Effects of Drugs on Sleep Architecture
The sleep patterns of patients with insomnia vary widely but are often characterized by reduced amounts of slow-wave sleep and by one or more awakenings during the night. The time required to fall asleep (sleep latency) is usually prolonged, and the total sleep time is decreased in most patients with insomnia. Elderly adults often have a similar sleep pattern.
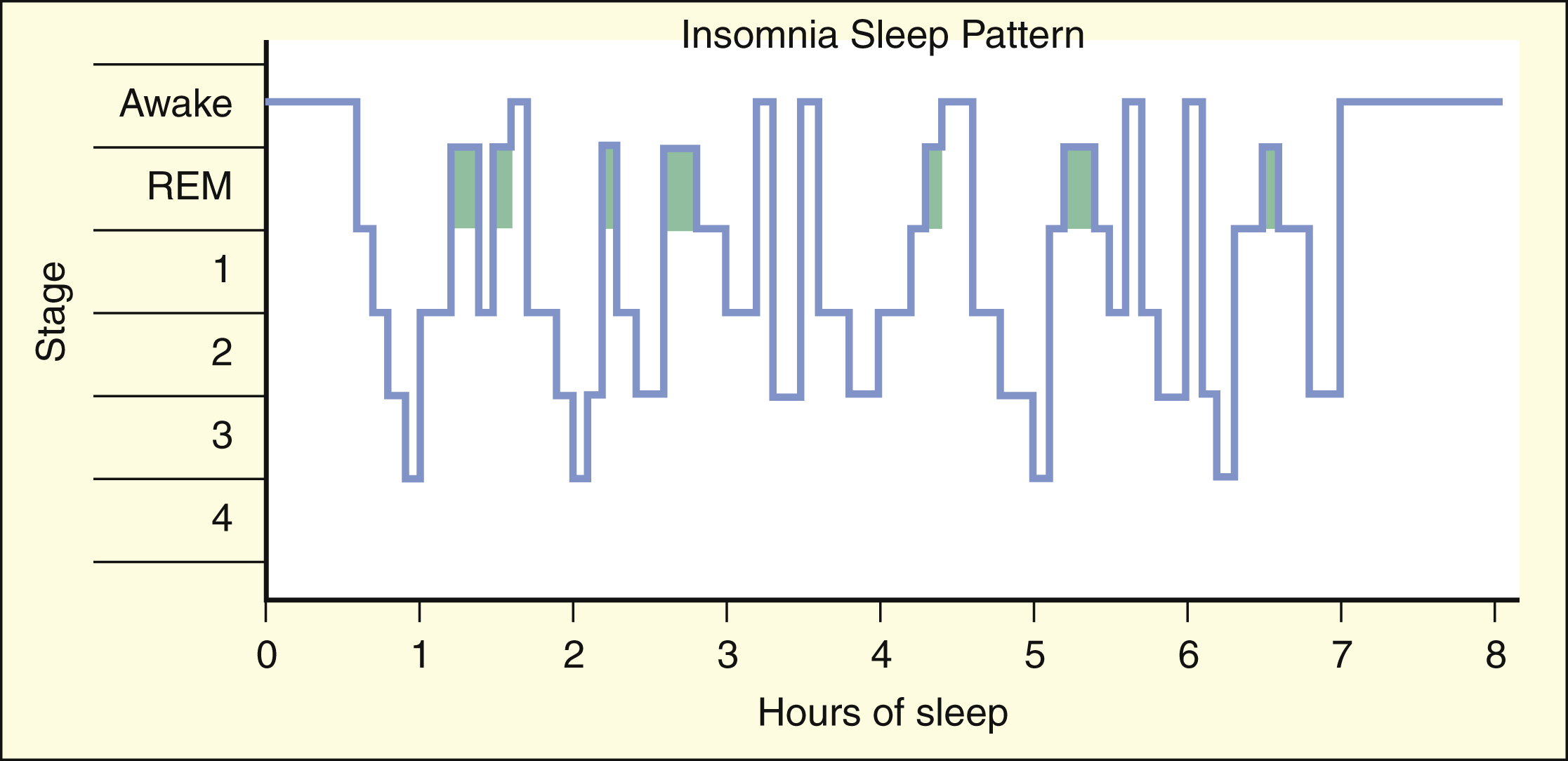
Benzodiazepines and most other hypnotics suppress the stages of 3, slow-wave sleep, and REM sleep. In contrast, zolpidem, zaleplon, eszopiclone, and ramelteon (new agents) have little effect on sleep architecture. Therefore, use of these new agents may better restore the sleep pattern to normal.

Sleep patterns change with age and are altered by sedative-hypnotic and other CNS drugs.
Neurologic Basis of Sleep
The neuronal systems involved in sleep include the basal forebrain nuclei and the reticular formation. Projecting from the basal forebrain to the cortex are cholinergic fibers that are believed to be involved in the induction of sleep. The basal forebrain is the only region of the brain that is active during slow-wave sleep and quiescent at other stages. The reticular formation facilitates the flow of sensory information from the thalamus to the cortex. When reticular nuclei are quiescent, the thalamus does not transfer information to the cortex, and this facilitates the onset of sleep.
Classification and Treatment of Sleep Disorders
Insomnia
Some patients with insomnia find it difficult to go to sleep or to stay asleep during the night, whereas others awaken too early in the morning. In general, the management of insomnia depends on whether the sleep disorder is caused by physiologic, psychologic, or medical conditions. As shown in Box 19–1, the patterns of sleep stages in patients with insomnia are irregular and include longer latency to fall asleep and frequent awakenings.
Occasional sleeplessness caused by acute stress or a minor illness is usually self-limiting and may not require treatment. More severe insomnia caused by medical conditions whose symptoms interfere with sleep is effectively treated with benzodiazepines or other sedative-hypnotic drugs, such as zolpidem and zaleplon, whereas insomnia related to psychologic and psychiatric disturbances is best managed with a combination of psychotherapy and sedative-hypnotic drugs. Benzodiazepines and most other hypnotic drugs decrease sleep latency (the time required to go to sleep) and increase sleep duration. The newest agents, zolpidem, zaleplon, eszopiclone, and ramelteon, have the advantages of not significantly affecting sleep architecture and not causing as much tolerance and dependence as do the older drugs. For these reasons, zolpidem, zaleplon, eszopiclone, and ramelteon have become the drugs of choice to treat most types of insomnia.
Other Sleep Disorders
Other sleep disorders include hypersomnia (difficulty in awakening), narcolepsy (sleep attacks), enuresis (bedwetting during sleep), somnambulism (sleepwalking), sleep apnea (episodes of hypoventilation during sleep), and nightmares and night terrors. Most of these disorders are managed with a combination of psychotherapy and antidepressant drugs or CNS stimulants. Sodium oxybate (Xyrem), a form of the abused drug γ-hydroxybutyrate, was recently approved for the treatment of cataplexy associated with narcoleptic attacks. CNS stimulants used to treat narcolepsy and other sleep disorders are discussed in Chapter 22.
SEDATIVE-HYPNOTIC DRUGS
The sedative-hypnotic drugs include benzodiazepines, barbiturates, some antihistamines, and a few nonbenzodiazepine agents, such as zolpidem, zaleplon, eszopiclone, and ramelteon. The properties of these drugs are summarized in Table 19–1, and their adverse effects, and drug interactions are listed in Table 19–2.
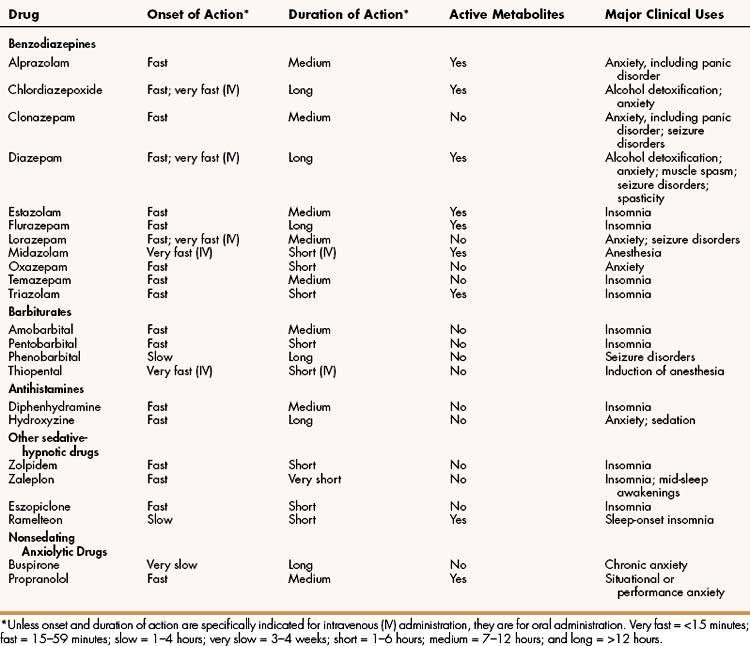
TABLE 19–2 Adverse Effects and Drug Interactions of Sedative-Hypnotic and Anxiolytic Drugs
| Drug | Common Adverse Effects | Common Drug Interactions |
|---|---|---|
| Benzodiazepines | ||
| Alprazolam | Arrhythmia, CNS depression, drug dependence, hypotension, and mild respiratory depression | Alcohol and other CNS depressants potentiate effects. Fluoxetine and fluvoxamine increase serum levels and effects |
| Chlordiazepoxide | Same as alprazolam | Alcohol and other CNS depressants potentiate effects. Cimetidine increases and rifampin decreases serum levels |
| Clonazepam | Same as alprazolam | Same as chlordiazepoxide |
| Diazepam | Same as alprazolam | Same as chlordiazepoxide |
| Estazolam | Same as alprazolam | Same as chlordiazepoxide |
| Flurazepam | Same as alprazolam | Same as chlordiazepoxide |
| Lorazepam | Same as alprazolam | Alcohol and other CNS depressants potentiate effects. Rifampin decreases serum levels |
| Midazolam | Same as alprazolam | Alcohol and other CNS depressants potentiate effects. Calcium channel blockers, erythromycin, and ketoconazole increase serum levels |
| Oxazepam | Same as alprazolam | Same as lorazepam |
| Temazepam | Same as alprazolam | Same as chlordiazepoxide |
| Triazolam | Amnesia, confusion, and delirium. Other adverse effects same as alprazolam | Alcohol and other CNS depressants potentiate effects. Cimetidine, erythromycin, ketoconazole, and oral contraceptives increase serum levels |
| Barbiturates | ||
| Amobarbital | CNS depression, drug dependence, and respiratory depression | Induces cytochrome P450 enzymes and increases metabolism of many drugs. Potentiates effects of other CNS depressants |
| Pentobarbital | Same as amobarbital | Same as amobarbital |
| Phenobarbital | Same as amobarbital | Same as amobarbital |
| Thiopental | Same as amobarbital | Same as amobarbital |
| Antihistamines | ||
| Diphenhydramine | Anticholinergic effects, such as blurred vision, dry mouth, and urinary retention; dizziness; and drowsiness | Alcohol, barbiturates, and other CNS depressants potentiate CNS effects |
| Hydroxyzine | Same as diphenhydramine | Same as diphenhydramine |
| Other sedative-hypnotic drugs | ||
| Zolpidem | Dizziness, drowsiness, and headache | Potentiation by alcohol and other CNSdepressants |
| Zaleplon | Same as zolpidem | Same as zolpidem |
| Eszopiclone | Drowsiness, dizziness, difficulty with coordination | Potentiation by alcohol and other CNS depressants |
| Ramelteon | Dizziness; drowsiness; alteration of reproductive hormone levels | Potentiation by alcohol and other CNS depressants. Fluvoxamine increases plasma ramelteon levels |
| Nonsedating Anxiolytic Drugs | ||
| Buspirone | Dizziness, headache, and nervousness | None |
| Propranolol | Bradycardia, bronchoconstriction, depression, fatigue, hypersensitivity, hypotension, impaired glycogenolysis, and vivid dreams | Cardiac depression increased by calcium channel blockers |
CNS = central nervous system.
Because the benzodiazepines have fewer adverse reactions and drug interactions and are safer in cases of overdose, they have largely replaced the barbiturates and other older drugs. Nevertheless, barbiturates are still used when benzodiazepines are ineffective or contraindicated. The sedating antihistamines are occasionally used to treat mild insomnia and anxiety and have less potential for abuse than do benzodiazepines and barbiturates. Many over-the-counter (nonprescription) sleep aids contain antihistamines as their effective ingredient.
Benzodiazepines
The benzodiazepines are a large group of drugs that have similar pharmacologic effects. They are so named because they share the common chemical structure of a benzene ring (benzo) joined to a seven-member ring containing two nitrogen molecules (diazepine). The particular use of specific drugs is largely determined by their pharmacokinetic properties and route of administration. Some benzodiazepines were developed and approved to treat anxiety, whereas others are approved for the management of insomnia or for other purposes.
Drug Properties
PHARMACOKINETICS
The pharmacokinetic properties of various benzodiazepines are compared in Table 19–1. The benzodiazepines are absorbed from the gut and distributed to the brain at rates that are proportional to their lipid solubility, which varies 50-fold among individual drugs in the class. As the plasma concentration of a benzodiazepine declines, the drug is redistributed from the brain to the blood, and this mechanism contributes significantly to the termination of its effects on the CNS.
All benzodiazepines are extensively metabolized in the liver. Most benzodiazepines are converted to active metabolites in phase I oxidative reactions catalyzed by cytochrome P450 enzymes. The active metabolites of chlordiazepoxide, diazepam, and flurazepam are long acting and contribute to the long duration of action of these agents. The active metabolites of alprazolam, estazolam, midazolam, and triazolam are shorter acting. Each of these active metabolites is eventually conjugated with glucuronate to form an inactive polar metabolite that is excreted in the urine. Other drugs, namely oxazepam, temazepam, and lorazepam, bypass phase I oxidation and are metabolized only by phase II conjugation (Fig. 19–1). These three drugs may be safer for use by elderly patients, because the capacity to conjugate drugs does not decline with age as much as the capacity for oxidative biotransformation does. Hence, these three drugs are less likely to accumulate to toxic levels in elderly patients.
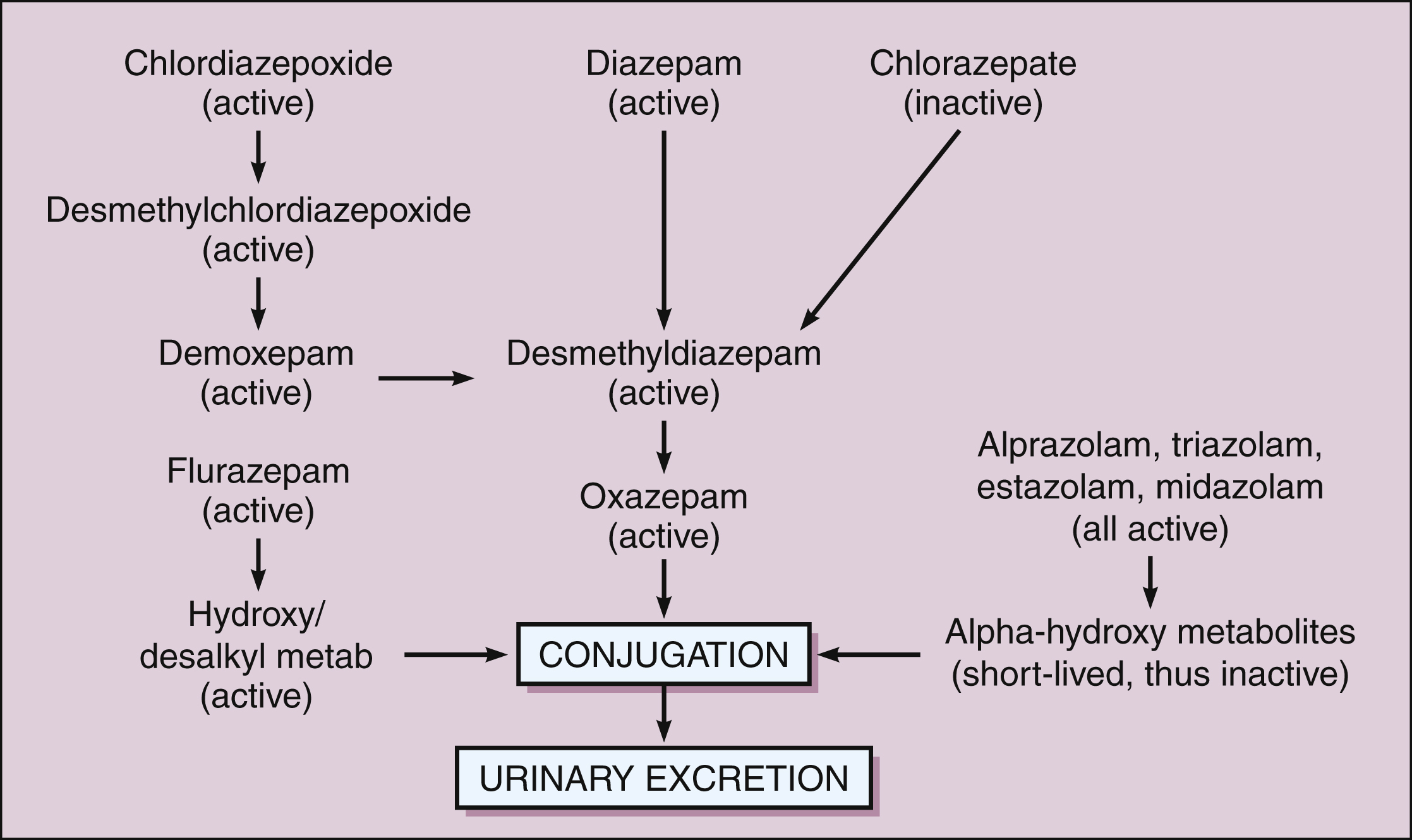
Figure 19–1 Major biotransformation pathways of benzodiazepines. Chlordiazepoxide and diazepam are converted to long-acting active metabolites. Alprazolam, midazolam, and triazolam are converted to a short-acting active metabolite. All benzodiazepines, including those with no active metabolites, are eventually converted to glucuronide compounds that are pharmacologically inactive and are excreted in the urine. The benzodiazepines that have no active metabolites include oxazepam, temazepam, and lorazepam (“out the liver”), and these may be the safest benzodiazepines to use in treating elderly patients.
Benzodiazepines also undergo some degree of enterohepatic cycling that prolongs their duration of action. In fact, some patients who are taking a drug such as diazepam may notice an increased sedative effect after eating a high-fat meal. The fatty meal causes the gallbladder to empty and thereby delivers bile containing diazepam to the intestines for reabsorption into the circulation.
MECHANISM OF ACTION
The benzodiazepines and barbiturates are believed to exert their effects on consciousness and sleep by facilitating the activity of γ-aminobutyric acid (GABA) at various sites in the neuraxis. GABA, the most ubiquitous inhibitory neurotransmitter in the CNS, regulates the excitability of neurons in almost every neuronal tract. As shown in Figure 19–2, the GABAA receptor–chloride ion channel has binding sites for benzodiazepines and barbiturates, as well as alcohols, steroids, and inhalational anesthetics. This receptor–ion channel complex is made up of five subunits with the major form of the complex containing α, β, and γ subunits. However, there are at least 16 types of these protein subunits (e.g., α1, α2, β1, β2, etc.), and it is not entirely clear which types of subunits make up the GABAA receptors that mediate the clinical and adverse effects of drugs that bind to them.
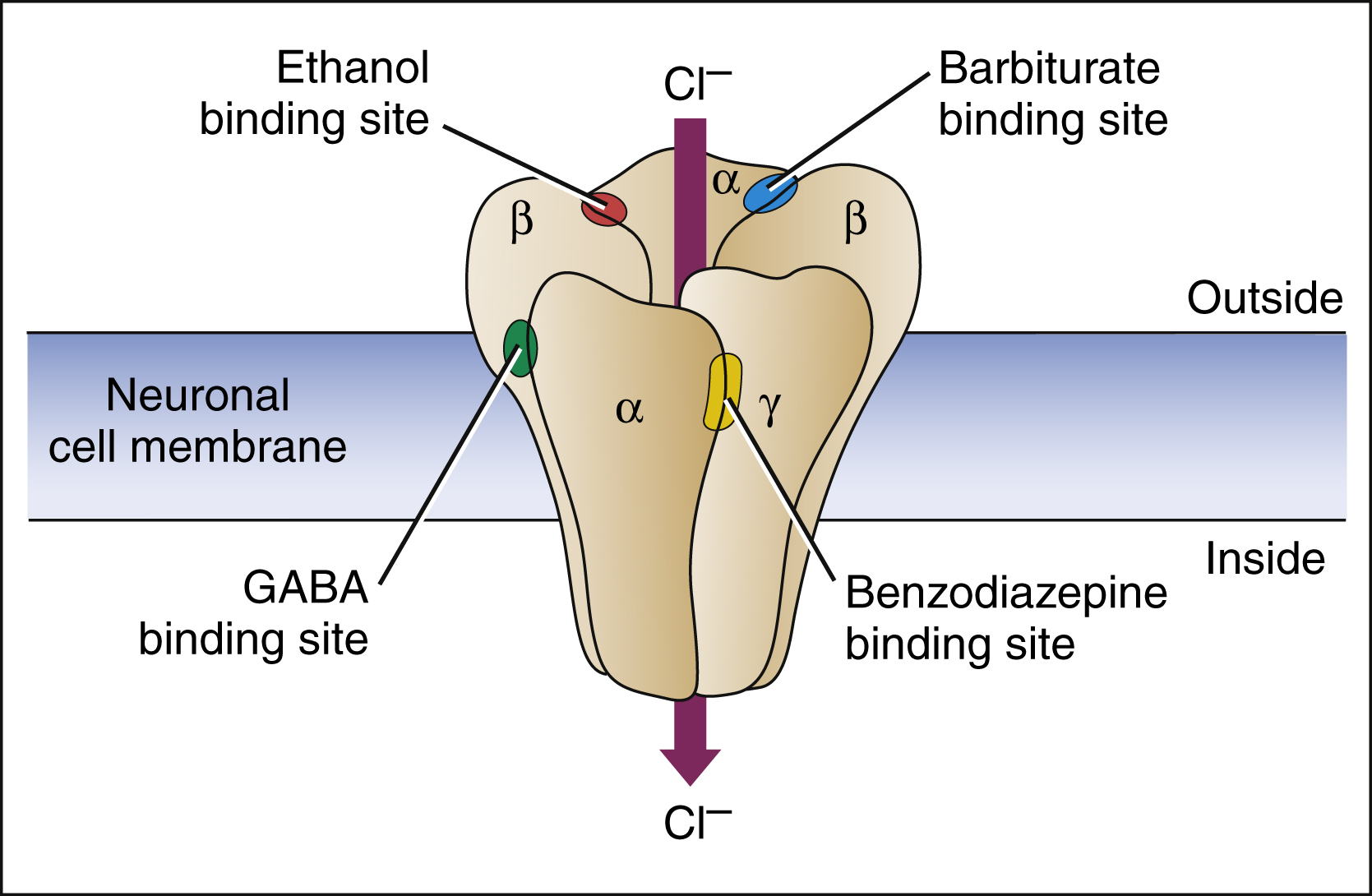
Figure 19–2 Receptor sites for GABA, benzodiazepines, barbiturates, and ethanol on the GABAA chloride ion channel. The GABAA chloride ion channel is a protein complex pentameric form that has varying combinations of α, β, and γ subunits. GABA binds to a site near the junction of α and β subunits, and this causes conformational changes that open the chloride ion channel and lead to neuronal membrane hyperpolarization. Benzodiazepines bind to an allosteric site formed by the cleft between α and γ subunits, and this facilitates GABA binding and increases the frequency of chloride channel opening. Barbiturates bind adjacent to α and β subunits and increase the duration of chloride channel opening, both in the presence and in the absence of GABA. Ethanol (ethyl alcohol) binds to a distinct site on the ionophore and enhances chloride influx. The ionophore also contains binding sites for steroids and inhalational anesthetics.
BOX 19–2 THE CASE OF THE GROGGY GRANDMOTHER
CASE PRESENTATION:
A 68-year-old female was prescribed 5 mg diazepam for anxiety by her primary care physician after her husband died. She was told to take one in the morning, along with her breakfast, and was advised to increase her activities to “take her mind off her worries.” After following the prescribed regimen for a week, she finds that she has problems going to sleep and without consulting her physician, takes another diazepam pill at night. She continues taking one diazepam pill in the morning and one pill at night for the next few days. Her friends note that she is not coming out of her room at the assisted living center as much as she used to, and they knock on her door to check up on her. They find her still in her nightgown and disorientated. They convince her to call her doctor who tells her to stop taking the second pill of diazepam at night; the doctor prescribes zolpidem, at a dose of 5 mg, to be taken at night to help her sleep. Over the next few days, the patient is more alert during the day and resumes her activities.
CASE DISCUSSION:
The use of benzodiazepines for anxiety disorders has largely replaced any other type of sedative-hypnotic agent, such as barbiturates, for this indication because of their increased safety and effectiveness. However, benzodiazepines are metabolized into many active metabolites, some that have longer durations of actions than their parent drug. Phase 1 or oxidative drug biotransformation (metabolism) in the elderly is reduced and this patient population is especially sensitive to the buildup of active metabolites of many benzodiazepines, including diazepam. Three agents are better suited in elderly patients; oxazepam, temazepam, and lorazepam do not undergo Phase 1 metabolism but are directly conjugated to inactive metabolites. Phase 2 metabolic reactions, such as conjugation, are less affected with aging. The use of zolpidem, a nonbenzodiazepine drug used for insomnia, has a short elimination half-life and leads to little hangover effect the next day, allowing for resumption of normal daytime activities for the elderly patient.
The benzodiazepine binding site is located at the interface between α and γ subunits at a site different than the binding site of GABA. Benzodiazepines bind to this site and increase the affinity of GABA for its binding site on the GABAA receptor–chloride ion channel complex. This is an example of an allosteric binding site. Benzodiazepines bind to receptors made up of both α1 and α2 subunits, whereas the newer nonbenzodiazepine agents (see below) are selective for receptors containing α1 subunits.
Benzodiazepines increase the frequency with which the channel opens, whereas barbiturates increase the length of time that the channel remains open. By increasing chloride conductance, these drugs cause neuronal membrane hyperpolarization and this, in turn, counteracts the depolarizing effect of excitatory neurotransmitters. Barbiturates increase chloride conductance independent of the presence of GABA, which is why they are capable of causing greater CNS depression and toxicity than the benzodiazepines.
In addition to their effects on GABA, benzodiazepines also inhibit the neuronal reuptake of adenosine. This action increases the inhibitory effect of adenosine on neurons that release acetylcholine from the pedunculopontine nucleus of the reticular formation, which is a brain structure mediating arousal. The effect of benzodiazepines on adenosine may also explain why these drugs dilate coronary arteries and decrease total peripheral resistance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


