Tumor type
Heritable retinoblastoma
Nonheritable retinoblastoma
Eyes affected
Bilateral in 90–95 % of cases
Unilateral in 100 % of cases
Unilateral in remaining cases
Age of onset
Usually < 1 year
Variable — < 1–5 years
Presence of family history
10–20 % of newly diagnosed cases
Negative
Risk of second malignancy
5–15 %
0.5 %
Recurrence risk
50 % risk of passing on an RB1 mutation to an offspring
Almost zero. Very low risk of passing on an RB1 mutation due to presence of germline mosaicism
Molecular Basis of Disease
RB is caused by biallelic inactivation of the RB1 gene. Friend and colleagues identified the RB1 gene in 1986, the first tumor suppressor gene to be cloned [9]. The gene consists of 27 exons that span 183 kilobases of genomic DNA on chromosome 13q14 (Fig. 29.1). RB1 encodes a 928 amino acid protein known as RB1, which is a nuclear phosphoprotein that plays a critical role in regulating cell cycle progression at the G1 to S phase transition. The regulatory activity of RB1 results from its ability to bind to and inhibit the function of the E2F families of transcription factors, which regulate multiple genes involved in S-phase entry. Unphosphorylated RB1 binds to the E2F/DP (dimerization partners of E2F) complex through a critical functional region containing RB1 A/B pocket domains. The pocket domains are encoded by exons 12 through 17 and 20 through 22 of the RB1 gene and are hotspots for inactivating mutations in RB and other cancers. The C-terminal region of RB1 contains the nuclear localization signal and the interaction motif that is the target site of cyclin-CDK (cyclin-dependent kinase) complexes. The C-terminal region can also bind to the nuclear c-Abl tyrosine kinase and MDM2 (Mdm2, TP53 E3 ubiquitin protein ligase homolog [mouse]), which have oncogenic properties (Fig. 29.1). In the presence of mitogenic or oncogenic stimuli, RB1 is phosphorylated by CDKs (CDK4 and CDK6) and dissociates from E2F/DP complexes, which leads to the release of inhibition of E2F target genes and progression through the cell cycle (Fig. 29.2) [10, 11].
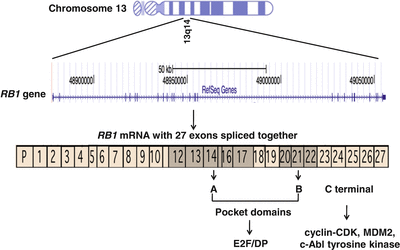
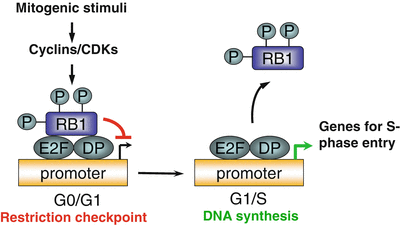
Figure 29.1
Genomic location and structure of the RB1 gene on chromosome 13q14 and spanning 183,000 bases. The 27 exons encode the 928 amino acid RB1 protein. The shaded exons encode the pocket domains A and B of RB1, which mediate interactions with the E2F and DP families of transcription factors
Figure 29.2
A simplified model for RB1 function. RB1 is hypophosphorylated during the G0/G1 phase of the cell cycle and interacts with E2F/DP family of transcription factors that inhibit transcription of downstream genes. In the presence of mitogenic stimuli, the cyclin-dependent kinase 4 (CDK4) protein phosphorylates RB1, which releases the E2F/DP factors and induces gene expression leading to the G1/S transition and progression through the cell cycle
Inactivation of the RB1 pathway through direct perturbation of the RB1 gene (deletion, mutation, or epigenetic mechanisms) leads to deregulated cell proliferation and ultimately tumorigenesis. Recent evidence suggests that RB1 also plays important roles in multiple cellular processes beyond cell cycle regulation, such as mitochondria biogenesis, cell senescence, and others [12]. Inactivation of the RB1 pathway also can cause a mitotic defect, and genomic instability and aneuploidy [12, 13].
RB has served as the paradigm for the “two-hit” model of tumorigenesis [14]. The Knudson hypothesis is that inactivation of both alleles of RB1 (two “hits”) is necessary to develop RB. In children with hereditary RB, the first hit is the germline mutation on one allele of RB1. The second hit is a somatic inactivation event on the remaining allele which includes point mutations, intragenic rearrangements, or hypermethylation of the promoter region and causes inactivation of RB1 protein and initiation of RB. In about 65 % of the RB cases, the second hit to the RB1 gene is accomplished by the mechanism of loss of heterozygosity (LOH), which can result from deletions or chromosomal rearrangements affecting RB1 [15]. In children with nonhereditary RB, the tumors develop as the result of two somatic RB1 inactivation events in a retinal progenitor or precursor cell during embryonic development.
The presentation of RB can be classified in three categories: familial hereditary (5–10 %), de novo hereditary (20–30 %), or sporadic (60–70 %) [16]. Familial hereditary cases are characterized by a positive family history and germline mutations in RB1 in individuals that present with bilateral disease in most cases and unilateral disease in 10–15 % of cases. However, only 20 % of bilateral cases and 15 % of unilateral cases have a positive family history and inherit the mutation from one of the parents. For individuals carrying a germline mutation in RB1, the risk of transmission of the mutation to an offspring is 50 %. In 80 % of newly diagnosed bilateral cases, the family history is negative and RB is caused by a de novo germline mutation on one of the parental germ cells. These represent the 20–30 % of sporadic hereditary cases with de novo mutations. In 90 % of these sporadic bilateral cases, the de novo mutation occurs on the paternal allele [17]. Approximately 60 % of RB cases are sporadic and nonheritable, with a unilateral presentation. Germline mutations are not present in the sporadic unilateral cases, and the risk of passing on the mutant allele to offspring is very low. There are reports of gonadal mosaicism in the parents of children with RB. In this setting, instead of being a single de novo event in one germ cell, a subset of the parental gametes carries the mutant RB1 allele. The degree of mosaicism determines the risk of transmitting the mutant allele to future offspring. Thus, the risk of RB in the siblings of a sporadic heritable RB case should not be underestimated.
Almost all types of mutations have been identified in the RB1 gene, including point mutations, deletions, translocations, insertions, and epigenetic changes. Conventional cytogenetic analysis of peripheral blood lymphocytes can identify deletions or rearrangements involving 13q14 in approximately 8 % of bilateral cases and 1–5 % of sporadic unilateral cases [15]. Smaller deletions in the RB1 region are identified in approximately 10 % of bilateral RB cases by Southern blot analysis [15]. Approximately half of all defined RB1 mutations are caused by single-base substitutions in coding exons, one third by small frameshift mutations, and another 10 % by rearrangements, such as intragenic or intergenic deletions or duplications involving chromosome 13q. These rearrangements may be limited to the RB1 gene or include genes flanking the RB1 locus [15, 18–21]. Nichols et al. carried out an investigation of a total of 180 RB cases, and the frequencies of germline mutations detected in bilateral, familial unilateral, and sporadic unilateral cases were 91 %, 70 %, and 7 %, respectively. Of the identified mutations, 76 % were coding sequence mutations, and 14 % were deletions of individual or multiple exons or the whole RB1 gene [22].
The majority (70 %) of mutations identified in RB1 are nonsense mutations or frameshift mutations, which lead to premature termination codons. Approximately 76 % of these nonsense mutations are recurrent CGA > TGA transitions affecting one of the 12 CGA codons within the open reading frame of RB1. Approximately 10 % of identified single-base substitutions are missense mutations, most of which localize to the pocket domains of RB1. Very few mutations have been identified in the promoter region of RB1 or exons 25–27, although this region contains two CGA codons [15]. A recent study reported the presence of a germline frameshift mutation in exon 27 in an unaffected father of a child with unilateral RB. Interestingly, the mutant transcript showed reduced expression, which might explain the milder unilateral disease manifestation [23]. Thus, unlike similar mutations located elsewhere in the gene, truncating mutations in the last exon of RB1 may be unique in their clinical presentations.
In addition to coding sequence variations, mutations affecting the conserved nucleotides in 5′ and 3′ splice sites within RB1 introns are observed in approximately 18 % of RB cases [15]. RB1 has 26 introns and the sizes of the introns range from small (intron 15 with 80 base pairs [bp]) to very large (intron 17 with 71,500 bp). While splice site mutations at the exon–intron junctions can be detected by routine sequence analysis, mutations located deep within the introns can be missed without analysis of the RB1 mRNA. A few cases harboring deep intronic mutations have been documented by cDNA analysis. These deep intronic mutations usually generate cryptic splice sites and cause retention of intronic sequences, which lead to subsequent frameshift and premature termination of translation [22, 24, 25]. Furthermore, missense or nonsense mutations that flank the consensus sequences of splice sites also may give rise to aberrant splicing [22]. Hypermethylation of CpG dinucleotides within the promoter region of RB1 can lead to gene silencing and is a somatic event in approximately 10 % of RB tumors and is almost never observed in constitutional cells [26, 27].
As described earlier, RB has a high rate of de novo mutations, which can originate at the preconception or post-conception stage leading to germline or somatic mosaicism, respectively. Preconception mutation events occur mainly during spermatogenesis [17, 28]. Postzygotic events can occur at earlier stages of embryo development, and the mosaicism may extend to various organs and tissues, including retina or even gonads. Postzygotic germline mosaicism can occur on either paternal or maternal alleles, and the mosaic mutations can be detected in lymphocyte DNA in some cases [28, 29]. Mosaicism for RB1 mutation should always be taken into account in genetic diagnosis, counseling, and disease management. In 1998, Sippel and colleagues reported that germline mosaicism occurs in up to 10 % of families with newly diagnosed RB using combined technologies of single-strand conformational polymorphism and pedigree analysis [30].
Approximately 10 % of RB cases are caused by heterozygous deletions of chromosome 13q that include the RB1 gene. The sizes of the deletions vary from small (4,000 bases) to large (34 million bases) and the breakpoints are usually nonrecurrent [31]. Because larger deletions may involve many other genes besides RB1, individuals with deletions of 13q14 can exhibit other clinical manifestations in addition to RB. As might be expected, the length of the deletion seems to define the spectrum of clinical manifestations, with smaller deletions (those confined to 13q14) associated with macrocephaly, tall stature, obesity, and motor and/or speech delays, and larger deletions manifesting with characteristic craniofacial features, microcephaly, psychomotor delays, and other findings such as constipation and feeding problems [3, 31–33]. Interestingly, deletions larger than 1 Mb and encompassing the MED4 gene, which encodes a mediator of RNA polymerase type II, are associated with a milder phenotype (i.e., unilateral eye involvement and less penetrant disease) [31]. The mechanism by which deletion of MED4 lessens the severity of RB disease is unclear; however, reduced expression and nonpenetrance in carriers of large deletions might result from the loss of one or more genes contiguous with RB1 that are important for basic cellular functions. In a susceptible retinal cell, loss of the second copy of these critical genes may lead to cell death as opposed to tumor formation.
Finally, interfamilial or intrafamilial variations of phenotypic expression of identical RB1 mutations have been observed among RB families. This indicates that additional genetic factors modify genotype–phenotype correlations in RB [3, 34, 35]. For example, Klutz et al. reported the parent-of-origin effect in two unrelated families that segregate an identical base substitution within intron 6 of RB1 gene. The substitution leads to exon skipping and a frameshift mutation that creates a premature termination codon. In addition to the fact that both families show incomplete penetrance, carriers who inherited the mutant allele from the maternal germline are unaffected, while carriers who inherited the mutant allele from the paternal germline are affected [3, 34, 35].
The identification and characterization of the RB1 gene has provided critical insights into the biology of RB, and also many other tumor types, including lung, brain, breast, and bladder cancers. In the latter cases, somatic inactivation of the RB1 pathway is essential for tumorigenesis [36]. Since tumor development is associated with the sequential acquisition of multiple mutations that drive unregulated proliferation and/or reduced cell death [37, 38], it is apparent that alterations in other critical signaling pathways underlie RB tumorigenesis. For example, in addition to deletions flanking the RB1 gene, karyotype analysis has identified various recurrent chromosome imbalances in RB, which include isochromosome 6p, trisomy 1q, monosomy 16, add(1p), double-minute chromosomes, and homogeneously staining regions involving the MYCN and INT1 oncogenes [39].
The tumor suppressor protein, TP53, is a master regulator of cell cycle, apoptosis, and DNA damage and is the most frequently mutated gene in human cancer. But many studies have shown that the TP53 gene is not mutated in RB [40, 41]. In 2006, Laurie and colleagues shed light on this issue by demonstrating that the TP53 pathway is also inactivated by the MDM (murine double minute) family of proteins following loss of RB1 during retina development [42]. The MDM family of proteins, such as MDM2 and MDM4 (MDMX), are known inhibitors of TP53 and both are expressed in RB cells [43]. Additionally, a single-nucleotide polymorphism (SNP) located in the promoter region of MDM2 (rs2279744, SNP309, 309 T> G) is correlated with enhanced MDM2 expression and attenuation of the TP53 pathway [44]. It has been postulated that the MDM2 309 T>G SNP behaves as a modifier gene in RB [45]. Another similar study pointed out that the TP53 p.Arg72Pro SNP (rs1042522), which correlates with increased expression of MDM2 and compromised TP53 pro-apoptotic activity, is significantly associated with onset of RB [46]. Collectively, these data strongly support that inactivation of the TP53 pathway is an important cooperating event in the development of RB.
In addition to TP53, genetic lesions in other signaling pathways might contribute to RB malignancy. A recent report implicated the involvement of oncogenic microRNA cluster miR-17~92 in RB tumorigenesis in a mouse model and possibly also in humans [47]. In another study, Zhang and his colleagues performed whole-genome sequencing of RB tumor samples and matched normal DNA utilizing next-generation sequencing technology. Surprisingly, they observed very few recurrent genetic lesions, except in RB1 and MYCN. But gene expression and epigenetic analyses identified changes in other cancer-related genes, such as the protooncogene SYK [48].
Clinical Utility of Testing
The diagnosis of RB is essentially based on clinical examination of the fundus of the eye, which is confirmed by imaging technologies including B scan ultrasonography and computerized tomography or magnetic resonance imaging scans. Once diagnosis is established, RB1 genetic testing is indicated. For individuals with bilateral or familial RB, the goal of genetic testing is to identify the predisposing germline RB1 mutation, which can be detected in approximately 94 % of cases [22]. The identification of the germline RB1 mutation enables appropriate primary tumor treatment and surveillance for additional ocular tumors and/or non-ocular malignancies. Furthermore, the identification of a germline mutation allows risk estimation for siblings and offspring. One of the main benefits of genetic testing is to rule out germline mutations in other young at-risk relatives of the proband to avoid unnecessary screening of children under anesthesia for early detection of RB. For individuals with sporadic unilateral RB, genetic testing focuses on identification of somatic mutations in the tumor (when available), followed by germline testing. Once mutations are identified in the tumor, peripheral blood DNA should be tested to determine whether one of the mutations is actually a predisposing germline mutation, which is estimated to exist in 13 % of sporadic unilateral cases [49].
Prenatal diagnosis and preimplantation genetic diagnosis (PGD) are available for at-risk pregnancies, which require the prior identification of predisposing germline mutations within the families. Prenatal genetic testing options include the molecular analysis of DNA isolated from the cells of the developing fetus through chorionic villus sampling or amniocentesis. More recently, PGD is available at a limited number of institutions [50–52]. PGD is performed in combination with in vitro fertilization and offers a way to test an individual’s embryos for genetic disorders before transfer of unaffected embryos into the uterus. A recent report demonstrated the feasibility of PGD based on the inheritance of SNPs that are tightly associated with a wild-type or mutant RB1 allele [52]. This approach is used when genetic screening for mutation detection is not readily available or the disease-causing mutation has not been identified. However, this approach can lead to erroneous conclusions in the presence of gonadal mosaicism and should be used with caution [53].
Available Assays
A broad spectrum of mutations ranging from point mutations, deletions/insertions, chromosomal rearrangements, and epigenetic silencing has been documented in the RB1 gene. Accordingly, various types of molecular testing are used by clinical molecular laboratories for RB1 mutation analysis. The genetic tests to identify mutations involve multiple testing approaches, which should be adjusted according to the diagnosis, family history, and availability of genetic materials.
For individuals with bilateral or multifocal unilateral RB, or unilateral tumor with a positive family history, clinical genetic testing for germline mutations starts with sequence analysis of the RB1 coding exons, flanking introns, and the promoter region using DNA isolated from peripheral blood lymphocytes. Given the fact that larger deletions and insertion involving the whole RB1 gene or certain exons are not readily detectable by sequence analysis, alternative methods can be applied, including fluorescence in situ hybridization (FISH), multiplex ligation-dependent probe amplification, quantitative real-time PCR, targeted array-based comparative genomic hybridization, and heterozygosity testing based on informative polymorphic markers. Cytogenetically visible deletions or rearrangements involving RB1 can be detected by karyotype and FISH analyses, respectively, and are estimated to occur in 5 % of unilateral cases and 7.5 % of bilateral cases [49]. To detect deeper intronic mutations, reverse transcription-polymerase chain reaction methods can be used to test RNA isolated from RB tumor tissues or peripheral blood lymphocytes, followed by evaluation for abnormalities in the size and sequence of RB1 transcripts [25]. Through the use of multiple sequential testing methods, the sensitivity of mutation detection has reached 90–95 % for probands with heritable RB [22, 49].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


