1 Introduction
Sterilization is anessential stage in the processing of any product destined forparenteral administration, or for contact with broken skin, mucosal surfaces, or internal organs, where the threat of infection exists.In addition, the sterilization of microbiological materials, soiled dressings and other contaminated items is necessary to minimize the health hazard associated with these articles.
Sterilization processes involve the application of a biocidal agent or physical microbial removal process to a product or preparation with the object of killing or removing all microorganisms. These processes may involve elevated temperature,reactive gas, irradiation or filtration through a microorganism-proof filter. The success of the process depends on a suitable choice of treatment conditions, e.g. temperature and duration of exposure.It must be remembered, however, that with all articles to be sterilized there is a potential risk of product damage, which for a pharmaceutical preparation may result in reduced therapeutic efficacy, stability or patient acceptability. Thus, there is a need to achieve a balance between the maximum acceptable risk of failing to achieve sterility and the maximum level of product damage that is acceptable. This is best determined from a knowledge of the properties of the sterilizing agent, the properties of the product to be sterilized and the nature of the likely contaminants. A suitable sterilization process may then be selected to ensure maximum microbial kill/removal with minimum product deterioration.
2 Sensitivity of microorganisms
The general pattern of resistance of microorganisms to biocidal sterilization processes is independent of the type of agent employed (heat, radiation or gas), with vegetative forms of bacteria and fungi, along with the larger viruses, showing a greater sensitivity to sterilization processes than small viruses and bacterial or fungal spores. The choice of suitable reference organisms for testing the efficiency of sterilization processes (see section 12.3) is therefore made from the most durable bacterial spores; these are usually represented by Bacillus stearothermophilus for moist heat, certain strains of B.subtilis for dry heat and gaseous sterilization, and B.pumilus for ionizing radiation.
Ideally, when considering the level of treatment necessary to achieve sterility a knowledge of the type and total number of microorganisms present in a product, together with their likely response to the proposed treatment, is necessary. Without this information, however, it is usually assumed that organisms within the load are no more resistant than the reference spores or than specific resistant product isolates. In the latter case, it must be remembered that resistance may be altered or lost entirely by repeated laboratory subculture and the resistance characteristics of the maintained strain must be regularly checked.
A sterilization process may thus be developed without a full microbiological background to the product, instead being based on the ability to deal with a ‘worst case ’ condition. This is indeed the situation for official sterilization methods, which must be capable of general application, and modern pharmacopoeial recommendations are derived from a careful analysis of experimental data on bacterial spore survival following treatments with heat, ionizing radiation or gas.
However, the infectious agents responsible for spongiform encephalopathies (prions, see Chapters 5 and 20) such as bovine spongiform encephalopathy (BSE) and Creutzfeldt –Jakob disease (CJD) exhibit exceptional degrees of resistance to many lethal agents. Recent work has even cast doubt on the adequacy of the process of 18 minute exposure to steam at 134–138°C which has been recommended for the destruction of prions (and which far exceeds the lethal treatment required to achieve adequate destruction of bacterial spores).
2.1 Survivor curves
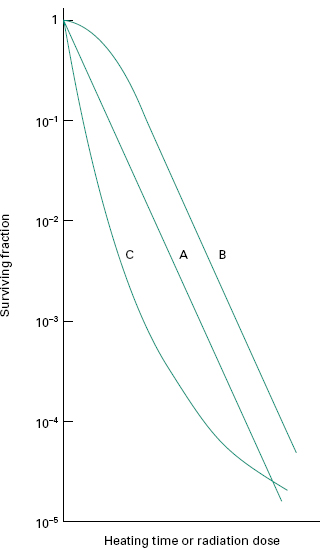
When exposed to a killing process, populations of micro-organisms generally lose their viability in an exponential fashion, independent of the initial number of organisms. This can be represented graphically with a ‘survivor curve ’ drawn from a plot of the logarithm of the fraction of survivors against the exposure time or dose (Figure 21.1). Of the typical curves obtained, all have a linear portion which may be continuous (plot A), or may be modified by an initial shoulder (B) or by a reduced rate of kill at low survivor levels (C). Furthermore, a short activation phase, representing an initial increase in viable count, may be seen during the heat treatment of certain bacterial spores. Survivor curves have been employed principally in the examination of heat sterilization methods, but can equally well be applied to any biocidal process.
2.2 Expressions of resistance
2.2.1 D-value
The resistance of an organism to a sterilizing agent can be described by means of the D-value. For heat and radiation treatments,respectively, this is defined as the time taken at a fixed temperature or the radiation dose required to achieve a 90% reduction in viable cells (i.e. a 1 log cycle reduction in survivors; Figure 21.2A). The calculation of the D-value assumes a linear type A survivor curve (Figure 21.1), and must be corrected to allow for any deviation from linearity with type B or C curves. Some typical D-values for resistant bacterial spores are given in Table 21.1.
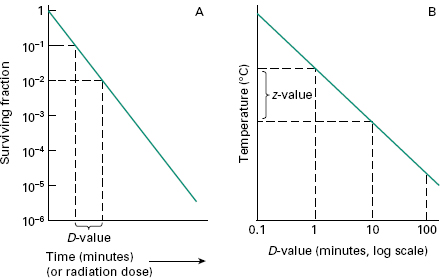
Table 21.1 Inactivation factors (IF) for selected sterilization protocols and their corresponding biological indicator (BI) organisms Sterilization protocol

2.2.2 Z-value
For heat treatment, a D-value only refers to the resistance of a microorganism at a particular temperature. In order to assess the influence of temperature changes on thermal resistance, a relationship between temperature and log D-value can be developed, leading to the expression of a Z-value, which represents the increase in temperature needed to reduce the D-value of an organism by 90% (i.e. 1 log cycle reduction; Figure 21.2 B). For bacterial spores used as biological indicators for moist heat (B. stearothermophilus) and dry heat (B. subtilis) sterilization processes, mean Z-values are given as 10 °C and 22 °C, respectively. The Z-value is not truly independent of temperature but may be considered essentially constant over the temperature ranges used in heat sterilization processes.
2.3 Sterility assurance
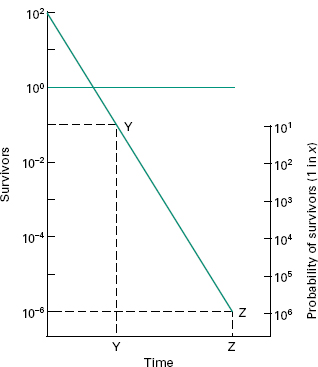
The term ‘sterile ’, in a microbiological context, means no surviving organisms whatsoever. Thus, there are no degrees of sterility; an item is either sterile or it is not, and so there are no levels of contamination which may be considered negligible or insignificant and therefore acceptable. From the survivor curves presented, it can be seen that the elimination of viable microorganisms from a product is a time-dependent process, and will be influenced by the rate and duration of biocidal action and the initial microbial contamination level. It is also evident from Figure 21.2 A that true sterility, represented by zero survivors, can only be achieved after an infinite exposure period or radiation dose. Clearly, then, it is illogical to claim, or expect, that a sterilization procedure will guarantee sterility. Thus, the likelihood of a product being produced free of microorganisms is best expressed in terms of the probability of an organism surviving the treatment process, a possibility not entertained in the absolute term ‘sterile ’. From this approach has arisen the concept of sterility assurance or a microbial safety index which gives a numerical value to the probability of a single surviving organism remaining to contaminate a processed product. For pharmaceutical products, the most frequently applied standard is that the probability, poststerilization, of a non-sterile unit is no more than 1 in 1 million units processed (i.e. ≤ 10− 6).The sterilization protocol necessary to achieve this with any given organism of known D-value can be established from the inactivation factor (IF) which may be defined as: where t is the contact time (for a heat or gaseous sterilization process) or dose (for ionizing radiation) and D is the D-value appropriate to the process employed.
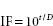
Thus, for an initial burden of 102 spores an inactivation factor of 108 will be needed to give the required sterility assurance of 10−6 (Figure 21.3). The sterilization process will therefore need to produce sufficient lethality to achieve an 8 log cycle reduction in viable organisms; this will require exposure of the product to eight times the D-value of the reference organism (8 D). In practice, it is generally assumed that the contaminant will have the same resistance as the relevant biological indicator spores unless full microbiological data are available to indicate otherwise. The inactivation factors associated with certain sterilization protocols and their biological indicator organisms are given in Table 21.1.
Figure 21.3 Sterility assurance. At Y, there is (literally) 10− 1 bacterium in one bottle, i.e. in 10 loads of single containers, there would be one chance in 10 that one load would be positive. Likewise, at Z, there is (literally) 10− 6 bacterium in one bottle, i.e. in 1 million (106) loads of single containers, there is one chance in 1 million that one load would be positive.
A sterilization process should always be considered a compromise between achieving good antimicrobial activity and maintaining product stability. It must, therefore, be validated against a suitable test organism and its efficacy continually monitored during use. Even so, a limit will exist as to the type and size of microbial challenge that can be handled by the process without signifi-cant loss of sterility assurance. Thus, sterilization must not be seen as a ‘catch-all ’ or as an alternative to Good Manufacturing Practice but must be considered as only the final stage in a programme of microbiological control. The European Pharmacopoeia recognizes five methods for the sterilization of pharmaceutical products: (1) steam sterilization (heating in an autoclave); (2) dry heat; (3) ionizing radiation; (4) gaseous sterilization; and (5) filtration. In addition, other approaches involving steam and formaldehyde and UV light have evolved for use in certain situations. For each method, the possible permutations of exposure conditions are numerous, but experience and product stability requirements have generally served to limit this choice. Nevertheless, it should be remembered that even the recommended methods and regimens do not necessarily demonstrate equivalent biocidal potential (see Table 21.1), but simply offer alternative strategies for application to a wide variety of product types. Thus, each should be validated in their application to demonstrate that the minimum required level of sterility assurance can be achieved (sections 2.3 and 9).
In the following sections, factors governing the successful use of these sterilizing methods will be covered and their application to pharmaceutical and medical products considered. Methods for monitoring the efficacy of these processes are discussed in section 12.
Heat is the most reliable and widely used means of sterilization, affording its antimicrobial activity through destruction of enzymes and other essential cell constituents. These lethal events proceed most rapidly in a fully hydrated state, thus requiring a lower heat input (temperature and time) under conditions of high humidity where denaturation and hydrolysis reactions predominate, rather than in the dry state where oxidative changes take place. This method of sterilization is limited to thermostable products, but can be applied to both moisture-sensitive and moisture-resistant items for which dry (160–180 °C) and moist (121–134 °C) heat sterilization procedures are respectively used. Where thermal degradation of a product might possibly occur, it can usually be minimized by selecting the higher temperature range, as the shorter exposure times employed generally result in a lower fractional degradation.
4.1 Sterilization p rocesses
In any heat sterilization process, the articles to be treated must first be raised to sterilization temperature and this involves a heating-up stage. In the traditional approach, timing for the process (the holding time) then begins. It has been recognized, however, that during both the heating-up and cooling-down stages of a sterilization cycle (Figure 21.4), the product is held at an elevated temperature and these stages may thus contribute to the overall biocidal potential of the process.
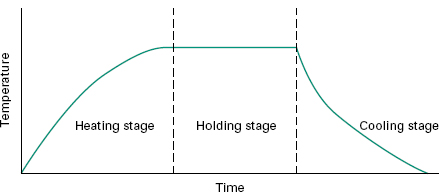
A method has been devised to convert all the temperature–time combinations occurring during the heating, sterilizing and cooling stages of a moist heat (steam) sterilization cycle to the equivalent time at 121 °C. This involves following the temperature profile of a load, integrating the heat input (as a measure of lethality), and converting it to the equivalent time at the standard temperature of 121 °C. Using this approach, the overall lethality of any process can be deduced and is defined as the F-value; this expresses heat treatment at any temperature as equal to that of a certain number of minutes at 121 °C. In other words, if a moist heat sterilization process has an F-value of x, then it has the same lethal effect on a given organism as heating at 121 °C for x minutes, irrespective of the actual temperature employed or of any fluctuations in the heating process due to heating and cooling stages. The F-value of a process will vary according to the moist heat resistance of the reference organism; when the reference spore is that of B. stearothermophilus with a Z-value of 10 °C, then the F-value is known as the F 0-value.
A relationship between F-and D-values, leading to an assessment of the probable number of survivors in a load following heat treatment, can be established from the following equation: where D is the D-value at 121 °C, and N 0 and N represent, respectively, the initial and final number of viable cells per unit volume.
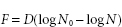
The F-concept has evolved from the food industry and principally relates to the sterilization of articles by moist heat. Because it permits calculation of the extent to which the heating and cooling phases contribute to the overall killing effect of the autoclaving cycle, the F-concept enables a sterilization process to be individually developed for a particular product. This means that adequate sterility assurance can be achieved in autoclaving cycles in which the traditional pharmacopoeial recommendation of 15 minutes at 121 °C is not achieved. The holding time may be reduced below 15 minutes if there is a substantial killing effect during the heating and cooling phases, and an adequate cycle can be achieved even if the ‘target ’ temperature of 121 °C is not reached. Thus, F-values offer both a means by which alternative sterilizing cycles can be compared in terms of their microbial killing efficiency, and a mechanism by which overprocessing of marginally thermolabile products can be reduced without compromising sterility assurance. The European Pharmacopoeia emphasizes that when a steam sterilization cycle is designed on the basis of F 0 data, it may be necessary to perform continuous and rigorous microbio-logical monitoring of the bioburden (section 10) during routine manufacturing in order consistently to achieve an acceptable sterility assurance level.
F 0 values may be calculated either from the area under the curve of a plot of autoclave temperature against time constructed using special chart paper on which the temperature scale is modified to take into account the progressively greater lethality of higher temperatures, or by use of the equation: where Δ t is the time interval between temperature measurements, T is the product temperature at time t, and Z is (assumed to be) 10 °C.
Thus, if temperatures were being recorded from a thermocouple at 1.00 minute intervals then Δ t 1.00, and a temperature of, for example, 115 °C maintained for 1 minute would give an F 0 value of 1 minute × 10 (115–121)/10, which is equal to 0.25 minutes. In practice, such calculations could easily be performed on the data from several thermocouples within an autoclave using suitable software, and, in a manufacturing situation, these would be part of the batch records. Application of the F-value concept has been largely restricted to steam sterilization processes, although there is a less frequently employed, but direct parallel in dry heat sterilization (see section 4.3).
4.2 Moist h eat sterilization
Moist heat has been recognized as an efficient biocidal agent from the early days of bacteriology, when it was principally developed for the sterilization of culture media. It now finds widespread application in the processing of many thermostable products and devices. In the pharmaceutical and medical sphere it is used in the sterilization of dressings, sheets, surgical and diagnostic equipment, containers and closures, and aqueous injections, ophthalmic preparations and irrigation fluids, in addition to the processing of soiled and contaminated items (Chapter 22).
Sterilization by moist heat usually involves the use of steam at temperatures in the range 121–134 °C, and while alternative strategies are available for the processing of products unstable at these high temperatures, they rarely offer the same degree of sterility assurance and should be avoided if at all possible. The elevated temperatures generally associated with moistheat sterilization methods can only be achieved by the generation of steam under pressure.
aCalculated for a spore suspension having a D 121 of 1.5 min and a Z-value of 10 °C.
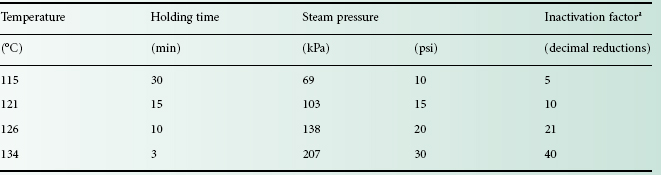
By far the most commonly employed standard temperature/time cycles for bottled fluids and porous loads (e.g. surgical dressings) are 121 °C for 15 minutes and 134 °C for 3 minutes, respectively. Not only do high-temperature–short time cycles often result in lower fractional degradation, they also afford the advantage of achieving higher levels of sterility assurance due to greater inactivation factors (Table 21.2). Before the publication of the 1988 British Pharmacopoeia the 115 °C for 30 minute cycle was considered an acceptable alternative to 121 °C for 15 minutes, but it is no longer considered sufficient to give the desired sterility assurance levels for products which may contain significant concentrations of thermophilic spores.
Table 21.2 Pressure–temperature relationships and antimicrobial efficacies of alternative steam sterilization cycles
4.2.1 Steam as a sterilizing a gent
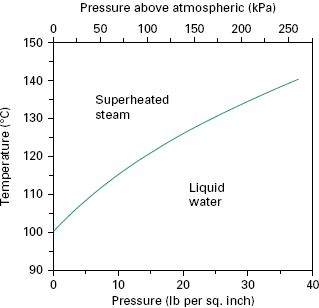
To act as an efficient sterilizing agent, steam should be able to provide moisture and heat efficiently to the article to be sterilized. This is most effectively done using saturated steam, which is steam in thermal equilibrium with the water from which it is derived, i.e. steam on the phase boundary (Figure 21.5). Under these circumstances, contact with a cooler surface causes condensation and contraction, drawing in fresh steam and leading to the immediate release of the latent heat, which represents approximately 80% of the total heat energy. In this way, heat and moisture are imparted rapidly to articles being sterilized and dry porous loads are quickly penetrated by the steam.
Steam for sterilization can either be generated within the sterilizer, as with portable bench or ‘instrument and utensil ’ sterilizers, in which case it is constantly in contact with water and is known as ‘wet ’ steam, or can be supplied under pressure (350–400 kPa) from a separate boiler as ‘dry ’ saturated steam with no entrained water droplets. The killing potential of ‘wet ’ steam is the same as that of ‘dry ’ saturated steam at the same temperature, but it is more likely to soak a porous load, creating phys ical difficulties for further steam penetration. Thus, major industrial and hospital sterilizers are usually supplied with ‘dry ’ saturated steam and attention is paid to the removal of entrained water droplets within the supply line to prevent introduction of a water ‘fog ’ into the sterilizer.
If the temperature of ‘dry ’ saturated steam is increased, then, in the absence of entrained moisture, the relative humidity or degree of saturation is reduced and the steam becomes superheated (Figure 21.5). During sterilization this can arise in a number of ways, for example by overheating the steam-jacket (see section 4.2.2), by using too dry a steam supply, by excessive pressure reduction during passage of steam from the boiler to the sterilizer chamber, and by evolution of heat of hydration when steaming overdried cotton fabrics. Superheated steam behaves in the same manner as hot air as condensation and release of latent heat will not occur unless the steam is cooled to the phase boundary temperature. Thus, it proves to be an inefficient sterilizing agent and, although a small degree of transient superheating can be tolerated, a maximum acceptable level of 5 °C superheat is set, i.e. the temperature of the steam is never greater than 5 °C above the phase boundary temperature at that pressure.
The relationship between temperature and pressure holds true only in the presence of pure steam; adulteration with air contributes to a partial pressure but not to the temperature of the steam. Thus, in the presence of air the temperature achieved will reflect the contribution made by the steam and will be lower than that normally attributed to the total pressure recorded. Addition of further steam will raise the temperature but residual air surrounding articles may delay heat penetration or, if a large amount of air is present, it may collect at the bottom of the sterilizer, completely altering the temperature profile of the sterilizer chamber. It is for these reasons that efficient air removal is a major aim in the design and operation of a boiler-fed steam sterilizer.
4.2.2 Sterilizer design and operation
Steam sterilizers, or autoclaves as they are also known, are stainless steel vessels designed to withstand the steam pressures employed in sterilization. They can be: ‘portable ’ sterilizers, which generally have internal electric heaters to produce steam and are used for small pilot or laboratory-scale sterilization and for the treatment of instruments and utensils; or large-scale sterilizers for routine hospital or industrial use, operating on ‘dry ’ saturated steam from a separate boiler (Figure 21.6). Because of their widespread use within pharmacy this latter type will be considered in greatest detail.
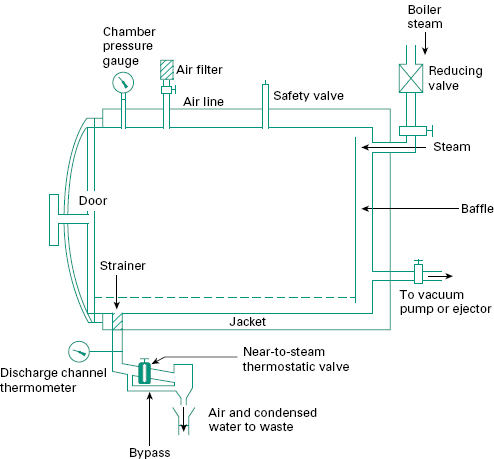
There are two main types of large sterilizers, those designed for use with porous loads (i.e. dressings) and generally operated at a minimum temperature of 134 °C, and those designed as bottled fluid sterilizers employing a minimum temperature of 121 °C. The stages of operation are common to both and can be summarized as air removal and steam admission, heating-up and exposure, and drying or cooling. Many modifications of design exist and in this section only general features will be considered. Fuller treatments of sterilizer design and operation can be found in the relevant Department of Health technical memorandums (DH 1995, DH 1997).
4.2.2.1 General design features
Steam sterilizers are constructed with either cylindrical or rectangular chambers, with preferred capacities ranging from 400 to 800 L. They can be sealed by either a single door or by doors at both ends (to allow through-passage of processed materials; see Chapter 23). During sterilization the doors are held closed by a locking mechanism which prevents opening when the chamber is under pressure and until the chamber has cooled to a preset temperature, typically 80 °C.
In the larger sterilizers the chamber may be surround ed by a steam jacket which can be used to heat the autoclave chamber and promote a more uniform temperature throughout the load. The same jacket can also be filled with water at the end of the cycle to facilitate cooling and thus reduce the overall cycle time. The chamber floor slopes towards a discharge channel through which air and condensate can be removed. Temperature is monitored within the opening of the discharge channel and by thermocouples in dummy packages; jacket and chamber pressures are followed using pressure gauges. In hospitals and industry, it is common practice to operate sterilizers on an automatic cycle, each stage of operation being controlled by a timer responding to temperature-or pressure-sensing devices.
The stages of operation are as follows.
1 Air removal and steam admission. Air can be removed from steam sterilizers either by downward displacement with steam, evacuation or a combination of the two. In the downward displacement sterilizer, the heavier cool air is forced out of the discharge channel by incoming hot steam. This has the benefit of warming the load during air removal, which aids the heating-up process. It finds widest application in the sterilization of bottled fluids where bottle breakage may occur under the combined stresses of evacuation and high temperature. For more air-retentive loads (i.e. dressings), however, this technique of air removal is unsatisfactory and mechanical evacuation of the air is essential before admission of the steam. This can either be to an extremely high level (e.g.2.5 kPa) or can involve a period of pulsed evacuation and steam admission, the latter approach improving air extraction from dressings packs. After evacuation, steam penetration into the load is very rapid and heating-up is almost instantaneous. It is axiomatic that packaging and loading of articles within a sterilizer be so organized as to facilitate air removal.
During the sterilization process, small pockets of entrained air may still be released, especially from packages, and this air must be removed. This is achieved in a porous load autoclave with a near-to-steam thermostatic valve incorporated in the discharge channel. The valve operates on the principle of an expandable bellows containing a volatile liquid which vaporizes at the temperature of saturated steam thereby closing the valve, and condenses on the passage of a cooler air-steam mixture, thus reopening the valve and discharging the air. Condensate generated during the sterilization process can also be removed by this device. Small quantities of air will not, however, lower the temperature sufficiently to operate the valve and so a continual slight flow of steam is maintained through a bypass around the device in order to flush away residual air.
It is common practice to package sterile fluids, especially intravenous fluids, in flexible plastic containers. During sterilization these can develop a considerable internal pressure in the airspace above the fluid and it is therefore necessary to maintain a proportion of air within the sterilizing chamber to produce sufficient overpressure to prevent these containers from bursting (air ballasting). In sterilizers modified or designed to process this type of product, air removal is therefore unnecessary but special attention must be paid to the prevention of air ‘layering ’ within the chamber. This is overcome by the inclusion of a fan or through a continuous spray of hot water within the chamber to mix the air and steam. Air ballasting can also be employed to prevent bottle breakage.
2 Heating-up and exposure. When the sterilizer reaches its operating temperature and pressure the sterilization stage begins. The duration of exposure may include a heating-up time in addition to the holding time and this will normally be established using thermocouples in dummy articles.
3 Drying or cooling. Dressings packs and other porous loads may become dampened during the sterilization process and must be dried before removal from the chamber. This is achieved by steam exhaust and application of a vacuum, often assisted by heat from the steam-filled jacket if fitted. After drying, atmospheric pressure within the chamber is restored by admission of sterile filtered air.
For bottled fluids the final stage of the sterilization process is cooling, and this needs to be achieved as rapidly as possible to minimize thermal degradation of the product and to reduce processing time. In modem sterilizers, this is achieved by circulating water in the jacket that surrounds the chamber or by spray-cooling with retained condensate delivered to the surface of the load by nozzles fitted into the roof of the sterilizer chamber. This is often accompanied by the introduction of filtered, compressed air to minimize container breakage due to high internal pressures (air ballasting). Containers must not be removed from the sterilizer until the internal pressure has dropped to a safe level, usually indicated by a temperature of less than 80 °C. Occasionally, spray-cooling water may be a source of bacterial contamination and its microbiological quality must be carefully monitored.
4.3 Dry heat sterilization
The lethal effects of dry heat on microorganisms are due largely to oxidative processes, which are less effective than the hydrolytic damage which results from exposure to steam. Thus, dry heat sterilization usually employs higher temperatures in the range 160–180 °C and requires exposure times of up to 2 hours depending on the temperature employed.
Again, bacterial spores are much more resistant than vegetative cells and their recorded resistance varies markedly depending on their degree of dryness. In many early studies on dry heat resistance of spores their water content was not adequately controlled, so conflicting data arose regarding the exposure conditions necessary to achieve effective sterilization. This was partly responsible for variations in recommended exposure temperatures and times in different pharmacopoeias.
Dry heat application is generally restricted to glassware and metal surgical instruments (where its good penetrability and non-corrosive nature are of benefit), non-aqueous thermostable liquids and thermostable powders (see Chapter 22). In practice, the range of materials that are actually subjected to dry heat sterilization is quite limited, and consists largely of items used in hospitals. The major industrial application is in the sterilization of glass bottles which are to be filled aseptically, and here the attraction of the process is that it not only achieves an adequate sterility assurance level, but that it may also destroy bacterial endotoxins (products of Gram-negative bacteria, also known as pyrogens, that cause fever when injected into the body). These are difficult to eliminate by other means. For the purposes of depyrogenation of glass, temperatures of approximately 250 °C are used.
The F-value concept that was developed for steam sterilization processes has an equivalent in dry heat sterilization although its application has been limited. The FH designation describes the lethality of a dry heat process in terms of the equivalent number of minutes exposure at 170 °C, and in this case a Z-value of 20 °C has been found empirically to be appropriate for calculation purposes; this contrasts with the value of 10 °C which is typically employed to describe moist heat resistance.
4.3.1 Sterilizer design
Dry heat sterilization is usually carried out in a hot-air oven which comprises an insulated polished stainless steel chamber, with a usual capacity of up to 250 L, surrounded by an outer case containing electric heaters located in positions to prevent cool spots developing inside the chamber. A fan is fitted to the rear of the oven to provide circulating air, thus ensuring more rapid equilibration of temperature. Shelves within the chamber are perforated to allow good airflow. Thermocouples can be used to monitor the temperature of both the oven air and articles contained within. A fixed temperature sensor connected to a chart or digital recorder provides a permanent record of the sterilization cycle. Appropriate door-locking controls should be incorporated to prevent interruption of a sterilization cycle once begun.
Recent sterilizer developments have led to the use of dry heat sterilizing tunnels where heat transfer is achieved by infrared irradiation or by forced convection in filtered laminar airflow tunnels. Items to be sterilized are placed on a conveyer belt and pass through a high-temperature zone (250–300 + °C) over a period of several minutes.
4.3.2 Sterilizer operation
Articles to be sterilized must be wrapped or enclosed in containers of sufficient strength and integrity to provide good poststerilization protection against contamination. Suitable materials are paper, cardboard tubes or aluminium containers. Container shape and design must be such that heat penetration is encouraged in order to shorten the heating-up stage; this can be achieved by using narrow containers with dull, non-reflecting surfaces. In a hot-air oven, heat is delivered to articles principally by radiation and convection; thus, they must be carefully arranged within the chamber to avoid obscuring centrally placed articles from wall radiation or impending air flow. The temperature variation within the chamber should not exceed ± 5 °C of the recorded temperature. Heating-up times, which may be as long as 4 hours for articles with poor heat-conducting properties, can be reduced by preheating the oven before loading. Following sterilization, the chamber temperature is usually allowed to fall to around 40 °C before removal of sterilized articles; this can be accelerated by the use of forced cooling with filtered air.
The chemically reactive gases ethylene oxide [(CH 2) 2 O] and formaldehyde [(methanal, H.CHO)] possess broad-spectrum biocidal activity, and have found application in the sterilization of reusable surgical instruments, certain medical, diagnostic and electrical equipment, and the surface sterilization of powders. Sterilization processes using ethylene oxide sterilization are far more commonly used on an international basis than those employing formaldehyde.
Ethylene oxide treatment can also be considered as an alternative to radiation sterilization in the commercial production of disposable medical devices (Chapter 22). These techniques do not, however, offer the same degree of sterility assurance as heat methods and are generally reserved for temperature-sensitive items.
The mechanism of antimicrobial action of the two gases is assumed to be through alkylation of sulphydryl, amino, hydroxyl and carboxyl groups on proteins and imino groups of nucleic acids. At the concentrations employed in sterilization protocols, type A survivor curves (section 2.1, Figure 21.1) are produced, the lethality of these gases increasing in a non-uniform manner with increasing concentration, exposure temperature and humidity. For this reason, sterilization protocols have generally been established by an empirical approach using a standard product load containing suitable biological indicator test strips (section 12.3). Concentration ranges (given as weight of gas per unit chamber volume) are usually of the order of 800–1200 mg/L for ethylene oxide and 15–100 mg/L for for maldehyde, with operating temperatures in the region of 45–63 °C and 70–75 °C, respectively. Even at the higher concentrations and temperatures, the sterilization processes are lengthy and therefore unsuitable for the resterilization of high-turnover articles. Further delays occur because of the need to remove toxic residues of the gases before release of the items for use. In addition, because recovery of survivors in sterility tests is more protracted with gaseous sterilization methods than with other processes, an extended quarantine period may also be required.
As alkylating agents, both gases are potentially mutagenic and carcinogenic (as is the ethylene chlorohydrin that results from ethylene oxide reaction with chlorine); they also produce symptoms of acute toxicity including irritation of the skin, conjunctiva and nasal mucosa. Consequently, strict control of their atmospheric concentrations is necessary and safe working protocols are required to protect personnel. Table 21.3 summarizes the comparative advantages afforded by ethylene oxide and low-temperature steam and formaldehyde (LTSF) processes.
Table 21.3 Relative merits of ethylene oxide and low-temperature steam and formaldehyde (LTSF) processes
| Advantages of ethylene oxide over LTSF | Advantages of LTSF over ethylene oxide |
| Wider international regulatory acceptance | Less hazardous because formaldehyde is not flammable and is more readily detected by smell |
| Better gas penetration into plastics and rubber | The gas is obtained readily from aqueous solution (formalin) which is a more convenient source than gas in cylinders |
| Cycle times may be shorter | |
| Relatively slow to form solid polymers (with the potential to block pipes, etc.) | |
| With long exposure times it is possible to sterilize at ambient temperatures | |
| Very low incidence of product deterioration |
5.1 Ethylene oxide
Ethylene oxide gas is highly explosive in mixtures of more than 3.6% v/v in air; in order to reduce this explosion hazard it is usually supplied for sterilization purposes as a 10% mix with carbon dioxide, or as an 8.6% mixture with HFC 124 (2-chloro-1,1,1,2 tetrafluoroethane), which has replaced fluorinated hydrocarbons (freons). Alternatively, pure ethylene oxide gas can be used below atmospheric pressure in sterilizer chambers from which all air has been removed.
The efficacy of ethylene oxide treatment depends on achieving a suitable concentration in each article and this is assisted greatly by the good penetrating powers of the gas, which diffuses readily into many packaging materials including rubber, plastics, fabric and paper. This is not without its drawbacks, however, as the level of ethylene oxide in a sterilizer will decrease due to absorption during the process and the treated articles must undergo a desorption stage to remove toxic residues. Desorption can be allowed to occur naturally on open shelves, in which case complete desorption may take many days, e.g. for materials like PVC, or it may be encouraged by special forced-aeration cabinets where flowing, heated air assists gas removal, reducing desorption times to between 2 and 24 hours.
Organisms are more resistant to ethylene oxide treatment in a dried state, as are those protected from the gas by inclusion in crystalline or dried organic deposits. Thus, a further condition to be satisfied in ethylene oxide sterilization is attainment of a minimum level of moisture in the immediate product environment. This requires a sterilizer humidity of 30–70% and frequently a preconditioning of the load at relative humidities of more than 50%.
5.1.1 Sterilizer design and operation
An ethylene oxide sterilizer consists of a leak-proof and explosion-proof steel chamber, normally of 100–300 L capacity, which can be surrounded by a hot-water jacket to provide a uniform chamber temperature. Successful operation of the sterilizer requires removal of air from the chamber by evacuation, humidification and conditioning of the load by passage of subatmospheric-pressure steam followed by a further evacuation period and the admission of preheated vaporized ethylene oxide from external pressurized canisters or single-charge cartridges. Forced gas circulation is often employed to minimize variations in conditions throughout the sterilizer chamber. Packaging materials must be air-, steam-and gas-permeable to permit suitable conditions for sterilization to be achieved within individual articles in the load. Absorption of ethylene oxide by the load is compensated for by the introduction of excess gas at the beginning or by the addition of more gas as the pressure drops during the sterilization process. The same may also be true for moisture absorption, which is compensated for by supplementary addition of water to maintain appropriate relative humidity.
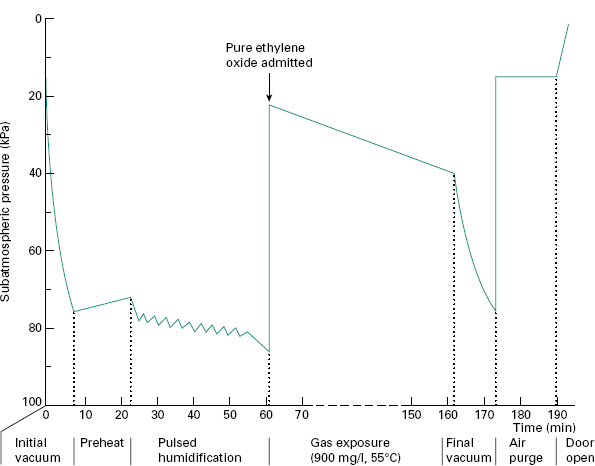
After treatment, the gases are evacuated either directly to the outside atmosphere or through a special exhaust system. Filtered, sterile air is then admitted either for a repeat of the vacuum/air cycle or for air purging until the chamber is opened. In this way, safe removal of the ethylene oxide is achieved, reducing the toxic hazard to the operator. Sterilized articles are removed directly from the chamber and arranged for desorption. The operation of an ethylene oxide sterilizer should be monitored and controlled automatically. A typical operating cycle for pure ethylene oxide gas is shown in Figure 21.7.
< div class='tao-gold-member'>