Principles of Virus Structure
Stephen C. Harrison
Virus particles are carriers of genetic material from one cell to another. They are, in effect, extracellular organelles. They contain most or all of the molecular machinery necessary for efficient and specific packaging of viral genomes, escape from an infected cell, survival of transfer to a new host cell, attachment, penetration, and initiation of a new replication cycle. In many cases, the molecular machinery works in part by subverting more elaborate elements of a host cell’s apparatus for carrying out related processes.
A number of organizational modes have evolved to perform the functions just outlined. The most critical distinction, from a structural perspective, is between enveloped viruses—those with lipid-bilayer membranes—and nonenveloped viruses—those without such membranes. Both categories include well-known human pathogens. Examples of the former are human immunodeficiency virus (HIV) and influenza virus; examples of the latter, poliovirus and papillomavirus. Enveloped viruses have, in their lipid bilayer, an impermeable barrier between their genomes and the outside environment, reducing the need for continuity of any protein layer. Nonenveloped viruses require a tightly packed shell to exclude nucleases or other sources of genomic damage.
For the structure of any virus particle, a central constraint is that the information needed to specify its macromolecular components must not exhaust the genetic capacity of the packaged genome. This requirement for genetic economy is in practice quite stringent. For example, consider a very simple genome of 5 kb, enough to encode about 1,600 amino acid residues, if reading frames do not overlap. A tightly condensed single-stranded RNA or DNA of this size will occupy a spherical volume about 90 Å in radius. To protect it with a gap-free protein shell, 30 Å thick, would require roughly 25,000 amino acid residues—far more than the viral nucleic acid can encode. The shell of a nonenveloped virus with even a very small genome must therefore contain a large number of identical protein subunits—at least 60, if the coat-protein gene is to use up less than 25% of the coding capacity in the enclosed nucleic acid. As explained later, an important consequence of this observation (first made by Crick and Watson56 even before a triplet code had been established) is that virus particles, or their substructures, are usually highly symmetric.
How Virus Structures are Studied
Electron microscopy is the most direct way to determine the general morphology of a virus particle. Traditional thin-sectioning methods are useful for examining infected cells and larger, isolated particles. The thickness of a section and the coarseness of staining methods limit resolution to about 50 to 75 Å, even in the best cases. (Resolution means the approximate minimum size of a substructure that can be separated in an image from its neighbor. Recall that one atomic diameter is 2.3 Å; an α-helix, 10 Å; and a DNA double helix, 20 Å.) Negative staining, with uranyl acetate, potassium phosphotungstate, or related electron-dense compounds, gives somewhat more detailed images of isolated and purified virus particles. Viruses embedded in negative stain are often relatively well preserved. The electron beam destroys the particle itself very rapidly, but it leaves the dense “cast” of stain undamaged for much longer. If the particle is fully covered by the negative stain, the image contains contrast from both the upper and the lower surface of the particle, and visual interpretation of finer aspects of the image can be difficult.57
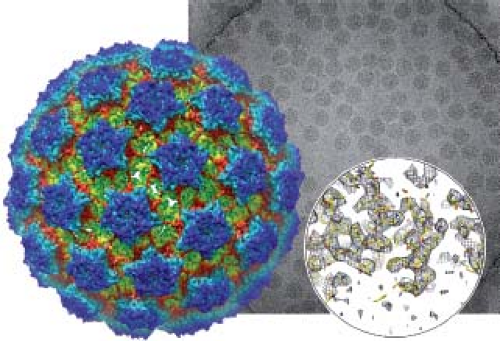 Figure 3.1. Bovine papillomavirus (BPV), as seen by electron cryomicroscopy (cryoEM). In the foreground is a color rendering of the three-dimensional image reconstruction, based on the kinds of micrographs shown in the background picture. The circular inset at lower right illustrates that this reconstruction provides information that extends to a nearly atomic level of detail (resolution); it shows a small part of the density map that resulted from the image analysis and the fit to that map of parts of the L1 polypeptide chain. (See Grigorieff and Harrison94 and Wolf et al.246) |
Methods for preserving viruses and other macromolecular assemblies by rapid freezing to liquid nitrogen or liquid helium temperatures have permitted visualization of electron-scattering contrast from the structures in the particle itself and not just from the cast created by a surrounding layer of negative stain.10 Moreover, quantitative methods for image analysis, originally developed for studying negatively stained particles, have been applied effectively to such images. An advantage of such electron cryomicroscopy (cryoEM) is that regular images can be selected from a heterogeneous field, allowing study of unstable or relatively impure preparations. Advances during the decade preceding the current revision of this chapter have enabled cryoEM three-dimensional density maps at resolutions that reveal molecular details—the tracing of a polypeptide chain and the orientations of large amino acid side chains.94 One example is illustrated in Figure 3.1.246 Such image reconstructions are obtained by combining information from hundreds or thousands of different images of individual particles. The combination is possible because the particles of these viruses are all the same. When such uniformity is not present, for example, as in the case of a complete herpesvirus particle rather than an isolated nucleocapsid, then information from different particles cannot be combined. A tomographic tilt series of images from a single particle can be obtained (analogous to a computed tomography [CT] scan in medical radiography), but the resulting three-dimensional image is of much lower resolution, as electron damage limits its quality, even when the data are taken at liquid nitrogen or liquid helium temperatures (electron cryotomography, or cryoET). Tomographic reconstructions can nonetheless be very useful, as illustrated in Figure 3.2. In some cases, averaging the images of defined substructures within a tomogram or among many tomograms (e.g., the “spikes” on the surface of certain enveloped viruses) can yield a more detailed representation.
The information obtained from even the most elegant of electron microscopy methods still falls short of the atomic detail that often can be obtained by x-ray diffraction methods, if single crystals of the relevant structure can be prepared. It has been known since the 1930s that simple plant viruses, such as tomato bushy stunt virus (TBSV), can be crystallized,13 and the first x-ray diffraction patterns of such crystals were recorded as early as 1938.17 Crystallization of poliovirus and other important animal viruses showed that the approach could be extended to human pathogens.213 The first complete high-resolution structure of a crystalline virus was obtained from TBSV in 1978,107 and since then the structures of a number of animal, plant, and insect pathogens have been determined (for a compilation, see the VIPER website: http://viperdb.scripps.edu). Only very regular structures can form single crystals, and in order to study the molecular details of larger and more complex virus particles, it is necessary to “dissect” them into well-defined subunits or substructures. This dissection was originally done with proteases, by disassembly, or by isolation of substructures from infected cells. For example, the structure of the influenza virus hemagglutinin244—the first viral glycoprotein for which atomic details were visualized—was obtained from crystals of protein cleaved from the surface of purified virions243; the structure of the adenovirus hexon was obtained from excess unassembled protein derived from adenovirus-infected cells.189 In the past two decades, this dissection has more commonly been carried out using recombinant expression (e.g., of a fragment of gp120 from HIV-1131). Most of the high-resolution structures of enveloped virus components described in this chapter—both surface glycoproteins and internal proteins—come from x-ray crystallographic analysis of recombinant gene products, often suitably truncated or otherwise modified to enable crystallization. A handful of atomic-level structures of virus components have come from nuclear magnetic resonance (NMR) spectroscopy,178,200 but application of that technique is limited to relatively small proteins or protein complexes.
Symmetry of Viruses
Virus particles must assemble specifically and rapidly in an infected cell, as directed by the mutual interactions among their component protein subunits. Specificity requires a defined stereochemical relationship between contacting proteins. Because there are many copies of the same subunit, there must also be many repeating instances of the same kind of contact. This repetition—a consequence of the requirement for genetic economy described in the introductory section of this chapter—implies symmetry.
A rigorous definition of symmetry involves an operation, such as a rotation, that brings an object into self-coincidence. For example, if the ring of three commas in Figure 3.3A is rotated by 120 or 240 degrees, it will not be possible to recognize that a rotation has occurred (assuming that the commas are truly indistinguishable). The full symmetry of an object is defined by the collection of such operations that apply to it. In the case of protein assemblies, these operations can be rotations, translations, or combinations of the two. A symmetry axis that includes rotation by 180 degrees is called a twofold axis or a dyad; one with a 120-degree rotation (and, of course,
a 240-degree rotation as well) is called a threefold axis; and so forth. Note the distinction between shape and symmetry: the shape of an object refers to the geometry of its outline, whereas its symmetry refers to the operations that describe it. The set of commas in Figure 3.3A has threefold symmetry; so does an equilateral triangle, the beer-company symbol with three interlocked rings, and countless other objects with unrelated shapes.
a 240-degree rotation as well) is called a threefold axis; and so forth. Note the distinction between shape and symmetry: the shape of an object refers to the geometry of its outline, whereas its symmetry refers to the operations that describe it. The set of commas in Figure 3.3A has threefold symmetry; so does an equilateral triangle, the beer-company symbol with three interlocked rings, and countless other objects with unrelated shapes.
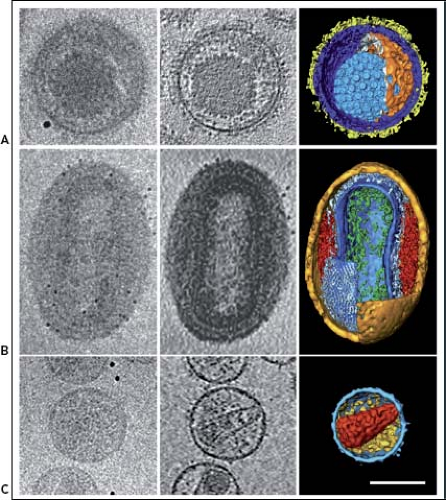 Figure 3.2. Electron cryotomography (cryoET) of herpes simplex virus type 1 (A),98 vaccinia intracellular mature virion (B),58 and HIV-1 (C).23 Images in the left-hand column are single, projected images; those in the middle column, slices through the reconstructed tomogram; those on the right, cut-away surface renderings of the three-dimensional tomographic reconstructions. (Adapted from Cyrklaff M, Risco C, Fernandez JJ, et al. Cryo-electron tomography of vaccinia virus. Proc Natl Acad Sci U S A 2005;102:2772–2777.) |
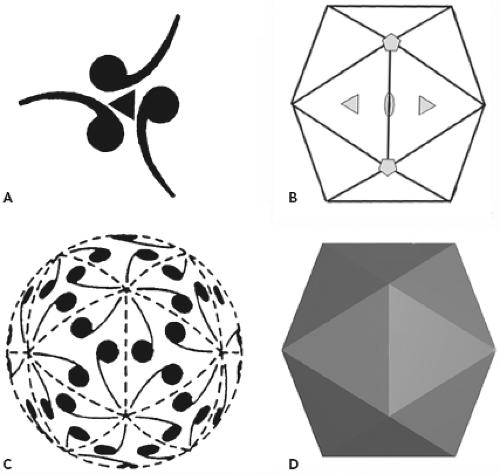 Figure 3.3. Icosahedral symmetry. A: Threefold symmetry: the three commas are related to each other by 120-degree rotations about the central axis, marked by a small triangle. B: Outline of an icosahedron, showing positions of some of the symmetry axes (imagined to extend from the center of the icosahedral to the point on the surface marked by the symbol): fivefold, threefold, and twofold axes are marked by pentagons, triangles, and an oval, respectively. C: An icosahedrally symmetric arrangement of commas on the surface of a sphere. For locations of symmetry axes, compare with panel B. D: Shaded surface view of an icosahedron. |
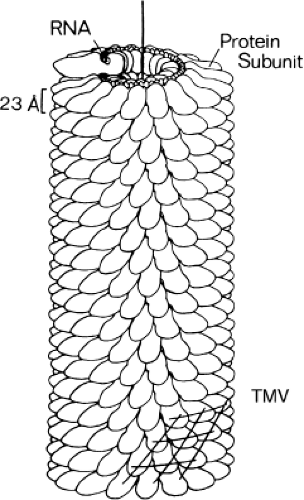 Figure 3.4. Diagram of the tobacco mosaic virus (TMV) particle. The elongated “loaves,” with a groove for the RNA, represent the protein subunits. Three RNA nucleotides fit into the groove on each subunit. There are 16 ¹/³ subunits per turn of the right-handed helix (i.e., 49 subunits in three turns), with a rise of 23 Å as indicated. At the lower right, the surface lattice is drawn onto the outer particle. (Adapted from Caspar DL. Assembly and stability of the tobacco mosaic virus particle. Adv Protein Chem 1963;18:37–121.) |
As a first example, consider the rod-like coat of tobacco mosaic virus (TMV)125 (Fig. 3.4). The helical arrangement of its protein subunits illustrates that symmetry is an important consequence of its assembly from many identical building blocks. If we look at the model of TMV, we find that a rotation of 22 degrees and a translation of 1.4 Å along the particle axis will superpose subunit 1 on subunit 2. But if the surfaces of subunit 2 are the same as those of subunit 1, the same rotation and translation must superpose subunit 2 on subunit 3, and so forth. The combination of rotation and translation that effects this superposition is a screw axis. Strictly speaking, the screw axis of TMV would only be an ideal symmetry operation if the helix were infinite. In practice, it is so long that we can neglect end effects.
In TMV, and probably in the nucleocapsids of negative-strand RNA viruses such as influenza and vesicular stomatitis virus (VSV), the RNA winds in a helical path that follows the protein.125 That is, the tubular package does not simply contain the RNA; it co-incorporates it. There are exactly three nucleotides per subunit in TMV, and they fit into a defined groove between the helically arrayed proteins. By contrast, the protein coat of a filamentous, single-stranded DNA (ssDNA) phage, such as M13, forms a sleeve that surrounds and constrains the closed, circular genome, without there being a specific way in which each subunit contacts one or more nucleotides.87 Thus, there can be a nonintegral ratio of nucleotides to protein monomers.
The length of the packaged nucleic acid determines the length of virus particles such as TMV or M13. Structures such as the tail of bacteriophage lambda or T4 have a protein component that extends from the initiating structure at the base of the tail to the end connected to the head.3 The number of such polypeptide chains corresponds to the rotational symmetry of the tail.
Rod-like structures are not very efficient ways to package long genomes. At least one dimension of a helical assembly such as TMV grows linearly with the length of the packaged viral DNA or RNA, leading to awkwardly elongated particles. The number of subunits is likewise proportional to length. Isometric (i.e., essentially spherical) particles are more compact and more economical: if the nucleic acid condenses into the interior of the particle, then the diameter increases as the cube root of the genome length, and the number of required subunits as the genome length to the two-thirds power. Most animal viruses are roughly isometric.
Closed, isometric shells composed of identical subunits that interact through conserved, specific interfaces can have one of only three symmetries: the symmetry of the regular tetrahedron, the cube, or the regular icosahedron. These shells will accommodate 12, 24, or 60 subunits, respectively. The icosahedral shells are obviously the most efficient of the three designs: they use the largest number of subunits to make a container of a given size, and hence they use subunits of the smallest size and the smallest coding requirement. Tetrahedral and cubic symmetries have not appeared in any naturally occurring virus assemblies. Note the distinction between icosahedral symmetry and icosahedral shape. Not all objects with icosahedral symmetry have even the vague outline of an icosahedron; conversely, painting a single asymmetric object, such as a comma, on each face of an icosahedron, rather than three such objects related by the threefold axis through the middle of the face, would destroy the symmetry of the decorated object but would not affect its shape.
The diagram in Figure 3.3B shows the operations that belong to an icosahedrally symmetric object. They are a collection of twofold, threefold, and fivefold rotation axes. Placement of a single, asymmetric object on a surface governed by this symmetry leads to the generation of 59 others, when the various rotations are applied (Fig. 3.3C). One such object, one-sixtieth of the total shell, can therefore be designated as an icosahedral asymmetric unit, the fundamental piece of structure from which all the rest can be produced by the operations of icosahedral symmetry.
Structures of Closed Shells
With a typical, compact protein domain of 250 to 300 amino acid residues, close to the upper limit for most single-protein
domains, what sort of icosahedrally symmetric container can we construct? Suppose that the protein is so shaped that 60 copies fit together into a 30-Å thick shell with no significant gaps. Then the cavity within that shell will have a radius of about 80 Å, which can contain a 3- to 4-kb piece of single-stranded DNA or RNA, tightly condensed. A few, very simple virus particles indeed conform to this description. The parvoviruses (see Chapter 57) contain a 5.3-kb ssDNA genome, and their shells have 60 copies of a protein of approximately 520 residues (Fig. 3.5). The capsid protein therefore uses up about one-third of the genome. (“Capsid,” from the Latin capsa, “box,” designates the protein shell that directly packages DNA or RNA; “nucleocapsid” refers to the shell plus its nucleic acid contents.) Likewise, the satellite of tobacco necrosis virus (STNV) contains 60 copies of a 195-residue subunit and a 1,120-bp single-stranded RNA (ssRNA) genome, of which over half is used for the coat protein.141 As the name implies, however, STNV is actually a defective virus, and it requires tobacco necrosis virus co-infection to propagate.
domains, what sort of icosahedrally symmetric container can we construct? Suppose that the protein is so shaped that 60 copies fit together into a 30-Å thick shell with no significant gaps. Then the cavity within that shell will have a radius of about 80 Å, which can contain a 3- to 4-kb piece of single-stranded DNA or RNA, tightly condensed. A few, very simple virus particles indeed conform to this description. The parvoviruses (see Chapter 57) contain a 5.3-kb ssDNA genome, and their shells have 60 copies of a protein of approximately 520 residues (Fig. 3.5). The capsid protein therefore uses up about one-third of the genome. (“Capsid,” from the Latin capsa, “box,” designates the protein shell that directly packages DNA or RNA; “nucleocapsid” refers to the shell plus its nucleic acid contents.) Likewise, the satellite of tobacco necrosis virus (STNV) contains 60 copies of a 195-residue subunit and a 1,120-bp single-stranded RNA (ssRNA) genome, of which over half is used for the coat protein.141 As the name implies, however, STNV is actually a defective virus, and it requires tobacco necrosis virus co-infection to propagate.
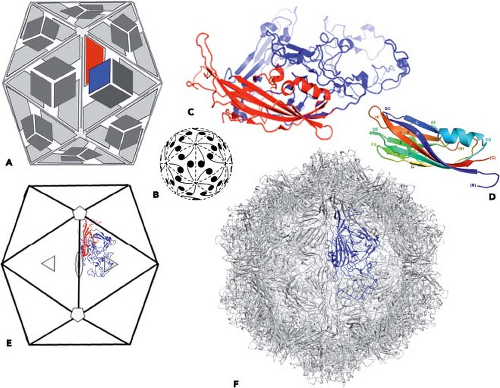 Figure 3.5. Canine parvovirus (CPV): a simple, icosahedrally symmetric virion. A: Icosahedron, viewed along a twofold axis, with diagrammatic representations of a protein subunit with a core domain (colored red on one of the subunits) and a projecting region (blue). Compare the subunits with the representation of commas in B, repeated from Figure 3.3 C. C: Ribbon diagram of the CPV protein subunit; the core domain (red) is a β-jelly-roll, from which emanate several loops that cluster to form a complex projecting region (blue). The simplified representation of the β-jelly-roll in D is in rainbow coloring, from blue at its N-terminus to red at its C-terminus. The eight strands are lettered B–I; the loops have the letters of the strands they connect. The projecting region of the CPV subunit comprises loops BC, EF, and GH. E: Icosahedron, as in A, but with a ribbon representation of one subunit; symbols for symmetry axes as in Figure 3.3B. F: Ribbon representation of all 60 subunits, with the subunit from E in blue and all others in gray. |
More complex viruses have evolved ways to make larger, icosahedrally symmetric shells without expending unnecessary genetic resources. The simplest, but least economical, is just to use several different subunits, each of “garden variety” size, to make up one icosahedral asymmetric unit. The picornaviruses (polioviruses, rhinoviruses, etc.) have 60 copies of three distinct proteins, VP1, VP2, and VP3, each between 230 and 300 amino acid residues, as well as 60 copies of a small internal peptide, VP4 (see Fig. 3.6). The shell has a cavity about 95 Å
in radius, which holds an RNA genome of 7.5 to 8 kb. The picornaviruses thus expend about one-third of their genome to encode the structural proteins of the virion. (The term virion means virus particle, generally implying the mature, infectious structure.) We note here two other important features of picornavirus molecular architecture. First, the folded structures of VP1, VP2, and VP3 all have the same kernel—a domain known as a jelly-roll β-barrel (Figs. 3.5 and 3.6). The single subunits of the parvoviruses and of STNV have the same basic fold. It is a module particularly well suited to the formation of closed, spherical shells because of its block-like, trapezoidal outline, but its prevalence among viral subunits may be evidence of a deeper evolutionary relationship. A second noteworthy feature of picornavirus design is that arm-like extensions of the subunits tie together the assembled particle (Fig. 3.6). The importance of scaffold-like intertwining of subunit arms was first discovered in the simple plant viruses.107 In effect, folding of part of the subunit and assembly of the shell are concerted processes.
in radius, which holds an RNA genome of 7.5 to 8 kb. The picornaviruses thus expend about one-third of their genome to encode the structural proteins of the virion. (The term virion means virus particle, generally implying the mature, infectious structure.) We note here two other important features of picornavirus molecular architecture. First, the folded structures of VP1, VP2, and VP3 all have the same kernel—a domain known as a jelly-roll β-barrel (Figs. 3.5 and 3.6). The single subunits of the parvoviruses and of STNV have the same basic fold. It is a module particularly well suited to the formation of closed, spherical shells because of its block-like, trapezoidal outline, but its prevalence among viral subunits may be evidence of a deeper evolutionary relationship. A second noteworthy feature of picornavirus design is that arm-like extensions of the subunits tie together the assembled particle (Fig. 3.6). The importance of scaffold-like intertwining of subunit arms was first discovered in the simple plant viruses.107 In effect, folding of part of the subunit and assembly of the shell are concerted processes.
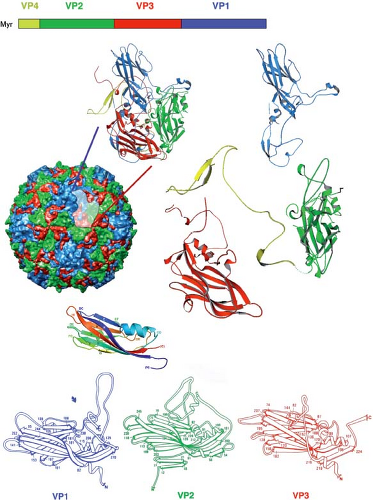 Figure 3.6. Poliovirus. Top: The order of structural proteins in the polyprotein encoded by the viral RNA. These domains are at the N-terminal end of the polyprotein, which is modified by myristoylation (Myr). The viral protease that cleaves between VP0 (= VP4 + VP2) and VP3 and between VP3 and VP1 is encoded by a region 3′ to the region that encodes the structural proteins; the VP4-VP2 cleavage is autolytic and occurs only after assembly of the virion precursor. Middle: Surface representation of the virus particle, with colors as in the diagram at the top. Two successively “exploded” views of an icosahedral asymmetric unit (protomer) are shown next to the surface rendering. VP1, VP2, and VP3 each have a central β-jelly-roll, with variable interstrand loops and variable N- and C-terminal extensions. The rainbow-colored β-jelly-roll below the surface view is repeated from Figure 3.5D. Bottom: Side-by-side views of the β-jelly-roll domains of VP1, VP2, and VP3 to illustrate their congruence. |
Quasiequivalent Icosahedral Arrangements
A more economical way to build shells from more than 60 average-sized, identical subunits was described by Caspar and Klug35 in 1961. It is illustrated by the diagram of 180 commas in Figure 3.7. The commas have similar interactions (head-to-head in pairs; neck-to-neck in rings of three; tail-to-tail in rings of five or six), but they fall into three sets, designated A, B, and C. If the commas are taken to represent proteins, then the conformational differences between A and B positions, for example, involve the differences between rings of five and rings of six, for contacts involving the parts of the proteins symbolized by the tails. Caspar and Klug35 suggested that protein subunits might have the sort of flexibility or capacity for conformational switching needed to accommodate somewhat different packing environments without sacrificing specificity. They postulated that viruses with more than 60 chemically and genetically identical subunits might exhibit the sort of near equivalence seen in the A, B, and C conformers in the comma illustration. They called this sort of local distortability, which might conserve much of the specificity and character of the protein contacts, quasi-equivalence.
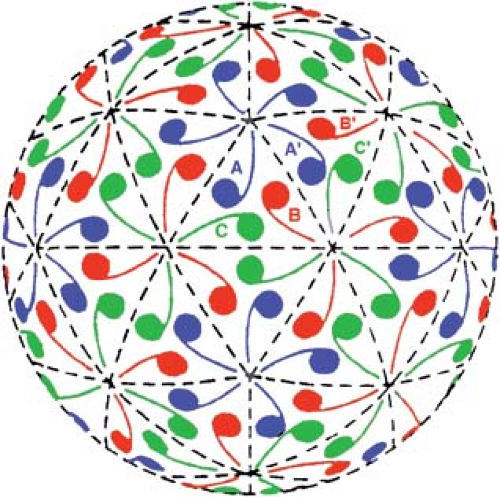 Figure 3.7. Quasiequivalent arrangement of 180 commas, in a T = 3 icosahedral surface lattice on a sphere. Compare Figure 3.3C, a T = 1 arrangement of 60 commas with icosahedral axes oriented similarly. The three quasiequivalent positions within a single icosahedral asymmetric unit are shown in blue, red, and green and labeled A, B, and C, respectively, in two of the asymmetric units. |
A number of plant and animal viruses, such as TBSV107 and Norwalk virus,182 conform to this description of quasiequivalent arrangements (Fig. 3.8). In TBSV and Norwalk virus, there are 180 genetically and chemically identical subunits in the capsid. The subunits are actually larger than those of the picornaviruses, but most of the extra size comes from a second, projecting domain that serves functions other than the construction of a closed shell. The size of the shell domain (S domain) in both cases is just about 200 residues, and the folded structure of the domain is again a jelly-roll β-barrel. The important feature of the packing of these 180 S domains is illustrated by the TBSV diagram in Figure 3.8. The contents of an icosahedral asymmetric unit can be described as three chemically identical subunits, with somewhat different conformations. These conformers are denoted A, B, and C, echoing the designation of commas in Figure 3.7. The differences among the conformers reside principally in an ordered or disordered conformation for part of the N-terminal arm and in the angle of the hinge between the S domain and the projecting, P domain. The A and B conformations are nearly identical, with disordered arms and similar hinge angles. The C conformation has an ordered arm and a different hinge angle from A and B. The ordered arms extend along the base of the S domain and intertwine with two others around the icosahedral threefold axis. Thus, the whole collection of 60 C-subunit arms forms a coherent inner scaffold.
How equivalent or nonequivalent are the actual intersubunit contacts in TBSV and related structures? Most of the interfaces are well conserved, with very modest local distortions that do not significantly change the way individual amino acid side chains contact each other. The interfaces between conformers that do exhibit noteworthy differences are those that include the ordered arms in one of the quasiequivalent locations (the C-conformer). At these interfaces, there is a discrete switch between two states, with ordering and disordering of the arm as the toggle. Nonetheless, many side chain contacts are conserved around the fulcrum that relates an A/B dimer to a C/C dimer (Fig. 3.8).
Only certain multiples of 60 subunits can pack with quasiequivalent contacts; they are given by the formula T = h2 + hk + k2, where h and k are any integer or zero.125 The multiple T is known as the triangulation number, because, as illustrated by comparison of the 60- and 180-comma structures in Figures 3.3 and 3.7, they correspond to subtriangulations of an icosahedral net on the surface of a sphere. Such nets are known as surface lattices. If we think of an icosahedrally symmetric structure as a folded-up hexagonal net (Fig. 3.9), then 12 uniformly spaced sixfold vertices are transformed into fivefold vertices.
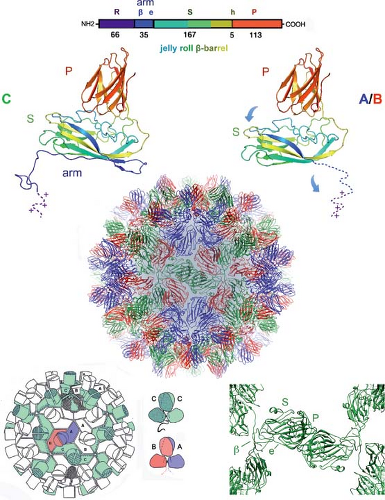 Figure 3.8. Tomato bushy stunt virus (TBSV), a T = 3 icosahedral structure. Top: Modular organization of the TBSV coat-protein polypeptide chain. R: unstructured, positively charged N-terminal region. β, e: segments of the “arm,” ordered on the C-conformation subunits and unstructured on the A- and B-conformation subunits; when ordered, the β segment forms an interdigitated β-annulus with corresponding segments from two other chains, and the e segment extends along the base of the subunit (see panel at bottom, left). S: shell domain, a β-jelly-roll. P: projecting domain, a β-sandwich of somewhat different fold from the jelly-roll S domain. h: hinge between the S and P domains. The color coding in the bar representation of the chain is repeated in the ribbon diagrams of the C (left) and A/B (right) conformations. Note that the two conformations differ in two respects: the ordering of the arm and the hinge angle between S and P domains (curved arrows on the right-hand ribbon diagram). Center: Ribbon representation of the entire protein coat of the virus; the colors of the A-, B-, and C-conformation subunits are as in Figure 3.7. Bottom left: Schematic figure, showing that the arms of the C-subunits (green) interdigitate around threefold axes of the icosahedral symmetry, forming a coherent inner framework. Bottom right: Magnified view of some of the C-subunits from the coat seen in the central part of the figure, illustrating the β-annulus (β) and the extended part of the arm (e). In the bottom center are schematic views of the C-C and A-B dimers, showing how the hinge between S and P domains correlates with the ordering of the arms (inserted into the slot between S domains, which have rotated away from the contact that they have when the arms are unfolded into the particle interior). |
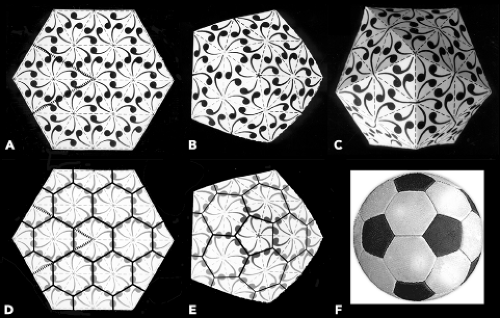 Figure 3.9. Generation of curved structures from planar lattices. A: Portion of a hexagonal lattice. Six triangular cells of the lattice meet at each lattice point, and each triangular cell contains three “subunits” (commas). Thus, there is a sixfold symmetry axis at each lattice point, a threefold symmetry axis at the center of each triangle, and a twofold axis at the midpoint of each edge. Imagine that the lattice extends indefinitely in all directions. B: Curvature can be introduced by transforming one of the sixfold positions into a fivefold (center). A 60-degree “pie slice” has been removed from the object in A by cutting along the heavy dotted lines, and the cut edges have been joined to generate the curved lattice shown here. C: If further cuts are made at regular intervals in an extended lattice, such as the one in A, and the edges joined as in B, a closed solid can be produced. In the case of the icosahedral solid shown here, vertices of the lattice separated by two cell edges have been transformed into fivefolds, while the intervening lattice points have been left as local sixfolds, producing a T = 4 (h = 2, k = 0) structure. Notice that the local sixfolds are actually only approximately sixfold in character; they correspond strictly to the twofold axes of the icosahedral object. D: Lines joining the centers of the triangular cells in A create a pattern of hexagons. E: When a sixfold is transformed into a fivefold, a hexagon becomes a pentagon. F: If second nearest-neighbor lattice points are all transformed into pentagons, a soccer-ball figure results. This is a T = 3 structure. A description of the lattice as a network of hexagons and pentagons is complementary to its description as a network of triangles. The representations in Figures 3.3, 3.5, 3.13, and 3.16 (left) use triangles. The representation in Figure 3.16 (right) uses hexagons and pentagons. One representation for a given lattice can easily be derived from the other. |
Nonequivalent Icosahedral Surface Packings
Hexagonal packing is an efficient way to tile a surface (think of hexagonal floor tiles), even if the building blocks themselves do not have sixfold symmetry and hence do not interact identically with their neighbors. In many larger, icosahedrally symmetric virus particles, the outer-shell building blocks are centered at the vertices of an icosahedral surface lattice, subtriangulated as anticipated by Caspar and Klug, but the oligomeric building blocks themselves are not hexamers. In some cases, for example, adenoviruses (Fig. 3.10), they are trimers, with a chemically distinct, pentameric building block on the fivefold vertices; in other cases, for example, the polyoma- and papillomaviruses, the building blocks are all identical pentamers (Fig. 3.1). Viewed at low resolution (e.g., by negative-stain electron microscopy), all of these viruses have globular “lumps” at the vertices of a lattice with one of the allowed triangulation numbers (T = 25 for the adenoviruses: Fig. 3.10; T = 7 for the polyoma- and papillomaviruses: Fig. 3.1), but when seen at higher resolution, the six-coordinated lumps are actually trimers or pentamers, and in the former case, the five-coordinated lumps are pentamers of a related but distinct polypeptide chain. Special mechanisms (either involving other structural proteins or flexible intersubunit connections) are needed to hold the particle together because a single set of repeating, quasiequivalent intersubunit contacts is not possible. Before the molecular principles of virus structure were fully understood, the globular lumps seen by low-resolution electron microscopy were called capsomeres, meaning the structural units of the capsid. This word is still used when referring to apparent morphologic units on the surface of a virus shell, but it is best reserved for cases where all capsomeres are the same and hence represent a defined oligomer, as in the pentameric units of papovaviruses (see later).
The flaviviruses and picobirnaviruses illustrate yet another adaptation to icosahedral packing. As illustrated in Figure 3.7, the asymmetric unit of an icosahedral surface lattice can be represented by a (spherical) triangle with a fivefold axis and two adjacent threefold axes as its vertices. The flavivirus envelope protein (E) is a flat, elongated dimer; three such dimers neatly fill a twofold-related pair of asymmetric-unit triangles, with the dyad of the central dimer coincident with the icosahedral twofold (Fig. 3.11).128 The shell contains 180 subunits, but not in a T = 3 arrangement. The picobirnavirus coat protein
is so shaped that two dimers can fill a similar (smaller) rhombic unit; the icosahedral twofold lies between the two dimers, and the complete coat contains 120 subunits.66 Recombinant brome mosaic virus coat-protein dimer, expressed in yeast cells, packs in a closely related way when it assembles into 120-subunit virus-like particles.126
is so shaped that two dimers can fill a similar (smaller) rhombic unit; the icosahedral twofold lies between the two dimers, and the complete coat contains 120 subunits.66 Recombinant brome mosaic virus coat-protein dimer, expressed in yeast cells, packs in a closely related way when it assembles into 120-subunit virus-like particles.126
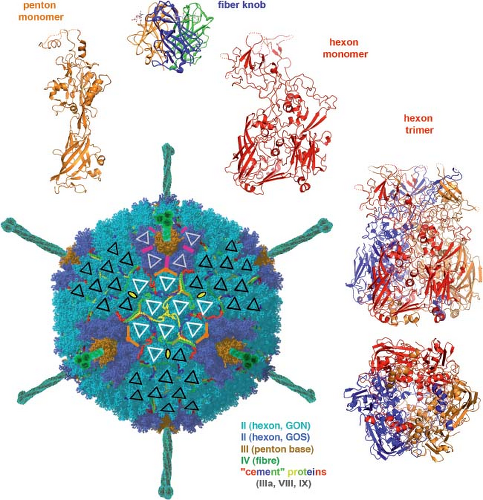 Figure 3.10. Adenovirus structure. A representation of the complete particle, based on a high-resolution electron cryomicroscopy (cryoEM) image reconstruction,142 is at the lower left, surrounded by ribbon representations of a number of the component proteins. The view of the particle is along a threefold symmetry axis. The hexons (light and medium blue) and the pentons (brown) lie on vertices of a T = 25 icosahedral lattice, but the hexons are actually trimers with a pseudohexameric character, as illustrated by the “bottom view” (as if from the particle interior) at the lower right. Three species of so-called cement proteins (IIIa, VIII, and IX) retain the hexons and pentons in the shell and determine its fixed geometry. One of them (various chains in red, dark blue, yellow, and light green) fits into the crevices between the hexons and organizes them into groups of nine (GON)—as shown by the sets of white and black triangles on the hexon surfaces. The other two are on the inner surface of the hexon–penton shell and cement five “peripentonal” hexons and the penton base into a group of six (GOS); locations of some of them are shown here simply as magenta and orange lines, because they are not visible from the outside of the particle. The trimeric fibers project from each penton base, with a receptor-binding knob (top of figure) at their tip. Each hexon monomer (see red ribbon diagram, upper right) has two jelly-roll β-barrels, in parallel orientation, imparting a pseudohexagonal character to the trimer. The penton base (upper left) has a single β-jelly roll. (Image reconstruction courtesy Z. H. Zhou; see also Harrison103). |
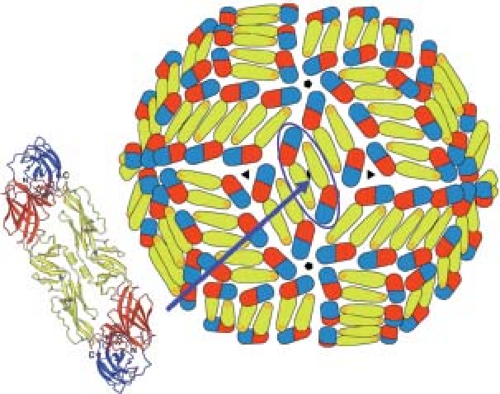 Figure 3.11. Organization of a flavivirus particle. Ninety dimers of the E protein tile the surface as shown. E is an elongated, three-domain protein (lower left), oriented with its long axis parallel to the surface of the virion. At the tip of domain II (yellow) is a hydrophobic fusion loop (orange, shown also as an asterisk on the larger schematic). |
The arrangement of 120 copies of the inner- (core-) shell protein in double-stranded RNA (dsRNA) viruses is a particularly striking example of nonequivalent packing (Fig. 3.12). There are two completely distinct environments for this protein (designated A and B in Fig. 3.12, center): two is not a permitted triangulation number, and quasiequivalent packing of 120 proteins in an icosahedral array is not possible. The amino acid side chains on the lateral surface of the core-shell protein have different partners, depending on the interface in which they lie. The distortion of the subunit itself, when the two environments are compared, is quite small.
Frameworks and Scaffolds
The protein subunits of TBSV or picornaviruses have extended N- or C-terminal arms augmenting a central jelly-roll β-barrel. These arms are essential for building a stable coat. They form an internal framework, such as the one illustrated for TBSV in Figure 3.9. In TBSV, the assembly unit—the oligomer of the coat subunit that forms spontaneously in solution (and by inference, in the cell following its synthesis)—is a dimer, which can have two conformations: an “A/B” dimer, with disordered N-terminal arms, and a “C/C” dimer, with folded arms.105 The local curvature of those two conformations is different, and the framework of C/C arms fixes the overall diameter of the particle. Removal of the N-terminal arms of TBSV-like subunits leads to self-assembly of a small, 60-subunit icosahedrally symmetric particle that cannot package RNA.88 That is, without the arms, there is no mechanism for a conformational switch.
In the papilloma- and polyomaviruses, N- and C-terminal extensions (principally the latter) of the subunit globular domains tie together the pentameric building blocks, which have almost no contacts except through these extensions (Fig. 3.13).139,246 Flexibility of the arms allows formation of the different kinds of contacts required to surround a pentamer with six other pentamers (i.e., to position a pentamer at the
six- as well as at the five-coordinated vertices of a T = 7 subtriangulated icosahedral lattice). The C-terminal arms emanate from one pentamer and dock into another. The way they dock is the same for all 360 arms, with identical interactions locking them in place; their configurations differ, however, between the point at which they emerge from the globular domain of their subunit of origin and the point at which they dock into their target subunit.
six- as well as at the five-coordinated vertices of a T = 7 subtriangulated icosahedral lattice). The C-terminal arms emanate from one pentamer and dock into another. The way they dock is the same for all 360 arms, with identical interactions locking them in place; their configurations differ, however, between the point at which they emerge from the globular domain of their subunit of origin and the point at which they dock into their target subunit.
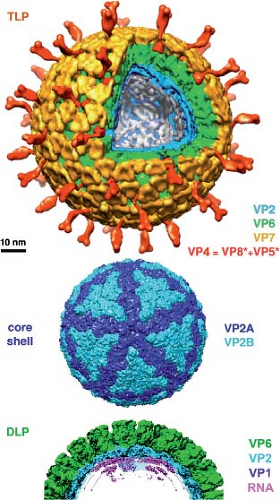 Figure 3.12. Molecular organization of a rotavirus particle, illustrating the multiple concentric protein shells.42,202,259 The complete virion (top) or triple-layered particle (TLP) has an outer layer composed of VP7 (yellow) and VP4 (red: cleaved during maturation into two parts, VP8* and VP5*, which remain associated). The double-layered particle or DLP (bottom) has a core shell (center) with 120 VP2 subunits (blue) surrounded by a layer of 290 VP6 trimers (green) in a T = 13 icosahedral lattice. The VP6 layer in turn dictates the organization of the VP7 layer, which clamps into place 60 VP4 trimers projecting from a particular set of six-coordinated positions. The locations of the VP1 polymerase (purple, ribbon representation)72 and of tightly wound, double-stranded RNA (dsRNA) (magenta)151 are also shown in the bottom cutaway. The icosahedrally symmetric core shell has 120 VP2 subunits in two sets (designated A and B, dark blue and light blue, respectively), with completely nonequivalent contacts and only slightly different conformations. This type of shell is characteristic of many groups of dsRNA viruses. |
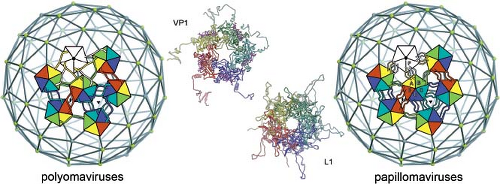 Figure 3.13. Packing of pentamers in the capsids of polyoma- and papillomaviruses. The ribbon diagrams in the center show pentamers of VP1 (polyomaviruses) and L1 (papillomaviruses), viewed from their outward-facing surfaces. Note the C-terminal arms of the subunits, which extend away from the pentamers in VP1 but loop back to it in L1. The schematic diagrams to the left and right illustrate the packing of these pentamers in the virion shell. The framework shows a T = 7 icosahedral lattice; VP1 or L1 pentamers are centered on both six- and five-coordinated positions. |
Larger and more complex structures, such as adenoviruses, have separate framework proteins. The principal outer-shell components of adenoviruses are hexons (trimers of a subunit with two similar jelly-roll β-barrel domains) and pentons (pentamers of a subunit with a single jelly-roll β-barrel domain); a set of additional proteins cement the structure together and determine its size (Fig. 3.10).80,214,215 The elaborate interaction patterns of these cement proteins stabilize a group of nine hexons, centered on the icosahedral threefold axis, and a group of six (five hexons and a penton), centered on the icosahedral fivefold axis.134,142 The structure of an adenovirus-like bacteriophage, PRD1,16 shows a somewhat simpler size-determining and stabilizing framework: a tape-measure protein extends from the penton toward the icosahedral twofold axis, where it interacts with an identical protein running toward it from the twofold-related penton (Fig. 3.14).2 Unlike adenoviruses, PRD1 has a lipid-bilayer membrane between the P3 layer and the internally coiled DNA.11,49
During assembly of the heads of most double-stranded DNA (dsDNA) bacteriophages, an internal scaffold protein directs formation of a prohead.33 Signals related to initiation of DNA packaging trigger release and recycling (P22) or degradation (T4) of the scaffold, accompanied by a reorganization and expansion of the head (Fig. 3.15A,B).68,119 DNA is pumped into the empty head until it reaches a tightly coiled state, as illustrated in Figure 3.15C.69,70,211 In these examples, scaffold is a good description of the internal protein, because it is removed once the structure is complete.
The fundamental principle embodied in all the various structures just described is one of mass production. One or more standard building blocks assemble into the larger structure. In simple (T = 1) cases, such as the parvoviruses and picornaviruses, a repeating set of identical interactions determines the final structure. Even in many of these cases, however, extended arms form an interconnecting framework. In more elaborate cases, framework elements, either permanent or transient, ensure a unique outcome.
Elongated Shells
The examples in Figure 3.16 illustrate elongated particles with caps at either end. In many of the dsDNA bacteriophages, the shell looks like a familiar icosahedral design at the poles. As the lattice approaches the equator, however, the regular interspersion of fivefolds and local sixfolds gives way to local sixfolds only, so that there is a tubular region around the middle of the particle (Fig. 3.16A–C).224 The tubular region can be of varying extent; in extreme cases, it can be much longer than the caps themselves. A further variation on this theme is found in the shells formed by the CA fragment of the lentivirus Gag protein. Conical structures seen within HIV-1 particles have been shown to be based on the sort of arrangement shown in Figure 3.16D, where one cap has more than six fivefolds and the other has less, so that the diameters of the two caps are different.83 (Note that if there are only sixfold and fivefold vertices in a closed surface lattice, there will always be exactly 12 of the latter.)
Multishelled Particles
Most dsRNA viruses have a genuinely multishelled icosahedral organization, with some common features and some variation from group to group (see Chapters 44–46). In virions of the mammalian dsRNA virus groups (reoviruses, rotaviruses, and orbiviruses), the innermost protein shell contains 120 copies of
a large, rather plate-like protein96,151,186 (Fig. 3.12). Surrounding the inner shell is a second characteristic layer. In most cases, it contains 780 copies of a trimeric protein with a radially directed jelly-roll β-barrel and inwardly directed N- and C-termini, which together form an extensive and largely α-helical “base” domain.95,140,149 This second layer corresponds closely to a “classical,” quasiequivalently packed, T = 13 icosahedral shell—all the interactions between adjacent trimers are variations on the same set of contacts.
a large, rather plate-like protein96,151,186 (Fig. 3.12). Surrounding the inner shell is a second characteristic layer. In most cases, it contains 780 copies of a trimeric protein with a radially directed jelly-roll β-barrel and inwardly directed N- and C-termini, which together form an extensive and largely α-helical “base” domain.95,140,149 This second layer corresponds closely to a “classical,” quasiequivalently packed, T = 13 icosahedral shell—all the interactions between adjacent trimers are variations on the same set of contacts.
 Figure 3.14. Bacteriophage PRD1. Left: Side and bottom views of the hexon protein, P3. The colors correspond to those in the ribbon diagrams of the adenovirus hexon trimer in Figure 3.10. Like the adenovirus hexon, P3 has two jelly-roll β-barrels, but the loops that project outward are much less elaborate.16 (The variable adenovirus hexon loops probably evolved as a means of immune evasion, not relevant for a bacteriophage.) The image on the upper right, based on a crystal structure of the intact phage particle,2 is a view along a twofold axis. One threefold set of P3 trimers is highlighted by triangles. The pentons (P31) are in red. At the lower right is a view with the outer layer stripped away, to show the extended tape-measure protein, P30, which helps determine the size of the shell, and the lipid bilayer just beneath it. There are 60 copies of P30; each chain extends from a twofold axis (N-terminal end, blue) to the inner surface of a penton (C-terminal end, red). At the twofold axis, one P30 associates with a second, twofold-related P30, which projects toward the opposite icosahedral vertex. (Courtesy D. Stuart, Oxford University.) |
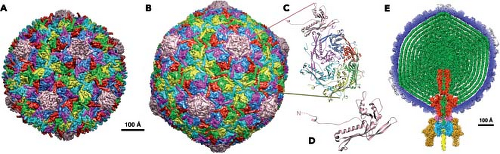 Figure 3.15. Capsid reorganization and DNA packaging in tailed bacteriophages.119 A: Surface of the HK97 procapsid. The surface organization is a locally distorted T = 7 arrangement, with fivefold symmetric association of the subunit at the fivefold positions (beige) but a skewed arrangement in the rings of six subunits that surround a local six-coordinated position (colored in magenta, blue, red, green, yellow, and cyan, in clockwise order).53 An N-terminal extension of the head subunit is the scaffold for prohead assembly; its cleavage by a co-assembled protease triggers rearrangement of the subunits into the expanded, thinner, more angular shell illustrated in B.65 B: Capsid (head) of the mature HK97 particle; molecular surface, based on crystallographic model, colored as in A.242 This view is oriented so that a fivefold axis is vertical. The image is derived from the structure of an empty capsid with 420 subunits in a T = 7 icosahedral lattice. In a wild-type bacteriophage particle, one of the rings of five subunits is replaced with a portal protein connected to a tail (see E). C: Expanded view in ribbon representation of one icosahedral asymmetric unit (i.e., one of the five subunits in the pentameric ring and one each of the quasiequivalent subunits in the hexameric ring). All subunits are chemically identical. In HK97, but not in many related bacteriophages, an intersubunit isopeptide bond, which forms during maturation, crosslinks the entire coat.65 D: A further enlarged view of a single, 31-kD subunit. The 105-residue N-terminal extension that functions as an assembly scaffold is indicated schematically by a dotted line. E: Cutaway representation of a three-dimensional electron cryomicroscopy (cryoEM) image reconstruction of bacteriophage P22. Its assembly is formally similar to that of HK97, but there is a distinct, recycled scaffold protein33 and no covalent crosslinking of the head.173 The packaged DNA (green) winds tightly around an internal extension of the portal protein (red).169,222 The axis of DNA winding is vertical in this view; averaging of many particles in the reconstruction produces concentric shells of density, because the exact register of the DNA coils varies from particle to particle. (Images in A–D from VIrus Particle ExploreR [VIPERdb] Web Site, http://viperdb.scripps.edu/.) |
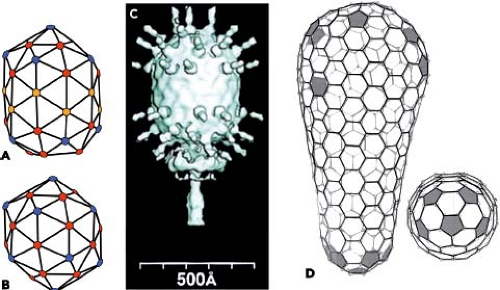 Figure 3.16. Elongated shells. A–C: Bacteriophage φ29.224 The surface lattice of the φ 29 capsid (A) has the equivalent of a T = 3 icosahedral cap (B) at either pole with an equatorial insertion of two rows of six-coordinated positions (i.e., six, locally sixfold-related, coat-protein subunits). The blue dots are at five-coordinated positions (five, locally fivefold-related, coat-protein subunits); the red dots are at the six-coordinated positions of a T = 3 lattice in the cap; the orange dots are at the inserted six-coordinated positions. The cap at the “south pole” is further modified by replacement of the axial pentameric cluster of coat subunits with the collar and tail structure, as shown in the surface view in C. D: The conical structure of the mature capsid of HIV-1.83 The capsid subunit, CA, cleaved from the Gag precursor, forms a structure with two unequal caps, one with seven five-coordinated positions and one with five. In the former, the five-coordinated positions have more intervening six-coordinated lattice points than in the latter, so that the radius of the one is larger than the radius of the other. The shaft of six-coordinated positions is wrapped in such a way that a circumference includes increasing numbers of subunits as one traces from the “bottom” to the “top” of the conical capsid, as illustrated here. The two caps have a five-coordinated lattice point at the apex, but immediately deviate from an icosahedral arrangement, as shown in the end-on view of the lower cap (bottom left). |
Various elaborations and simplifications of the two-layer design just described differentiate the families of dsRNA viruses. For example, in the reoviruses, the T = 13 layer has gaps, through which pentameric “turrets” of yet another protein, anchored on the inner shell, project; only 600 of the potential 780 subunits are actually present.27,64,186 The birnaviruses lack the 120-subunit layer altogether and have instead 780 copies of a single major capsid protein, with a shell domain that resembles those of plant and insect viruses and a trimer-clustered projecting domain that resembles the jelly-roll β-barrel in the T = 13 shell of rotaviruses and orbiviruses.54 The T = 13 packing of the shell domain so closely recalls that of its counterparts in T = 3 and T = 4 positive-strand RNA virus structures that a bridge between the two families seems plausible. Similarities in the RNA-dependent RNA polymerases of these viruses also suggest some common ancestry. The dsRNA bacteriophages such as φ6 contain the 120-subunit, inner-shell layer and a fenestrated, T = 13 layer (rather like reoviruses), contained within a lipid-bilayer membrane.27,112,114,236
Rearrangements in Surface Lattices
Icosahedral surface lattices can undergo rearrangements, which preserve the overall symmetry of the structure but change the pattern of specific intersubunit contacts. There can be an accompanying change in the diameter of the shell. These rearrangements are cooperative—that is, they occur more or less simultaneously across the whole structure. As illustrated in Figure 3.14, when dsDNA bacteriophages such as P22 insert their genomic DNA into a preformed prohead, the outer shell of the prohead expands as its subunits shift around to form the mature structure.33,53,117,133 Another well-characterized example is expansion of the T = 3 plant viruses, which occurs when the calcium ions that stabilize a particular set of subunit interfaces are removed190 (Fig. 3.17). This swelling is believed to be the first step in disassembly; plant viruses are injected by their vectors directly into the cytoplasm of the recipient cell, where they are exposed to a low CaR2+-3+ environment. A similar, but transient, expansion occurs when poliovirus binds its receptor.15 In both the T = 3 plant viruses and the picornaviruses, internally directed “arms” of the protein subunits move outward from the interior as expansion creates gaps in the shell. Exposure of the arms may be part of the uncoating process in the case of the plant viruses or of the penetration
process in the case of the picornaviruses. Cooperativity of these rearrangements implies that a few points of inhibition can prevent the change. For example, only a few intersubunit crosslinks from bound neutralizing antibodies are sufficient to block infection by a picornavirus particle.71 The same may be true of small molecules that inhibit the subunit conformational changes needed for the receptor-triggered expansion of picornaviruses.8,177,212
process in the case of the picornaviruses. Cooperativity of these rearrangements implies that a few points of inhibition can prevent the change. For example, only a few intersubunit crosslinks from bound neutralizing antibodies are sufficient to block infection by a picornavirus particle.71 The same may be true of small molecules that inhibit the subunit conformational changes needed for the receptor-triggered expansion of picornaviruses.8,177,212
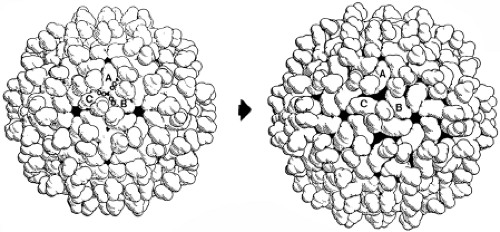 Figure 3.17. Expansion of tomato bushy stunt virus (TBSV).190 The mature, compact particle (upper left) expands when CaR2+-3+ ions (small circles) are removed. The expanded form (upper right) is reached by a smooth transition, in which many of the intersubunit contacts are conserved. The contacts that included the ions in the compact state have separated substantially, creating a fenestrated shell. |
Helical surface lattices can also rearrange without dissociating. Contraction of bacteriophage tail sheaths is a good example.
Two Recurring Globular Domains in Icosahedral Capsid Proteins
The icosahedrally symmetric shells of nearly all well-characterized, nonenveloped viruses contain one of two types of globular domain. (The known exceptions at the time of writing this chapter are the RNA bacteriophages—R17, Qβ, and their relatives229—and the dsRNA picobirnaviruses.66) One is the jelly-roll β-barrel in viruses of animals and plants, which we have described in various examples of viruses of animals and plants; it is also the principal component of icosahedral ssDNA bacteriophage capsids (e.g., φX174).152 The various ways this module can form a coat are quite different, of course, and we have emphasized earlier the importance of framework components (either as extensions of the polypeptide chain of the β-barrel or as separate protein species) in directing or regulating coat assembly. What sort of evolutionary parsimony resulted in such widespread appearance of a single kind of protein module is not evident. Viruses can jump from plants to insects and from insects to vertebrates, so the recurrence of the jelly-roll β-barrel is unlikely to reflect a common origin for all these viruses that antedates host divergence, but rather the result of more recent selection and genetic exchange. Cellular fusion proteins acquired from viral fusion proteins through retrotransposons illustrate one way in which such exchange can occur.
Figure 3.14 shows the second basic building block, discovered initially in the coat of dsDNA bacteriophages such as HK97 and subsequently found in most other dsDNA bacteriophages (T4, lambda, P22, etc.). This HK97 fold is also the core of the herpesvirus capsid subunit.9 Like their bacteriophage cousins, herpesviruses pump their genome DNA into a preformed shell through a specialized icosahedral vertex and a dodecameric portal protein.39,163,164 Adenoviruses, and probably their bacteriophage cousins like PRD1, with hexon-like capsid subunits, are also thought to insert DNA into a preassembled empty capsid, but the motors that effect the insertion seem to be different from those in the herpesviruses.113,170,256,257 Thus, the structures of the coat proteins of two major classes of dsDNA viruses appear to correlate with the machinery by which members of each of these classes package DNA.
Self-Assembly and Cleavage Steps
Some of the simplest virus particles can assemble spontaneously from their dissociated or recombinant components, in the absence of any further modifications or scaffolds. These particles are said to self-assemble, because they do not require additional activities (encoded either by the virus or by the host cell) to form. In an infected cell, however, host chaperones, such as Hsc70 and its paralogs, may enhance efficiency of subunit folding or subunit assembly, even when they are not absolutely essential.
Most viruses, and nearly all viruses that infect animal cells, cannot reassemble from dissociated particles, because one or more irreversible steps intervene in forming the mature, infections virion. The picornaviruses, already described, illustrate one kind of irreversible step. In an infected cell, the principal structural proteins are cleaved from a polyprotein precursor (by a viral protease) before particle assembly, but one final, autocatalytic cleavage step occurs after assembly—the scission of a peptide bond between VP4 and VP2 (see Chapter 16 and caption to Fig. 3.7). The cleavage depends on the three-dimensional arrangement of the scissile bond, as found in a newly assembled precursor particle. Rearrangements of parts of the subunits following the cleavage stabilize the now mature, infectious virion. Proteolytic cleavages by cellular or extracellular host proteases are critical steps in the maturation of many types of virus particles, even when processing of a precursor polyprotein is not involved. For example, many of the surface glycoproteins that facilitate membrane fusion during entry of enveloped viruses require activation by a furin-like protease late in the secretory pathway.
Specific, postassembly proteolytic cleavage usually has two consequences. First, as in poliovirus or many viral fusion
proteins (see later), it leads to a local rearrangement of polypeptide chains that stabilizes the structure. Second, it allows the structure to undergo a much larger reorganization when “triggered” by binding of a specific ligand. Thus, when a mature poliovirus particle binds its receptor, an expansion occurs that allows VP4 to escape and to interact with adjacent membrane—a critical first step for translocating the particle (or its genome) from an endocytic compartment into the cytosol.21,100,157 Likewise, many fusion proteins of enveloped viruses undergo large-scale, fusion-promoting conformational changes when they bind protons in acidic endosomes—but again, only if the critical cleavage has occurred.209 In physicochemical terms, the cleaved structure is metastable: a large kinetic barrier separates it from its true energy minimum. The barrier can be so large that the virus remains infectious for many weeks or months. Ligand binding (receptors, protons, etc.) lowers the kinetic barrier, leading to a rapid conformational rearrangement, coupled in most cases to an important step in viral entry.
proteins (see later), it leads to a local rearrangement of polypeptide chains that stabilizes the structure. Second, it allows the structure to undergo a much larger reorganization when “triggered” by binding of a specific ligand. Thus, when a mature poliovirus particle binds its receptor, an expansion occurs that allows VP4 to escape and to interact with adjacent membrane—a critical first step for translocating the particle (or its genome) from an endocytic compartment into the cytosol.21,100,157 Likewise, many fusion proteins of enveloped viruses undergo large-scale, fusion-promoting conformational changes when they bind protons in acidic endosomes—but again, only if the critical cleavage has occurred.209 In physicochemical terms, the cleaved structure is metastable: a large kinetic barrier separates it from its true energy minimum. The barrier can be so large that the virus remains infectious for many weeks or months. Ligand binding (receptors, protons, etc.) lowers the kinetic barrier, leading to a rapid conformational rearrangement, coupled in most cases to an important step in viral entry.
Genome Packaging
Incorporation of viral nucleic acid must be specific, but it must also be independent of most of the base sequence of the genome. Therefore, viral genomes generally have a packaging signal—a short sequence or set of sequences that directs encapsidation. Recognition of the packaging signal depends on the nature of the genome and on the complexity of the assembly mechanism. In many cases, there is a direct interaction between the packaging signal and the capsid protein. Some complex viruses insert genomic nucleic acid into a preformed shell, and genome recognition is a property of the packaging system. If replication and packaging are closely coupled, as they are in picornaviruses,168 flaviviruses,121 and at least some RNA plant viruses,5 a specific packaging signal may be less essential.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


