14.1
where η is the Newtonian viscosity (mPa·s), σ is the shear stress (Pa), and γ˙ is the rate of shear (s−1). Newtonian fluids obey Equation 14.1; that is, there is a direct, time-independent, constant proportionality between σ and γ˙
is the rate of shear (s−1). Newtonian fluids obey Equation 14.1; that is, there is a direct, time-independent, constant proportionality between σ and γ˙ , so that η is the same irrespective of σ or γ˙
, so that η is the same irrespective of σ or γ˙ . Macromolecular solutions, however, seldom exhibit true Newtonian behavior unless solution concentrations are very low. While non-Newtonian fluid rheology encompasses shear-thinning, shear-thickening, or plastic behavior, sometimes accompanied by time dependency (i.e., thixotropy or rheopexy) [5], most polymeric or macromolecular solutions exhibit shear-thinning behavior when sheared.
. Macromolecular solutions, however, seldom exhibit true Newtonian behavior unless solution concentrations are very low. While non-Newtonian fluid rheology encompasses shear-thinning, shear-thickening, or plastic behavior, sometimes accompanied by time dependency (i.e., thixotropy or rheopexy) [5], most polymeric or macromolecular solutions exhibit shear-thinning behavior when sheared.
Huang and Sorensen [6] evaluated the shear rate dependence of viscosity of 3% w/w aqueous solutions of gelatin during the course of gelation. They found that although the apparent viscosity of the solutions was shear rate dependent (i.e., shear thinning), it was independent of the shear rate history of the solution.
The application of stress to polymeric materials often results in a change in the molecular weight distribution of the polymer [7]. Staudinger, in 1930, was apparently the first to observe shear-induced scission of polymers in solution [8]. Since then, numerous studies have confirmed the potentially deleterious effect of shear stress on the degradation of polymers in solution and the corresponding reduction in polydispersity as shear stress is increased [9]. It should be noted that reductions in polymer solution viscosity are not necessarily always attributed to depolymerization or degradation. Reduced viscosity could also result from deaggregation of macromolecular aggregates [9]. Conversely, there are reports in the literature of negligible effects of mechanical shear on polymer molecular weight. For example, Powell et al. [10] observed minimal degradation of hydroxyethyl cellulose under all but the most rigorous conditions (e.g., shear rates of ~1.9 × 106 s−1). Given the unpredictable outcome when shear or stress is imposed on polymeric solutions—in the absence of prior experimental data—it would be advisable to estimate the shear stresses and rates of shear incurred in manufacturing or processing operations and then evaluate polymeric stability under those conditions, at relevant temperatures and concentrations, before any manufacturing proceeds. The spectrum of shear rates involved in capsule manufacturing operations ranges from those incurred in dip coating operations (10–1 to 102 s−1) [11], precision pumping through dosator nozzles (100 to 103 s−1) [12], to those encountered in soft gel capsule production that are orders of magnitude higher (104 to 105 s−1).
Viscometers—instruments used to measure the viscosity of Newtonian fluids—are generally inappropriate for measuring the viscosity of non-Newtonian solutions as σ or γ˙ cannot be varied accurately or precisely (e.g., sample geometry violates rheological principles). Furthermore, γ˙
cannot be varied accurately or precisely (e.g., sample geometry violates rheological principles). Furthermore, γ˙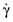 may vary substantially within the sample environment. The inability to control sample temperatures within a specific narrow range may also be problematic for some viscometers, given the temperature dependence of fluid or solution viscosity that often takes the form [13]
may vary substantially within the sample environment. The inability to control sample temperatures within a specific narrow range may also be problematic for some viscometers, given the temperature dependence of fluid or solution viscosity that often takes the form [13]
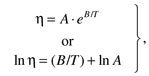 14.2
14.2where A and B are constants and T is absolute temperature (K).
Given the higher macromolecular concentrations that are used in gellant solutions (for dip coating, film forming, and injection molding processes) and the non-Newtonian behavior of the gellant solutions, the continued use of single point rheological methods intended for Newtonian fluids is potentially misleading and consequently unacceptable, especially for quality control/quality assurance (QA/QC) and quality-by-design (QbD) purposes, unless provisions can be made for varying the shear rate γ˙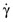 or shear stress σ. If that is done, an acceptable assessment of the gellant solution’s rheological behavior can be made. Viscometers that are suitable for shear viscosity measurements of non-Newtonian fluids typically accommodate multiple shear rates so that data for shear stress σ as a function of shear rate γ˙
or shear stress σ. If that is done, an acceptable assessment of the gellant solution’s rheological behavior can be made. Viscometers that are suitable for shear viscosity measurements of non-Newtonian fluids typically accommodate multiple shear rates so that data for shear stress σ as a function of shear rate γ˙ can be obtained, thereby defining the rheological behavior of a test fluid. These instruments include pressure-driven capillary and microfluidic slit viscometers, drag flow (i.e., rotational) viscometers, and falling or rolling ball viscometers [5]. Instruments capable of precise measurements of deformation and flow in a geometrically defined space are often described as rheometers. In general, viscometers and rheometers and the corresponding methods employed with them are suitable for non-Newtonian fluids and are appropriate as a part of a QA/QC program for gellant characterization. These instruments and methodologies are described in detail elsewhere [5,11,14,15].
can be obtained, thereby defining the rheological behavior of a test fluid. These instruments include pressure-driven capillary and microfluidic slit viscometers, drag flow (i.e., rotational) viscometers, and falling or rolling ball viscometers [5]. Instruments capable of precise measurements of deformation and flow in a geometrically defined space are often described as rheometers. In general, viscometers and rheometers and the corresponding methods employed with them are suitable for non-Newtonian fluids and are appropriate as a part of a QA/QC program for gellant characterization. These instruments and methodologies are described in detail elsewhere [5,11,14,15].
Solution Extensional Viscosity
While fluid flow and viscosity are often described in terms of steady shear of fluid laminae past one another, hard capsule manufacturing by a dip coating process necessitates consideration as well of the elongational or extensional viscosity of the film-forming solution occurring under non-shearing or “shear-free” conditions [16,17]. The thickness of the film coating the pins is a function of both the shear and extensional viscosities of the coating liquid, the rate of the pin’s immersion in and withdrawal from the gellant solution, the surface tension of the gellant solution, and the angle at which the pin is immersed and withdrawn from the gellant solution [16,17]. As the coated pin is withdrawn from the gellant solution, a filament forms between the hemispherical end of the pin and the solution bath. The efficiency of the capsule manufacturing process is reflected in the time required for the filament to break [16]. Simulations of the stretching and breaking of the filament show the potentially substantial influence of the elongational viscosity of the gellant solution on the filament breakage time [17].
As shear flow of macromolecular solutions proceeds, the macromolecules orient in the direction of flow but undergo some rotation, owing to the differential velocity across the flow field, thereby reducing the stretching of the macromolecules. The velocity gradient in shear flow is at right angles to the direction of flow. However, extensional flow in macromolecular solutions—sometimes referred to as “shear-free” flow—involves less rotation than shear flow because of the absence of competing forces and correspondingly more stretching and elongation of the macromolecules. In addition, the velocity gradient is in the direction of flow [15]. In extensional flow, the extensional stress (i.e., tension) is σe and the rate of extension, or elongation (i.e., extensional strain rate) is γ˙e . For Newtonian fluids, extensional viscosity, ηe, in uniaxial flow, can be characterized in an analogous manner to shear viscosity, that is
. For Newtonian fluids, extensional viscosity, ηe, in uniaxial flow, can be characterized in an analogous manner to shear viscosity, that is
 14.3
14.3For simple Newtonian or quasi-Newtonian fluids (at very low shear rates), ηe = 3η. This relationship between extensional and shear viscosities was first established in 1906 by Trouton [18]. However, for complex non-Newtonian fluids, extensional viscosities cannot be predicted readily on the basis of their shear flow behavior [19]. In such instances, experimentally determined extensional viscosities are best described as apparent extensional viscosities, that is, ηe,app. Just as the rheological behavior of macromolecular solutions can be described in terms of a power law relationship [5], the extensional rheological behavior of such solutions can be described in an analogous fashion. Thus, for extension-thinning fluids,
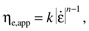 14.4
14.4where k is a proportionality constant and n is the power law index.
Research on extensional viscous behavior of macromolecular solutions was not the subject of substantial research efforts until the 1980s mainly because of the difficulty in making the measurements [20–23]. Only recently has commercial instrumentation for extensional viscosity measurements—that is, CaBER, an acronym for Capillary Breakup Extensional Rheometer—become available1 to supplant improvised in-house equipment.
CaBER instruments function by enabling the rapid separation (typically within 20–50 ms) of two discs between which a small volume of solution (generally <1 mL) has been placed. The instantaneous (user-selected) extensional strain rate imposed on the nearly cylindrical fluid sample by the rapid movement of the upper disc away from the lower stationary disc results in the formation of a uniform but unstable cylindrical filament of liquid that stretches between the discs. With the cessation of stretching, the fluid at the midpoint of the filament is subjected to an extensional strain rate and a corresponding drainage of fluid from the filament. The filament’s midpoint diameter is monitored—as it thins—by a laser micrometer as a function of time. The apparent simplicity of the filament formation and monitoring processes tends to minimize or obscure consideration of the potential effects of pre-shear history on the extensional behavior of some fluids, although research to date implicates wormlike micelle solutions and immiscible polymer blends rather than polymer solutions [24]. A prudent approach to CaBER experimentation and use warrants attention to the pre-shear history of the fluid sample and an evaluation of the effect of changes in the rate and extent of the rapid elongation2 imposed on the nearly cylindrical fluid sample at the outset of the procedure.
For viscous Newtonian fluids undergoing capillary thinning, the breakup process proceeds linearly with time [25,26]. Thus, the mid-filament diameter (Dmid) decreases with time, t, in accordance with
 14.5
14.5where α is a numerical coefficient, γ is the surface tension of the solution, ηs is solvent viscosity, and tb is the time to breakup [26].
However, for non-Newtonian fluids, the breakup process is nonlinear. For viscoelastic fluids, the mid-filament diameter decreases exponentially [26]:
 14.6
14.6where ηp = (η0 − ηs) is the solution viscosity owing to the polymeric component, η0 is the zero-shear rate viscosity of the solution, λ¯ is the relaxation time of the viscoelastic filament, and D1 is the mid-filament diameter after stretching has ceased.
is the relaxation time of the viscoelastic filament, and D1 is the mid-filament diameter after stretching has ceased.
Arnolds et al. [27] demonstrated the utility of a CaBER instrument for determining the extensional rheology of semi-dilute to concentrated aqueous solutions of polyethylene oxide. The authors noted that, in all instances, cylindrical filaments were formed that thinned down to a diameter of 5–10 μm before breaking. Filament breakage was immediately preceded by the formation of a beads-on-a-string structure that was apparently the result of flow-induced phase separation, with solvent predominating in the beads and the interbead filaments comprising highly extended polymer chains. This beads-on-a-string polymer solution behavior was consistent with the “blistering” phenomenon reported earlier by Sattler et al. [28] and further explicated by Sattler et al. in 2012 [29].
The extensional rheology of solutions of macromolecules that may be used as capsule shell excipients is, increasingly, the subject of ongoing research. Representative presentations and published reports that have focused on the extensional rheological behavior of biopolymer solutions include those on gelatin [30], ultrahigh-viscosity alginates [31], hydroxyethyl cellulose [32], methylhydroxyethyl celluloses [33], casein [34], and starches [34,35]. These citations provide further support for the use of extensional viscosity measurements of solutions of capsule shell excipients.3
Sol–Gel Transitions
Gel Formation
While many linear polymers undergo thermoreversible aggregation in dilute solution, most physical (non-covalent) gelation occurs in moderately concentrated polymer solutions as a result of the formation of supermolecular aggregates that extend continuously, as a network, throughout the system [36]. This description complements and extends the much earlier characterization—by P. H. Hermans [37]—of a gel as a “coherent system of at least two components, which exhibits mechanical properties characteristic of a solid, where both the dispersed component and the dispersion medium extend themselves continuously throughout the whole system”. Almdal et al. [38] reviewed the often ambiguous definitions and uses of the term gel in the literature and provided a further clarification based on the dynamic mechanical properties of these solid-like systems: Gels also exhibit a storage modulus G2, which exhibits a plateau extending to times at least of the order of seconds, and a loss modulus G3, which is considerably smaller than the storage modulus in the plateau region.
Non-covalent or physical gel network formation can be envisaged as resulting from macromolecular interactions as varied as van der Waals, dipole–dipole, Coulombic, charge transfer, and hydrophobic and hydrogen-bonding [39]. Data from rheological test measurements suggest that physical cross-links in gels move or break when a gel is stressed. Activation energies associated with cross-link breakage—estimated from the temperature dependence of viscoelastic parameters—range from 6 to 65 kcal/mol for gelatin and various polysaccharides [40].
Given the potential multiplicity of non-covalent interactions responsible for gel formation—particularly in systems with a substantial excess of solvent (typically water)—and limited information on pregel precursors, it is not surprising that the process of physical gelation is less completely understood than that of covalent gelation [41]. Nonetheless, Hermans [42] found that the cross-linking between N0 hydrocolloid molecules—each having f identical groups (f >> 1) capable of interacting with one group on another molecule—could be adequately described in terms of an equilibrium between the interactive surface sites. Hermans theorized that a fraction α of the f groups—distributed randomly over the macromolecules—has interacted to form intermolecular links and that the probability that any one group has interacted is independent of the number of groups on the same molecule that have interacted. Assuming the interactions comprise an equilibrium, the concentration of interacted groups is equal to a constant multiplied by the square of the concentration of the groups that have not interacted, that is,
 14.7
14.7where K′ is independent of molecular weight, that is, of f. When α << 1, this reduces to
 14.8
14.8where c is the concentration and K, as before, is independent of molecular weight. Thus, α ~ c for large values of f, and the fraction of the macromolecules in the gel phase, Ng, is related to α by
 14.9
14.9where αc = 1/f. Since Ng ≥ 0 only when α ≥ αc, then αc is the degree of interaction of the functional groups at the point when a gel forms. Combining Equations 14.8 and 14.9, the gel point occurs at a concentration cc inversely proportional to the length of the chain:
 14.10
14.10Thus, gelation occurs when one group on each macromolecule has interacted with another group on another macromolecule. Hermans posits that in cases of practical interest, α is of the same order of magnitude as αc and that α << 1. As Clark and Ross-Murphy [41] note, this leads to
 14.11
14.11where c0 = M/Kf2 is the critical (gel) concentration, that is, the concentration below which no macroscopic gel is formed under the prevailing conditions for a macromolecular segment of molecular weight M.
The literature is sometimes unclear in differentiating the critical concentration c0 from the overlap concentration c*, the latter term defined as the macromolecular concentration at which molecular overlap and cross-link formation occur for a time-averaged volume of rotation. The generally accepted hypothesis for physical cross-link formation is that c0 < c*; that is, cross-link formation does not require the persistence of the macromolecules in each others’ macromolecular space, except when the cross-link is formed [43]. Ferry [44] estimated that—given the average volume of solution pervaded by a polymer molecule with a molecular weight of 1–2 × 105Da—molecular overlap occurs at concentrations as low as 2–5% and that higher concentrations involve considerable molecular overlap and entanglement. Ferry hypothesized that the resultant structure behaves as a network (i.e., gel) and that its response to stress will depend on the extent to which entanglement and disentanglement can occur for a given duration and magnitude of stress.
Gelation, the critical transition from the sol (molecularly dispersed) state to the gel (intermolecularly connected) state, is best appreciated in terms of the transition variable—connectivity—among the system components. In physical terms, connectivity refers to the physical bonds that link the fundamental units of a system. The transition from the sol to the gel state is evidenced by the development—at the gel point—of connectivity throughout the system. During gelation, the system changes from one in which connectivity exists only on a very short-range scale to one in which connectivity is ultimately evident at every scale throughout the sample [45]. The infinite cluster formed, that is, a network that extends throughout the system from one end of the sample to the other, can be described as an infinitely large macromolecule [46].
Various theories have been advanced over the years to characterize gel formation and gellant connectivity at the gel point. Classical models developed by Flory [47] and Stockmayer [48] characterized gelation as a random process of multimolecular interaction that was also consistent in a number of ways with a model for percolation in a three-dimensional lattice. Herrmann et al. [49] proposed an alternative, kinetic model of gelation that more effectively dealt with the complexity of the sol–gel transition.
Nonetheless, theories and models aside, practical concerns remain: (a) the determination of the gel point for a given gellant concentration and temperature, (b) the determination of the mechanical or physical nature (e.g., rigidity, brittleness) of the resultant gel, and (c) the concentration dependence of gel strength and gel time. As Ross-Murphy [43] has noted, most research in the past has been more concerned with the temperature dependence rather than the concentration dependence of gel parameters. This bias needs to be corrected, particularly as new excipients are developed for hard and soft shell capsule manufacture. Gelation characterization and gel point determinations are essential to the setting of appropriate formulation and manufacturing parameters, whether for hard or soft shell capsules; assessment of gel rigidity and brittleness is critical to ensuring capsule integrity during the encapsulation process and post-manufacture.
Gel Point Determination
Gel points are frequently estimated by measurements of the fluidity of a solution by a test-tube inverting method or by a ball-drop method [50] but are relatively inaccurate, particularly as gellant solutions become more viscous (e.g., in the vicinity of the gel point). As noted above, the determination of solution viscosity as a function of temperature and gellant concentration has often been relied upon to determine the gel point for a system. The relatively abrupt divergence of solution viscosity behavior as temperature or gellant concentration change is indicative of the gel point having been reached. Nonetheless, the viscous behavior of these fluids as gel formation proceeds is almost invariably accompanied by elasticity, that is, viscoelasticity. The time dependence and temperature dependence of the viscoelastic properties of physical gels at and near the gel point have been studied extensively and shown to follow a power law function [51,52]. Therefore, gel point determinations based on viscous flow behavior alone can be misleading. Accordingly, the viscoelastic nature of gels warrants the use of rheological methods suitable for characterizing such materials and the processes of gelation and melting [53]. These are described in “Gel and Film Characterization” section.
Gel and Film Characterization
In an early overview of the rheological properties of industrial materials, Scott Blair [54] set out 8 objective rheological criteria and 24 rheological methods that could be used to establish the quality of materials. For “Jellies”—the most relevant of the 31 categories of substances tabulated by Scott Blair—seven rheological parameters were suggested: viscosity, elastic modulus, hysteresis, shear-thinning, yield value, thixotropy, and tensile strength. In the more than 70 years that have elapsed since Scott Blair’s critical review, rheological instrumentation has evolved dramatically—driven by marked improvements in engineering and computer hardware and software [55]—so that empirical tests [56] have increasingly given way to imitative tests that simulate the conditions to which material is subjected in use and to fundamental tests that measure well-defined physical properties.
Once a solution of a capsule shell excipient has gelled, rheological methods suitable for the rheological characterization of fluids are no longer practical or appropriate. The relationship between gel rigidity or elasticity and gellant concentration was recognized as far back as 1886 for gelatin gels [57,58]. But not until Ferry’s work in the 1940s [59] was the correlation more firmly established—at least for gelatin gels—as a consequence of Ferry’s use of gelatin samples of known average molecular weight and molecular size distribution.4
Deformation and penetrometer tests are among the oldest methods used to evaluate the potential gel strength afforded by capsule shell excipients. The Bloom gelometer test is one of the most widely employed empirical deformation procedures for evaluating the suitability of gellants (particularly gelatin) for capsule manufacture. It is rapid, is simple to perform, is suitable for routine quality control, and may correlate well with sensory test methods. Unfortunately, the Bloom test did not evolve from a fundamental understanding of gelation or gel rigidity, nor is its measurement convertible to fundamental physical parameters. Furthermore, as a one-point measurement, its replacement by quantitative fundamental and imitative rheological methods and instrumentation is warranted and long overdue.
Insofar as penetrometers are concerned, as Mitchell notes [40]: “rupture strength of a gel is not necessarily related to its elastic modulus and therefore single point measurements of ‘gel strength’ based on rupture [penetrometer] tests will not always rank a series of gels in the same order as tests which involve small deformations without rupture.”
Dynamic oscillatory methods employ rotational rheometers to apply either a small or a large sinusoidally oscillating stress or strain at an angular frequency to a fluid or semisolid sample contained within concentric cylinder, parallel plate, or cone and plate geometries [5]. These cyclical variations of stress and strain comprise the most commonly employed methods for the characterization of viscoelastic materials, that is, materials that exhibit both viscous and elastic behavior. The time scale is defined by the frequency of oscillation, ω, or the cycle time (rad s−1). By altering the time scales, one can establish the nature of the mechanisms underlying the material responses to the imposed strain.
The application of sinusoidal strain to a gel system can be represented by

and the linear response to low amplitudes of imposed strain, ~γ0, corresponds to

where δ is the phase shift or phase angle.
The stress response can also be represented as follows:

Quantifiable parameters of interest in this linear viscoelastic region5 include, among others, the storage modulus, G′, and the loss modulus, G″, the loss tangent or loss factor, tan δ, which corresponds to the tangent of the phase shift or phase angle6 δ or (G′/G″). The moduli can be defined as follows:


An additional term, the complex viscosity, η*, corresponds to (G*/ω), wherein G*—the complex shear modulus—is equal to

These various moduli—defined as ratios of stress and strain amplitudes—are the quantitative rheological parameters of gels most often evaluated [52]: the storage (elastic) modulus G′, based on the amplitude of in-phase stress, is indicative of the amount of recoverable energy stored elastically; the loss (viscous) modulus G″, based on the amplitude of out-of-phase stress, corresponds to the amount of energy dissipated as heat during shear; and the complex modulus G* defines the stiffness or rigidity of a sample.
Useful methods of representing the mechanical spectra of gellant solutions and characterizing the sol–gel transition include plotting tan δ, or both G′ and G″, as a function of ω. The approximate7 gel point corresponds to tan δ = 1, or when G′ = G″ (the crossover point8), that is, when the rheological behavior becomes predominantly solid-like, or elastic, rather than liquid-like [15]. Heuristically, the value of G′ correlates with the cross-link density of the gel network.
Dynamic oscillatory measurements involving small amplitude oscillatory shear (SAOS) can provide substantial information on semisolids via so-called “sweep” methods conducted in the linear viscoelastic region [60]: (a) frequency sweeps, wherein G′ and G″ are determined as a function of frequency ω at fixed temperatures; (b) temperature sweeps, in which G′ and G″ are determined as a function of temperature at fixed ω; and (c) time sweeps, in which G′ and G″ are determined as a function of time at fixed ω and temperature. Frequency sweep studies characterize the gel phase of a system wherein G′ is higher than G″ throughout a frequency range, that is, when molecular rearrangements within the network over time are minimal. Temperature sweep data can establish the temperature dependence of gelation while time sweep data can establish the temporal change in gel strength and the length of time required for recovery of sample mechanical strength. For a given concentration of gellant, temperature and time sweep data provide important parameters for the capsule manufacturing process by establishing an appropriate temperature and time range for unit operations.
As noted above, the power law dependence of the moduli G″ and G′ on frequency—in the linear viscoelastic region—characterizes the gel point at an intermediate state between a liquid and a solid:



