-strand breaks (SSB), which may be formed directly by ionizing radiation or indirectly due to enzymatic excision of a modified base by a DNA glycosylase, activate the enzyme poly(ADP-ribose) polymerase 1 (PARP1) (Fig. 39-7). At the site of the break, PARP1 transfers ADP-ribose moieties from nicotinamide adenine dinucleotide (NAD) to itself and to a number of other proteins involved in DNA and chromatin metabolism. The covalent addition of negatively charged ADP-ribose oligomers alters the interactions of these proteins with DNA and with other proteins. PARP1 recruits the BER protein XRCC1; together with DNA polymerase β and DNA ligase III, XRCC1 facilitates repair of the lesion. PARP1 has also been implicated in the recognition of DNA double-strand breaks (DSB) and in the recruitment of DNA-dependent protein kinase in DSB repair (see below), as well as in cell death pathways, modification of chromatin structure, transcriptional regulation, and mitotic apparatus function.
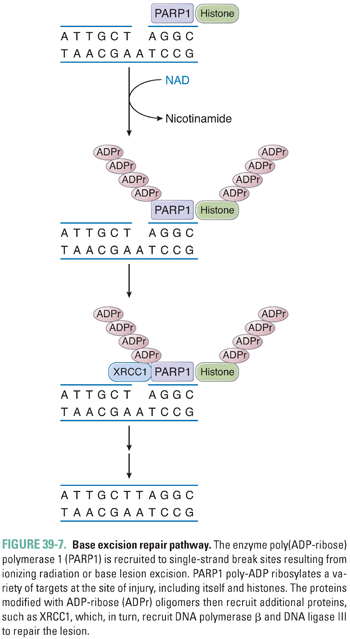
In response to the formation of bulky adducts that distort the DNA double helix, such as those induced by ultraviolet radiation and DNA-damaging chemotherapeutic agents, a complex set of proteins recognizes and initiates repair of the lesion via a process termed nucleotide excision repair (NER). Repair involves local opening of the double helix around the site of the damage, incision of the damaged strand on both sides of the lesion, excision of the oligonucleotide containing the lesion, and, finally, DNA repair synthesis and ligation. The endonuclease ERCC1 plays an important role in targeted excision of the DNA lesion. The genes involved in nucleotide excision repair were in part identified from study of the clinical syndromes xeroderma pigmentosa and Cockayne syndrome, which are rare photosensitivity disorders that exhibit defects in NER.
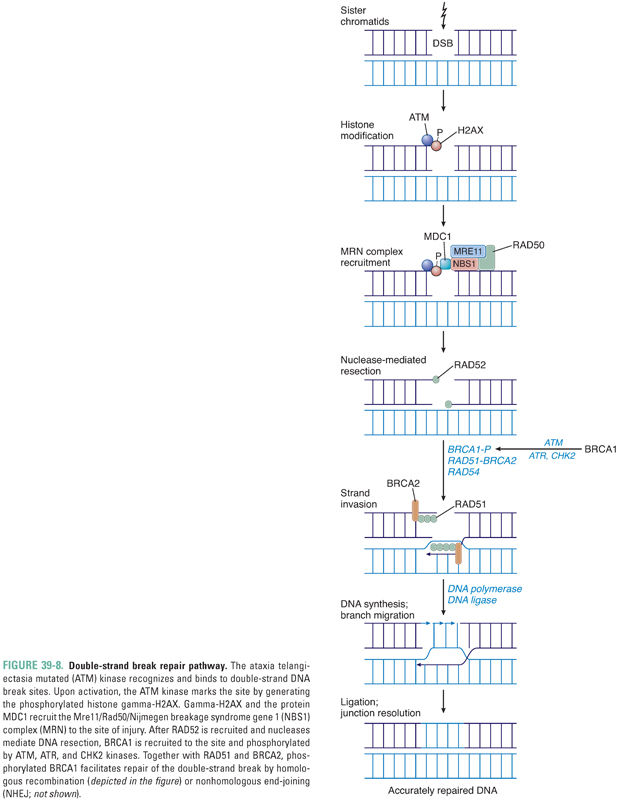
In response to a double-strand break, activation of the ataxia telangiectasia mutated (ATM) kinase results in generation of the phosphorylated histone gamma-H2AX at the site of the break. Together with the protein MDC1, gamma-H2AX recruits to the locus of DNA damage a complex (MRN) containing the proteins Mre11, Rad50, and Nijmegen breakage syndrome gene 1 (NBS1) (Fig. 39-8). The breast and ovarian cancer susceptibility gene product BRCA1 is also phosphorylated by the kinases ATM, ATR, and CHK2 in response to the double-strand break, and phosphorylated BRCA1, RAD51, and BRCA2 are also recruited to the break site. Subsequent repair is mediated either by homologous recombination, with formation and resolution of a Holliday junction (Fig. 39-8), or by nonhomologous end-joining (NHEJ), in which DNA-dependent protein kinase and a complex of proteins, including XRCC4, catalyze nucleolytic processes that allow end-joining by DNA ligase IV. The DNA repair effected by homologous recombination is more accurate than that mediated by NHEJ.
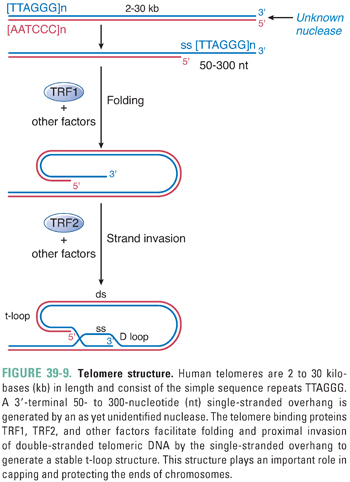
Human telomeres consist of the simple repeat sequence TTAGGG. These repeats are shaped, folded, and bound by a complex of proteins to form a unique structure termed a t-loop (Fig. 39-9). In the t-loop structure, a long single-stranded overhang at the 3′ end of the DNA invades the proximal double-stranded DNA component; this process is facilitated by TRF1, TRF2, and other protein factors. The t-loop and its associated complex of proteins are thought to play important roles in capping and protecting the chromosome end, as well as protecting telomeres from recognition by the DNA damage checkpoint machinery.
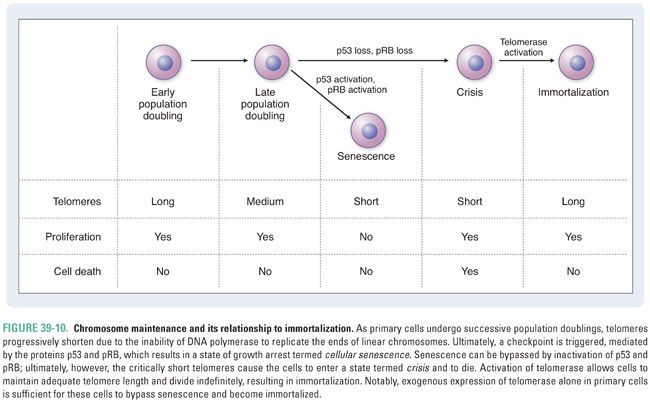
Because DNA polymerase is unable to replicate the ends of linear chromosomes completely, telomeres shorten with each division in normal cells. Telomere shortening ultimately results in disruption of the telomeric caps, activation of a DNA damage checkpoint, and a state of cell cycle arrest termed cellular senescence (Fig. 39-10). When cells are able to bypass this checkpoint via inactivation of the tumor suppressor protein p53, which normally regulates cell cycle arrest or apoptosis in response to DNA damage, chromosome fusions are observed. It is thought that the progressive shortening of telomeres with age promotes genomic instability and contributes to oncogenesis. However, cells also continue to die under these conditions. Activation of the enzyme telomerase, which is a reverse transcriptase that uses an RNA template to synthesize TTAGGG repeats, allows cells to restore telomere length and divide indefinitely. Telomerase activation is observed in normal germline cells and some stem cell populations and has been shown to maintain the presence of the 3′ overhang in normal cells. The immortalization process associated with telomerase activation is also essential for tumor formation and maintenance. In a minority of tumors, an alternative lengthening of telomeres (ALT) pathway is activated.
Once a cell has replicated its DNA, it is prepared to undergo mitosis. In this process, chromosomes condense and are segregated into two identical daughter cells. The cell cycle transitions from DNA replication (S phase) to G2 phase and then to mitosis (M phase) are complex and depend on the coordinated action of a number of so-called cyclin-dependent kinases (CDKs; see Chapter 33). Progression through mitosis is also facilitated by enzymes that include aurora kinases and polo-like kinases. Many cancer cells exhibit dysregulation of cell cycle timing and abnormalities in mitosis. Thus, pharmacologic inhibition of these regulatory kinases is an active area of cancer research. Currently, however, the microtubule machinery represents the primary target of agents that act in mitosis.
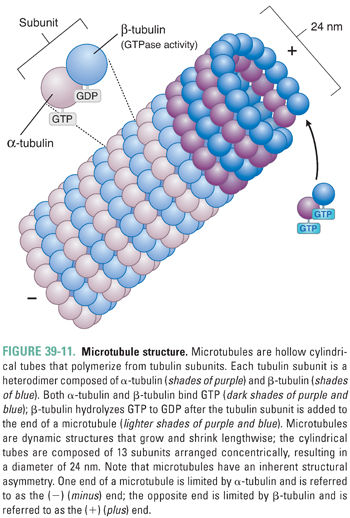
Microtubules are cylindrical, hollow fibers composed of polymers of tubulin, which is a heterodimeric protein consisting of α-tubulin and β-tubulin subunits (Fig. 39-11). α-Tubulin and β-tubulin are encoded by separate genes, but they have similar three-dimensional structures. Both α- and β-tubulin bind GTP; in addition, β-tubulin (but not α-tubulin) can hydrolyze GTP to GDP. Microtubules originate from a central microtubule organizing center (the centrosome, which includes two centrioles and associated proteins), where γ-tubulin (a protein with homology to α-tubulin and β-tubulin) nucleates tubulin polymerization. Nascent microtubules assemble into protofilaments, which are longitudinal polymers of tubulin subunits. Each protofilament interacts laterally with two other protofilaments to form a hollow-core tube, 24 nm in diameter, which consists of 13 protofilaments arranged concentrically. Because tubulin is a heterodimer, this tube has inherent asymmetry; the end of a microtubule nearest the centrosome is bordered by α-tubulin and is called the (−) (minus) end, while the end of a microtubule extending from the centrosome is bordered by β-tubulin and is called the (+) (plus) end (Fig. 39-11). Tubulin units are added at different rates to the (−) and (+) ends; the (+) end grows (adds tubulin) twice as fast as the (−) end.
Microtubules are not static structures. Rather, they possess an inherent property known as dynamic instability (Fig. 39-12). Tubulin heterodimers add to the end of the microtubule with GTP bound to both α-tubulin and β-tubulin subunits. As the microtubule grows, the β-tubulin of each tubulin heterodimer hydrolyzes its GTP to GDP. The hydrolysis of GTP to GDP introduces a conformational change in tubulin that destabilizes the microtubule. The exact mechanism of this destabilization is unknown, but it may be related to a decrease in the strength of lateral protofilament interactions or an increase in the tendency for protofilaments to “curve” away from the straight microtubule.
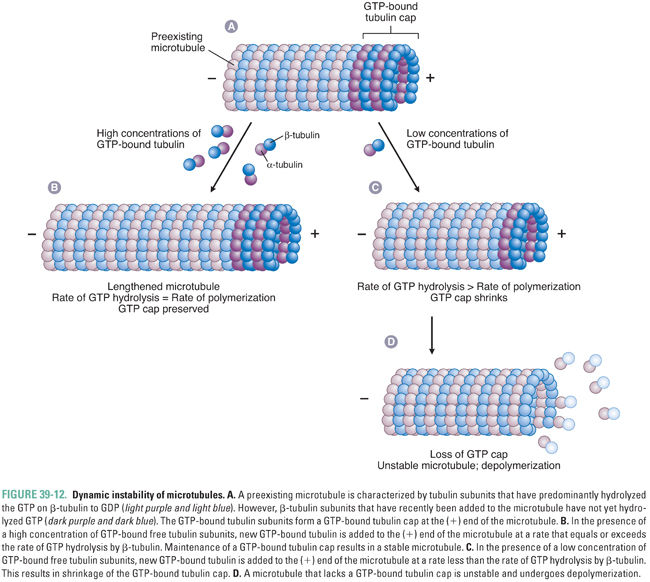
Therefore, microtubule stability is determined by the rate of microtubule polymerization relative to the rate of GTP hydrolysis by β-tubulin. If a microtubule polymerizes tubulin faster than β-tubulin hydrolyzes GTP to GDP, then, in the steady state, there is a cap of GTP-bound β-tubulin at the (+) end of the microtubule. This GTP cap provides stability to the microtubule structure, allowing further polymerization of the microtubule. Conversely, if tubulin polymerization proceeds more slowly than the hydrolysis of GTP to GDP by β-tubulin, then, in the steady state, the (+) end of the microtubule is enriched with GDP-bound β-tubulin. This GDP-bound tubulin conformation is unstable and causes rapid depolymerization of the microtubule. The ability of microtubules to assemble and disassemble rapidly is important for their many physiologic roles. Pharmacologic agents can disrupt microtubule function either by preventing the assembly of tubulin into microtubules or by stabilizing existing microtubules (and thereby preventing microtubule disassembly).
Microtubules have important physiologic roles in mitosis, intracellular protein trafficking, vesicular movement, and cell structure and shape. Mitosis is the physiologic role that is targeted pharmacologically; the other physiologic roles, however, predict many of the adverse effects of drugs that interrupt microtubule function.
Recall that microtubules nucleate from centrosomes, which consist of centrioles and other associated proteins. In mitosis, the two centrosomes align at opposite ends of the cell. Microtubules are extremely dynamic during M phase; they grow and shrink during M phase at rates much greater than during other phases of the cell cycle. This increased dynamic instability during M phase allows microtubules to locate and attach to the chromosomes. The microtubules emanating from each centrosome bind to kinetochores, which are proteins that attach to the centromere of a chromosome. Once the kinetochore of each chromosome is attached to a microtubule, microtubule-associated proteins act as motors to align the kinetochore-bound chromosomes at the equator of the cell (defined by the midpoint between the two centrosomes). When every chromosome has aligned at the equator, the microtubules shorten, separating a diploid pair of chromosomes into each half of the cell. Finally, cytokinesis (division of the cytoplasm) occurs, and two daughter cells are formed. Although many other proteins are involved in the regulation of mitosis, microtubules have a critical role in the process. Disruption of microtubule function freezes cells in M phase, leading eventually to the activation of programmed cell death (apoptosis).
 PHARMACOLOGIC CLASSES AND AGENTS
PHARMACOLOGIC CLASSES AND AGENTS
Traditional antineoplastic chemotherapy can be subdivided into several classes of agents. The antimetabolite drugs are compounds that either inhibit the enzymes involved in nucleotide synthesis and metabolism or are incorporated as analogues into DNA and result in chain termination or strand breaks. These drugs act primarily during the S phase of the cell cycle, when cells are undergoing DNA replication. Another broad class of agents, which induce cytotoxicity by modification of DNA structure and generation of DNA damage, includes alkylating agents, platinum compounds, bleomycin, and topoisomerase inhibitors. These drugs exert their effects during multiple phases of the cell cycle. The final category of agents inhibits microtubule assembly or depolymerization, disrupting the mitotic spindle and interfering with mitosis. For a summary of the major classes of chemotherapeutic agents, their cell cycle specificity, and major toxicities, see Table 41-2 (Chapter 41, Principles of Combination Chemotherapy).
Inhibitors of Thymidylate Synthase
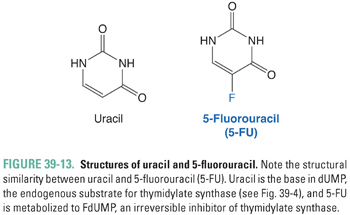
Thymidylate (dTMP) is synthesized by the methylation of 2′-deoxyuridylate (dUMP). This reaction, which is catalyzed by thymidylate synthase, requires MTHF as a cofactor (Fig. 39-4). 5-Fluorouracil (5-FU; Fig. 39-13) inhibits DNA synthesis, primarily by interfering with the biosynthesis of thymidylate. 5-FU is first converted to 5-fluoro-2′-deoxyuridylate (FdUMP) by the same pathways that convert uracil to dUMP. FdUMP then inhibits thymidylate synthase by forming, together with MTHF, a stable, covalent ternary enzyme–substrate–cofactor complex. Cells deprived of dTMP for a sufficient period of time undergo so-called thymineless death. 5-FU can also be metabolized to floxuridine triphosphate (FUTP), which can be incorporated into mRNA in place of uridylate and can thereby interfere with RNA processing. Either inhibition of thymidylate synthase by FdUMP or interference with RNA processing by FUTP, or a combination of the two mechanisms, could explain the toxic effect of 5-FU on cells. However, certain 5-FU congeners that inhibit thymidylate synthase but are not incorporated into RNA show antitumor efficacy similar to that of 5-FU. This finding points to thymidylate synthase inhibition as the dominant mechanism of 5-FU action.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


