Quentin J. Baca, Donald M. Coen, and David E. Golan
Minimum Inhibitory Concentration and Minimum Bactericidal Concentration | |
Types of Drug Interactions—Synergy, Additivity, and Antagonism | |
Co-administration of Penicillins with β-Lactamase Inhibitors | |

Many infections and some cancers can be successfully treated with single-drug therapies. Such regimens often fail, however, when pathogens or tumors develop resistance to chemotherapeutic agents, when multiple pathogens with different drug susceptibilities are simultaneously present, or when the dose of the therapeutic agent is limited by toxicity. Under these circumstances, combination chemotherapy may offer decisive advantages. The drugs in a multidrug regimen can interact synergistically to enhance the antimicrobial or antineoplastic effectiveness of the combination and can decrease the likelihood that resistance will emerge. Combinations are frequently used when treatment must be initiated before the definitive identification of the pathogen, and synergistic combinations can be used to reduce toxicity when the individual drugs in the combination have low therapeutic indices. Although combination chemotherapy opens new avenues for the expedient elimination of a pathogen or tumor, it also introduces the potential for multiple adverse effects and drug interactions. The goal of any combination drug regimen should be to efficiently remove the offending pathogen or tumor without incurring unacceptable host toxicity.
 Mr. M is a 27-year-old man from rural Haiti who presents to a clinic with a chronic cough. He cannot afford treatment at a private clinic, so he goes to a drugstore and asks the pharmacist for appropriate medication. The pharmacist thinks that Mr. M could have tuberculosis, and he sells Mr. M a 2-week supply of isoniazid and rifampin. Mr. M takes both drugs for a couple of days, but they make him nauseated, and he decides to take just the isoniazid for 2 weeks. His symptoms resolve.
Mr. M is a 27-year-old man from rural Haiti who presents to a clinic with a chronic cough. He cannot afford treatment at a private clinic, so he goes to a drugstore and asks the pharmacist for appropriate medication. The pharmacist thinks that Mr. M could have tuberculosis, and he sells Mr. M a 2-week supply of isoniazid and rifampin. Mr. M takes both drugs for a couple of days, but they make him nauseated, and he decides to take just the isoniazid for 2 weeks. His symptoms resolve.
Three months later, Mr. M’s cough returns. This time, he notices blood in his sputum and he has night sweats. He takes the remainder of the 2-week supply of rifampin and experiences a brief lull in his symptoms. Within a few days, however, his cough, bloody sputum, and night sweats return. Because he does not have enough money to buy additional drugs, he travels to the nearest government hospital to seek free care and medications. The government doctor takes three sputum samples, all of which are positive for acid-fast bacilli. The doctor also sends sputum to the laboratory for culture, but since Mycobacterium tuberculosis (the causative agent of tuberculosis) is slow-growing, she also starts Mr. M on a multidrug regimen consisting of isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifampin for 4 months.
Several weeks later, the culture reveals that Mr. M’s tuberculosis is not susceptible to either isoniazid or rifampin. Mr. M’s doctor is now seeking a new recommendation for treatment.
Questions
1. Why did the government doctor prescribe four different drugs for Mr. M?
2. How is resistance transferred from one generation of tubercle bacilli to the next? How does this resistance-transfer mechanism compare to the mechanism by which penicillin resistance is transferred?
3. Why were Mr. M’s initial efforts at treatment unsuccessful? What treatment strategy could have been employed to avoid Mr. M’s treatment failure?
4. Does Mr. M have multidrug-resistant tuberculosis (MDR-TB)? Should he stay on the four-drug regimen that includes isoniazid and rifampin? If not, how should his treatment be modified?
 ANTIMICROBIAL COMBINATION THERAPY
ANTIMICROBIAL COMBINATION THERAPY
In the treatment of microbial infections, drug combinations are used (1) to prevent the emergence of drug resistance, (2) to enhance the activity (efficacy) of the drug therapy against a specific infection (synergy), (3) to reduce toxicity to the host, (4) to treat multiple simultaneous infections (sometimes called polymicrobial infections), and (5) to treat a life-threatening infection empirically before the microorganism causing the infection has been identified. Because microbes are genetically distant from humans, antimicrobial drug combinations can target several different molecules that are specific to the microbe(s), potentially without a concomitant increase in adverse effects. In contrast, antineoplastic drug combinations are often limited by adverse effects (see below). The following section provides a conceptual framework for the different types of antimicrobial drug interactions and discusses specific examples of antimicrobial combination therapy.
Minimum Inhibitory Concentration and Minimum Bactericidal Concentration
Antimicrobial agents with activity against a pathogenic bacterial, protozoal, or fungal microorganism can be characterized by the minimum inhibitory concentration (MIC) and the minimum bactericidal concentration (MBC) for the drug–pathogen pair. The MIC is defined as the lowest concentration of drug that inhibits growth of a culture of the microorganism after 18–24 hours of incubation in vitro. The MBC is defined as the lowest concentration of drug that kills 99.9% of a culture of the microorganism after 18–24 hours of incubation in vitro. In general, the MBC is higher than the MIC. Comparisons between the MICs or MBCs and the clinically achievable concentrations of antimicrobial drugs allow these drugs to be grouped broadly into two categories: cidal and static (Table 41-1; also see Chapter 33, Principles of Antimicrobial and Antineoplastic Pharmacology). An antimicrobial agent is static (e.g., bacteriostatic, fungistatic) if its MIC is within the therapeutic range of the drug but its MBC is not. The agent is cidal (e.g., bactericidal, fungicidal) if its MBC is within the therapeutic range of the drug. Note that the MIC and MBC refer to a specific drug–microbe pair under a specific set of conditions. Many drugs with activity against an organism are static in one growth medium but cidal in another or are cidal at sufficiently high concentrations in vitro. Furthermore, for any particular drug, the MIC and MBC may differ from one microbe to the next. Indeed, a drug may be static against one organism and cidal against another. As an operational definition, we can state that, at therapeutic concentrations, cidal drugs kill the microorganism, while static drugs merely arrest microbial growth. In this definition, the therapeutic concentration refers to plasma drug levels that are sufficient for pharmacologic activity (here, killing or arresting the growth of the microorganism) without unacceptable toxicity to the patient. For example, most inhibitors of bacterial cell wall synthesis are bactericidal, whereas most inhibitors of bacterial protein synthesis are bacteriostatic (see Chapter 34, Pharmacology of Bacterial Infections: DNA Replication, Transcription, and Translation, and Chapter 35, Pharmacology of Bacterial and Mycobacterial Infections: Cell Wall Synthesis).

As noted in Chapter 33, an important distinction between static and cidal drugs lies in their clinical applications. In general, the successful use of static drugs to treat infections requires an intact host immune system. This is because static drugs do not kill microorganisms but only prevent them from multiplying. Accordingly, such drugs rely on the host’s immune and inflammatory mechanisms to effect clearance of organisms from the body. These drugs are more efficacious when initiated early in the course of an infection (i.e., when the infectious burden is lower). Furthermore, removal of a static drug before the immune system has completely cleared the infection can result in resumption of microbial growth and reappearance of infection (Fig. 41-1).
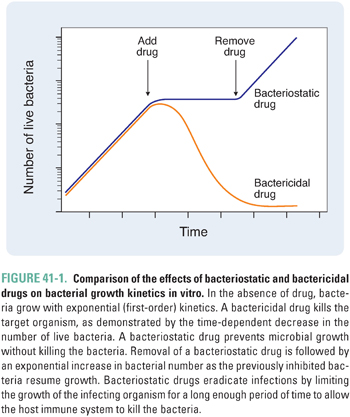
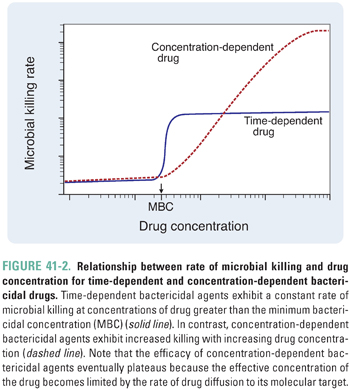
According to their mechanism of cell killing, bactericidal agents can be further characterized as time-dependent or concentration-dependent (Fig. 41-2). Time-dependent bactericidal agents exhibit a constant rate of killing that is independent of drug concentration, provided that the drug concentration is higher than the minimum bactericidal concentration (MBC). Thus, the overriding consideration for the clinical use of such agents is not the absolute drug concentration that is achieved, but the duration of time during which the drug concentration remains in the therapeutic range (which is defined as [drug] > MBC). In contrast, concentration-dependent bactericidal agents exhibit a rate of killing that increases with drug concentration for [drug] > MBC. For such agents, a single very large dose may be sufficient to eliminate the infection.
Types of Drug Interactions—Synergy, Additivity, and Antagonism
The discussion has thus far considered the general properties of drugs used as single agents to treat a microbial infection. When such drugs are used in combination with other agents, these effects can be modified (either enhanced or diminished). In fact, some drugs that have little or no activity against an organism when used as single agents can show high activity when used in combination with another agent. One example of this concept involves the treatment of Enterococcus faecalis, a Gram-positive organism that exhibits little susceptibility to aminoglycosides. Recall that aminoglycosides are thought to kill bacteria by inducing misreading of the genetic code and translation of defective proteins, which cause further cellular damage (see Chapter 34). In the case of E. faecalis, aminoglycosides are unable to penetrate the organism’s thick cell wall to reach their target, the 30S ribosomal subunit. However, when used in combination with a cell wall synthesis inhibitor such as vancomycin or a β-lactam antibiotic, aminoglycosides are able to reach the bacterial ribosomes and effectively kill the bacteria (see Chapter 35). The potentiating effect of the cell wall synthesis inhibitor on the activity of the aminoglycoside is an example of the important pharmacologic concept of synergy.
From this example, one could ask whether combining two drugs with individual activity against a particular microbe always results in a more efficacious drug combination. Surprisingly, for many combinations, this turns out not to be the case. In fact, when two drugs with activity against the same pathogen are combined, the drugs can interact to diminish the efficacy of the combination relative to each alone (antagonism). Alternatively, the drugs may not interact, and the effect of the combination is simply the sum of the effects of each drug used individually (additivity). The interaction between two antimicrobial drugs is often quantified by selecting a particular endpoint (e.g., inhibition of bacterial growth) and then measuring the effect of various combinations of the two drugs that reach this endpoint. When such data are plotted, additional information can be obtained (Fig. 41-3). The x- and y-intercepts correspond to the MICs of each of the two drugs, and the concavity of the curve indicates the nature of the interaction between the two drugs—concave-up is synergistic; concave-down is antagonistic; linear is additive. The following discussion provides a mathematical rationale for these relationships.
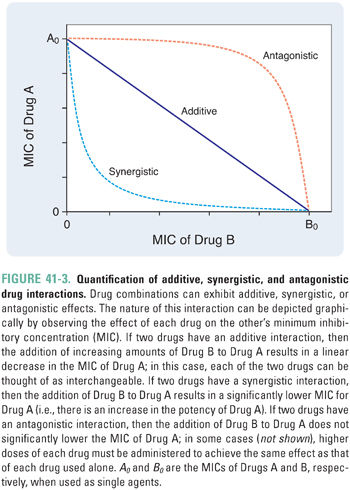
Suppose that Drugs A and B inhibit a particular enzyme required for bacterial growth. In this case, the ratio [A]/MICA would represent the fraction of bacterial growth inhibition that can be attributed to the presence of Drug A. This is known as the fractional inhibitory concentration of A (FICA). Similarly, FICB = [B]/MICB is the fraction of growth inhibition that can be attributed to Drug B. Now, suppose that the concentration of A is decreased by a small amount, −d[A]. To compensate for this loss of growth inhibition (dFICA = −d[A]/MICA), the concentration of B must be increased by an amount +d[B].
For additive drugs, the ratio −d[A]/d[B] (which is the same as the slope of the curve in Fig. 41-3) is a constant because one unit of A has exactly the same activity as (MICA/MICB) units of B. For example, A and B could bind to independent sites on the enzyme (i.e., each drug has no effect on the binding of the other drug).
In contrast, if A and B are synergistic, then the amount of B (d[B]) required to compensate for a decrease in A (−d[A]) depends on the amount of A that is already present. Because of the potentiating effect of Drug A on Drug B, d[B] is smaller for higher [A] (i.e., d2[A]/d[B]2 > 0, which corresponds to the concave-up curve in Fig. 41-3). For example, the binding of A could induce a conformational change in the enzyme that enhances the binding of B.
By extension, A and B are antagonistic if the amount of B required to compensate for a small decrease in the concentration of A is larger for higher [A] (i.e., d2[A]/d[B]2 < 0, which corresponds to the concave-down curve in Fig. 41-3). For example, the binding of A could induce a conformational change in the enzyme that reduces the binding of B.
Because of its intuitiveness and simplicity, the mathematical model described above is often used to define synergy, additivity, and antagonism. However, the experimental determination and quantitative analysis of the effects of multiple drugs are complex topics that are beyond the scope of this text. The interested reader is referred to the work of TC Chou (1984, 2006) for an extensive treatment of this subject.
Several generalizations can be made concerning the nature of drug interactions between different classes of antimicrobial agents. First, many bacteriostatic drugs (e.g., tetracycline, erythromycin, chloramphenicol) antagonize the action of bactericidal drugs (e.g., vancomycin, penicillin) by inhibiting cell growth and/or preventing the cellular processes that are required for cidal drugs to act (described below in more detail). Second, two bactericidal drugs usually act synergistically in combination. One notable exception to the latter generalization is that rifampin, a bactericidal inhibitor of RNA polymerase, antagonizes other bactericidal drugs by inhibiting cell growth. Finally, the interactions between two bacteriostatic drugs are often additive but cannot be predicted in all cases.
Examples of Antimicrobial Combination Therapy
The treatment of tuberculosis illustrates one of the principal reasons that combinations of drugs are used—to suppress the emergence of resistance. In the course of this illness, tuberculous bacilli (also called mycobacteria) are inhaled and phagocytosed by alveolar macrophages, in which the bacilli multiply within intracellular vacuoles. A predominantly T-cell–mediated lymphocytic response is elicited, and the macrophages and helper T cells form large granulomas that wall off the infected sites. Activated macrophages are usually able to keep the infection under control by killing the multiplying bacilli but are unfortunately unable to eradicate the infection completely. Tissue damage is caused by the release of neutral proteases and reactive oxygen intermediates from activated macrophages, with the end result that central necrosis occurs in the tuberculous cavities in the lungs. Inside each of these cavities, as many as 108 to 109 living bacilli may be held in check by macrophages and helper T cells.
Successful cure of tuberculosis infections typically requires the use of combinations of drugs with antimycobacterial activity. Commonly used drugs include isoniazid, rifampin, pyrazinamide, and ethambutol (see Chapter 35). As illustrated in the case of Mr. M, a standard regimen could consist of 2 months of isoniazid, rifampin, pyrazinamide, and ethambutol, followed by 4 months of isoniazid and rifampin. Kanamycin and other second-line drugs are sometimes substituted for one or two drugs in this regimen if resistance develops. Isoniazid and rifampin are often the preferred drugs in such regimens because of their ability to kill intracellular as well as extracellular mycobacteria.
As noted in Chapter 35, resistance to antimycobacterial drugs develops primarily through chromosomal mutations, and the frequency of resistance to any one of the drugs is about 1 in 106 bacteria. These mutations are passed on to daughter cells when the bacteria replicate, leading to establishment of a drug-resistant population. Chapter 35 discusses the implications of the fact that a tuberculous cavity contains 108 to 109 bacteria, while the frequency of mutants resistant to a single drug is about 1 in 106. On average, then, 100 bacteria will already be resistant to each drug in any single lesion, even before that drug is administered. Moreover, treatment with only one drug would result in selection for bacilli that are resistant to that drug. In the case of Mr. M, his initial 2 weeks of isoniazid treatment likely killed all the isoniazid-susceptible bacilli in his cavity. This accounts for the cessation in his symptoms after 2 weeks of treatment. However, the 100 or so isoniazid-resistant bacilli that were selected for by Mr. M’s use of monotherapy remained alive and multiplied. If he had taken rifampin as well as isoniazid, only 1 in 1012 bacilli would likely have been resistant to both drugs, and he might have eradicated the infection.
Over the 3 months during which Mr. M stopped taking isoniazid, the isoniazid-resistant bacilli remaining in his lungs multiplied, leading to a relapse in his symptoms. He then began taking rifampin. Of the 108 to 109 isoniazid-resistant bacilli in each lesion, again there was a 1 in 106 probability that a bacillus had mutated to acquire rifampin resistance. By taking rifampin for 2 weeks, he killed all the rifampin-susceptible bacilli but selected for rifampin-resistant organisms. He was therefore left with bacilli that were both isoniazid-resistant and rifampin-resistant—the phenotype of multidrug-resistant tuberculosis (MDR-TB).
Mr. M will require a new drug regimen for treatment of MDR-TB. Ideally, the regimen should be constructed using drugs that have been shown to be effective in susceptibility tests. Also, at least initially, drugs that are part of his current treatment plan should be avoided (i.e., pyrazinamide and ethambutol), because his bacilli may have developed resistance to these agents. Isoniazid and rifampin should certainly be avoided. Treatment for MDR-TB should begin with at least four new drugs to which Mr. M’s TB isolate has proven susceptibility. Such regimens usually include daily dosing of a parenteral aminoglycoside (kanamycin or amikacin) or peptide antibiotic (capreomycin), together with a fluoroquinolone (levofloxacin or moxifloxacin), for at least 4–6 months. Three to five oral drugs should be co-administered with the aminoglycoside and fluoroquinolone for 18–24 months after the sputum culture converts to negative. Ethionamide and clofazimine are second-line drugs that could be included in the regimen. Newer drugs that could be included are linezolid, bedaquiline, and delamanid. Note that, as a whole, the second-line regimen will be significantly more toxic than the first-line regimen. If bacilli isolated from Mr. M’s sputum prove to be susceptible to pyrazinamide and/or ethambutol, these agents could replace some of the more toxic drugs.
Considering all the issues discussed above, MDR-TB is to be avoided at all costs. Patients with drug-susceptible tuberculosis require access to combination therapy as well as help in adhering to the combination therapy to avoid the emergence of drug-resistant bacilli. This rationale is the basis for DOTS (Directly Observed Therapy Short Course), the World Health Organization (WHO)-recommended strategy for tuberculosis treatment. DOTS is a public health program that has five components: (1) political commitment and resources for TB control, (2) the use of sputum-smear microscopy for accurate diagnosis of TB infection, (3) a standardized 6- to 8-month treatment that is directly observed by a community health worker for at least the first 2 months, (4) a regular and uninterrupted supply of medicines, and (5) standardized recording and reporting of each patient’s treatment and progress to central authorities. When used in cases of drug-susceptible TB, DOTS has a remarkable cure rate and can prevent the development of resistance. As noted above, treatment of MDR-TB requires therapy that is more intensive, more invasive, more toxic, and of longer duration than the standard DOTS regimen.
The importance of controlling MDR-TB is highlighted by the growing prevalence of extensively drug-resistant TB (XDR-TB), which had been observed in all regions of the world by 2006. XDR-TB has extremely limited treatment options; by definition, clinical isolates display the phenotype of MDR-TB (resistance to isoniazid and rifampin) as well as resistance to fluoroquinolones and to at least one of the three commonly used parenteral anti-TB drugs (capreomycin, kanamycin, and amikacin). Because the prevalence of XDR-TB is generally higher in resource-poor countries and in populations that are co-infected with human immunodeficiency virus (HIV), world health organizations have recently emphasized the need for a coordinated global response to limit the spread of XDR-TB. Treatment of XDR-TB frequently involves the use of up to five drugs, guided by drug susceptibility testing.
A second reason for using a combination drug regimen is to take advantage of the synergy between the actions of the two drugs. This consideration is especially important in the setting of infections that are not readily handled by the immune defenses of immunocompromised patients. In the immunocompetent patient, bacteriostatic and bactericidal drugs are often equally efficacious in eliminating an infection. Bactericidal drugs are strongly preferred, however, in the setting of immunocompromised patients (e.g., HIV/AIDS patients, immunosuppressed transplant patients, and neutropenic cancer patients), endovascular infection (e.g., bacterial endocarditis), or meningitis. The reason for using bactericidal combinations in the immunocompromised patient should be obvious—the host does not have sufficient numbers of functioning lymphocytes and/or neutrophils to eliminate even a nondividing bacterial population. In the case of endocarditis, the reason is not so straightforward. In this case, although there is not a deficiency in the absolute number of leukocytes, the phagocytes are unable to efficiently penetrate the thick “vegetation”—composed of a meshwork of fibrin, platelets, and bacterial products—that surrounds the bacteria. Bactericidal drug combinations are sometimes indicated for meningitis to maximize the probability of overcoming the poor opsonization of bacteria by antibody and complement in the immunologically privileged site of the meninges, especially if the responsible organism is not known (see Chapter 9, Principles of Nervous System Physiology and Pharmacology).
One example of antibacterial synergy involves the use of a penicillin and an aminoglycoside to treat a common cause of subacute bacterial endocarditis, Streptococcus viridans. As described above, the mechanism of synergy relies on the penicillin inhibiting cell wall biosynthesis, which allows the aminoglycoside to penetrate the thick peptidoglycan layer of this Gram-positive organism.
Two other commonly used synergistic combinations include (1) the antifungal combination of amphotericin B and flucytosine and (2) the antibacterial and antiprotozoal combination of a sulfonamide and trimethoprim or pyrimethamine. These classic examples illustrate two basic mechanisms whereby one drug can potentiate the activity of another. It is thought that, analogous to the action of penicillins, which enhance the uptake of aminoglycosides by Gram-positive bacteria, amphotericin B enhances flucytosine uptake by fungal cells by damaging ergosterol-rich fungal cell membranes (see Chapter 36, Pharmacology of Fungal Infections). Only after penetrating the fungal membrane can flucytosine be converted into its active form (5-fluorouracil, which is converted to 5-FdUMP, an irreversible inhibitor of thymidylate synthase) by a fungal-specific deaminase. Amphotericin B has a low therapeutic index when used as a single agent (primarily as a consequence of its nephrotoxicity), but its synergistic effect in combination with flucytosine allows a reduction in the dose of amphotericin B required to treat a systemic fungal infection such as cryptococcal meningitis (with a corresponding reduction in toxicity).
Sulfamethoxazole and trimethoprim are commonly used in combination in the treatment of Pneumocystis jiroveci pneumonia, an opportunistic infection frequently encountered in patients with AIDS, as well as many urinary tract infections caused by Gram-negative enteric organisms. An analogous combination, sulfadoxine and pyrimethamine, is used in the treatment of malaria, toxoplasmosis, and other protozoal infections. These combinations illustrate a second mechanism whereby drugs can exert a synergistic effect. The mechanism of synergy is based on the sulfa drug’s inhibition of production of dihydrofolate, which ordinarily competes with the second drug for binding to its target, dihydrofolate reductase (see Chapter 33). The product of this enzyme, tetrahydrofolate, is a required substrate for purine biosynthesis and for many one-carbon transfer reactions and is thus necessary for microbial DNA replication and cell division (see Fig. 33-7).
Co-administration of Penicillins with β-Lactamase Inhibitors
The combination of a β-lactam antibiotic and a β-lactamase inhibitor (e.g., clavulanic acid, sulbactam, tazobactam) illustrates a mechanism of drug interaction that is not technically synergistic (because the β-lactamase inhibitor has no antibacterial activity on its own) but that shares a functional similarity with the drug combinations discussed above. Clavulanic acid is an inhibitor of β-lactamase, an enzyme used by many β-lactam-resistant Gram-positive and Gram-negative bacteria to inactivate penicillins (see Chapter 35). By preventing the hydrolysis and inactivation of penicillins, clavulanic acid (and other β-lactamase inhibitors) greatly increases the potency of penicillins (and other β-lactams) against bacteria that express β-lactamase. This combination has been effective in the treatment of infections due to penicillin-resistant Streptococcus pneumoniae, which is a common cause of otitis media in infants. Such organisms have typically acquired resistance to penicillins through a plasmid-encoded β-lactamase.
Polymicrobial and Life-Threatening Infections
Combinations of antimicrobial drugs are used not only to prevent the emergence of resistance and to act synergistically against a specific, known pathogen but also to treat polymicrobial infections and infections in which treatment must be initiated before the microbe causing the infection is identified. Consider, for example, the case of a ruptured appendix or colonic diverticulum that has leaked bacteria into the peritoneal cavity and formed an intra-abdominal abscess. Such an abscess is likely to contain a wide spectrum of microorganisms—much too broad to be targeted effectively by a single antibiotic. After draining the abscess, treatment with a combination of antibacterial agents such as a fluoroquinolone or β-lactam—to kill aerobic Gram-negative Enterobacteriaceae (e.g., Escherichia coli)—and clindamycin or metronidazole—to kill anaerobes (e.g., Bacteroides fragilis; see Chapter 37, Pharmacology of Parasitic Infections)—often results in clearance of the infection. (Note that it may sometimes be necessary to treat with antagonistic drug combinations in order to cover the spectrum of microorganisms that are likely to be present.) In cases where presumptive treatment is indicated before the causative microorganism is identified, body fluids such as blood, sputum, urine, and cerebrospinal fluid (CSF) should be submitted for culture before initiating therapy. A combination of drugs with activity against the microbes that are most likely to be involved in the infection (or that could result in the most serious outcome) is then administered until a positive bacteriologic identification is made and drug susceptibility results are obtained. At that point, it may be possible to discontinue unnecessary drugs and implement specific monotherapy.
As mentioned above, antagonistic drug combinations can sometimes be used in combination chemotherapy regimens, although this situation is to be avoided if possible. Antagonism is most commonly observed when static drugs are used in combination with cidal drugs. For example, tetracyclines are bacteriostatic antimicrobials that antagonize the bactericidal activity of penicillins (see Chapter 34). Recall that the bactericidal activity of penicillins depends on cell growth. By inhibiting the transpeptidation reaction involved in bacterial cell wall cross-linking, the penicillins create an imbalance between cell wall synthesis and autolysin-mediated cell wall degradation. If the bacterial cell continues to grow, this leads to spheroplast formation and eventually to osmotic lysis. A protein synthesis inhibitor such as tetracycline, which arrests cell growth, would therefore antagonize the effect of a β-lactam. Similarly, imidazoles and triazoles are fungistatic agents that antagonize the fungicidal activity of amphotericin B (see Chapter 36). The mechanism of antagonism can be appreciated by noting that amphotericin B acts by binding ergosterol and forming pores in the fungal membrane, whereas imidazoles and triazoles inhibit a microsomal cytochrome P450-dependent enzyme, 14α-sterol demethylase, which is involved in ergosterol biosynthesis. Thus, the imidazoles and triazoles oppose the action of amphotericin B by decreasing the concentration of the target for the latter drug. Despite these considerations, static and cidal antimicrobial drugs are sometimes used clinically in combination when no good alternatives exist. In such cases, it may be necessary to increase the dose of one or both drugs to compensate for the antagonistic drug–drug interaction. The resulting increase in the therapeutic drug concentration(s) can increase the likelihood of adverse effects.

For certain viruses, no drug provides long-term suppressive benefit when used as a single agent. This phenomenon, which is due largely to the development of drug resistance, has been most studied with HIV.
The viral life cycle is central to understanding the reason that monotherapy for HIV fails to suppress long-term viral replication (see Chapter 38, Pharmacology of Viral Infections; Fig. 38-2). After virus attachment, entry, and uncoating, the viral enzyme reverse transcriptase (RT) synthesizes double-stranded DNA from the single-stranded viral RNA genome. The DNA is then integrated into the host cell genome and transcribed over and over using the host cell’s transcription machinery. These complete genomic transcripts are eventually packaged into virions that infect new cells. However, HIV RT is relatively unfaithful, so replication error rates are quite high. In addition, transcription of the integrated DNA into RNA is also error prone. As a result, on average, every new HIV particle contains one mutation relative to its parental virus. The resulting error rate is not so high as to be intolerable to the virus, but it is sufficiently high that, after repeated cycles of infection, reverse transcription, and transcription, a substantial number of viruses encode altered targets of anti-HIV therapy and thereby acquire resistance, even prior to treatment.
In the setting of high mutation rates, combination antiviral therapy is beneficial. Combinations of RT inhibitors (as first shown for zidovudine and lamivudine) are more effective than one RT inhibitor alone, in part because resistance to one nucleoside analogue does not necessarily confer resistance to another. The current standard of care for treatment of HIV infection is “triple therapy.” Triple therapy can use many combinations—for example, two nucleoside analogue RT inhibitors and a nonnucleoside reverse transcriptase inhibitor (NNRTI), two nucleoside analogues and a protease inhibitor, or two nucleoside analogues and an integrase inhibitor. Clinical trials have shown that such combinations are able to reduce viral RNA plasma levels below the limit of detection (typically, 50 copies/mL). At such low levels of viral replication, the probability of resistance emerging to any one of the drugs is greatly reduced. Thus, for example, it has been shown that combinations remain effective for much longer periods of time than does any single agent.
Although combination formulations of common anti-HIV therapies have reduced “pill burden,” simplified treatment, and increased adherence, the adverse effects of some combinations can result in reduced adherence. Despite these concerns, data from randomized clinical trials now provide conclusive evidence for the use of combination antiretroviral therapy regardless of the stage of disease. Therefore, treatment guidelines in the United States as well as those developed by WHO now recommend universal treatment of all patients with HIV infection, regardless of CD4 T-cell counts.
Combination chemotherapy has also become common for treatment of other viruses that require prolonged therapy and have high mutation rates, such as hepatitis B virus (HBV) and hepatitis C virus (HCV). For HIV, HBV, and HCV, an important strategy in designing combination regimens is to include drugs that are less likely to select for resistant mutants because the mutants that are selected are also less fit.
 ANTINEOPLASTIC COMBINATION CHEMOTHERAPY
ANTINEOPLASTIC COMBINATION CHEMOTHERAPY
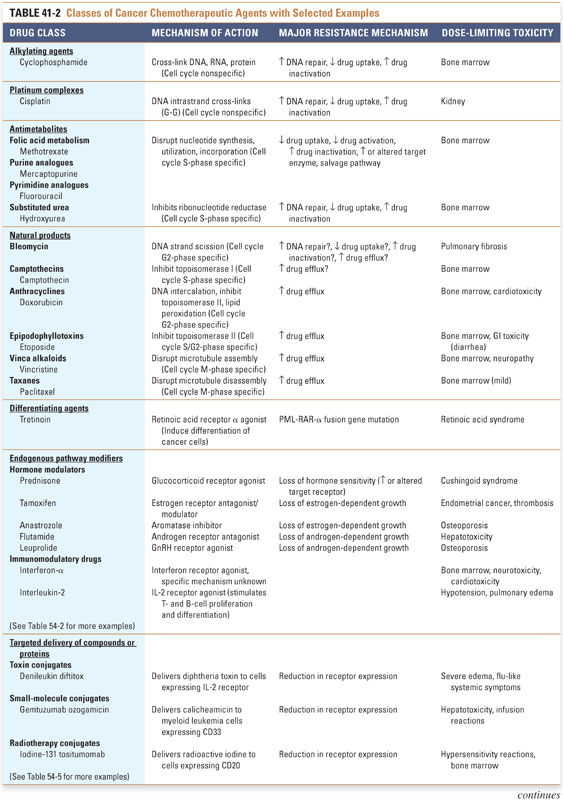
Several intrinsic difficulties are faced in the administration of antineoplastic chemotherapy. Cancer cells can be thought of as “altered self” cells that maintain similarities to normal, noncancerous cells, often making it difficult to target the cancer cells selectively. Also, many cancer chemotherapeutic agents have serious adverse effects that limit their dose and frequency of administration. Despite these hurdles, combination chemotherapy has led to remarkable advances in the treatment of cancer, including the examples of Hodgkin’s disease and testicular cancer discussed at the end of this section. Table 41-2 provides an overview of the major antineoplastic drug classes that are currently available, including their mechanisms of action, cell cycle specificities, major resistance mechanisms, and dose-limiting toxicities. Note that all these drug classes have been discussed in previous chapters; the following discussion integrates relevant information about the individual drugs in a clinical context.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


