INTRODUCTION
While infectious diseases and cancers have different underlying etiologies, from a pharmacologic perspective, the broad principles of treatment are similar. One common thread in these pharmacologic strategies is the targeting of selective differences between the microbe or cancer cell and the normal host cell. A second common thread is that the goal of treatment is complete inhibition of growth of the microbe or cancer cell. Because both microbes and cancer cells can evolve resistance to drug therapies, the development of new treatments is also a continually evolving process.
Infectious diseases and cancers are among the deadliest afflictions plaguing human societies. The World Health Organization (WHO) has estimated that, in the year 2012, communicable disease accounted for 23% of 56 million deaths worldwide, and malignant neoplasms accounted for a further 14.7%. Among the infectious diseases, the most common causes of mortality worldwide included lower respiratory infections (5.5%), diarrheal diseases (2.7%), HIV/AIDS (2.8%), tuberculosis (1.7%), and malaria (1.2%). In the developed world, cancer (along with heart disease and stroke) is a more significant cause of death than infectious disease. The deadliest cancers in the United States presently include lung cancer (159,260 estimated deaths in 2014), colon cancer (50,310), breast cancer (40,430), pancreatic cancer (39,590), and prostate cancer (29,480). Patterns of both infectious and neoplastic diseases will likely change as increasingly effective treatments and preventive measures are developed and distributed.
This chapter focuses on the principles of antimicrobial and antineoplastic pharmacology, but there are also many important and effective nonpharmacologic strategies to combat microbes and cancer. These strategies include public health measures, vaccinations, and screening. Most public health and vaccination programs aim to prevent infections rather than treat existing infections. Smallpox, for example, was eradicated worldwide in 1977 through aggressive vaccination programs, although concerns have been raised about the potential use of this virus as a bioterror agent. A similar campaign to eradicate polio is ongoing. Reductions in smoking and other environmental carcinogens have had a major impact on cancer mortality. Cancer screening, through regular mammograms, colonoscopy, and other tests, is widely used to detect cancer in its early and more treatable stages. Early detection through the widespread use of Papanicolaou cytologic tests (Pap smears) has caused the mortality of cervical cancer to decrease by more than two-thirds in the United States; cervical cancer has moved from the primary to the 15th leading cause of cancer deaths in women. It is hoped that widespread vaccination against specific types of human papilloma virus, the most common causative agent of cervical cancer, will further reduce the mortality of this cancer.
Effective strategies against disease, including drug therapy, also depend on socioeconomic factors. In affluent countries, the widespread use of antimicrobial drugs and improvements in sanitation and nutrition have markedly reduced mortality from infectious diseases. This progress has only begun to reach the developing world, where otherwise treatable infectious diseases remain major causes of mortality. Since 2012, a growing body of research has supported the use of combination therapy as preexposure prophylaxis (PrEP) in the prevention of HIV infection. In May 2014, the Centers for Disease Control and Prevention (CDC) released new guidelines on the use of tenofovir-emtricitabine to prevent HIV infection in populations at substantial risk, including men who have sex with men, heterosexual men and women with infected partners, and IV drug users. Despite the importance of public health measures, vaccinations, and screening procedures, drug therapy remains vital to the treatment of microbial disease and cancer. Understanding the general principles and mechanisms of antimicrobial and antineoplastic pharmacology is essential to the safe and effective prescribing of existing drugs and, especially in light of continually evolving mechanisms of resistance, to the discovery of new drugs.
 In 1935, in Wuppertal, Germany, 11-year-old Hildegard Domagk is desperately ill with a streptococcal infection that she contracted after an accidental pinprick with an embroidery needle. In desperation, Hildegard’s father, Dr. Gerhard Domagk, injects her with prontosil, a red dye with which he has been experimenting in his laboratory. In what seems like a miracle, she makes a complete recovery.
In 1935, in Wuppertal, Germany, 11-year-old Hildegard Domagk is desperately ill with a streptococcal infection that she contracted after an accidental pinprick with an embroidery needle. In desperation, Hildegard’s father, Dr. Gerhard Domagk, injects her with prontosil, a red dye with which he has been experimenting in his laboratory. In what seems like a miracle, she makes a complete recovery.
This story actually began 3 years earlier, when Dr. Domagk observed that prontosil protected mice and rabbits from lethal doses of staphylococci and streptococci. He discovered this by screening thousands of dyes (which are, in fact, chemicals that bind to proteins) for antibacterial activity. When his daughter became ill, however, Domagk was not sure whether prontosil’s antibacterial efficacy in mice would carry over to infections in humans. He kept his personal test of the drug a secret until data from other physicians indicated that the drug had been successful in curing other patients of their infections. In 1939, Gerhard Domagk was awarded the Nobel Prize in Physiology or Medicine for his discovery of the therapeutic benefit of prontosil.
Questions
1. What is the mechanism responsible for the antibacterial action of prontosil?
2. Why does prontosil kill bacteria but not human cells?
3. What has caused the utility of drugs such as prontosil to decline over the past 75 years?
4. Why are drugs of the same class as prontosil now used in combination with other antibacterial agents?
 MECHANISMS OF SELECTIVE TARGETING
MECHANISMS OF SELECTIVE TARGETING
The goal of antimicrobial and antineoplastic drug therapy is selective toxicity: inhibiting pathways or targets that are critical for pathogen or cancer cell survival and replication at concentrations of drug lower than those required to affect critical host pathways. Selectivity can be realized by attacking (1) targets unique to the pathogen or cancer cell that are not present in the host, (2) targets in the pathogen or cancer cell that are similar but not identical to those in the host, and (3) targets in the pathogen or cancer cell that are shared with the host—or are even host rather than pathogen gene products—but that vary in importance between pathogen and host and thus impart selectivity (Table 33-1). These selectively targeted differences range from structures that are unique to individual pathogens, such as the peptidoglycan cell wall of bacteria, to differences as slight as a single amino acid change in a signaling protein that is otherwise common to cancer cells and normal cells, such as the acquisition of activating mutations in the epidermal growth factor receptor (EGFR) in certain lung cancers. In principle, drugs exhibit the least toxicity to the host when they target unique differences and the most toxicity when they target common pathways. For a given drug, the ratio of the toxic dose to the therapeutic dose is termed the therapeutic index (see Chapter 2, Pharmacodynamics) and is an indication of how selective the drug is in producing the desired effects. A highly selective drug such as penicillin, which targets the peptidoglycan cell wall unique to bacteria, can be prescribed safely because of the large difference between its therapeutic and toxic concentrations. Drugs such as erlotinib, which inhibits mutated EGFRs but cannot fully discriminate between wild-type and mutant receptors, have a much lower therapeutic index and a higher incidence of adverse effects.

As we learn more about the biology of pathogens and cancer cells, more selective drugs are being developed. For example, imatinib is a highly selective anticancer agent that targets the kinase domain of a fusion protein that is produced by a novel gene rearrangement. This fusion protein kinase is constitutively active and highly expressed in chronic myelogenous leukemia cells and is important for the growth and survival of these cells. In contrast, the drug has relatively little effect on normal cells, in which the unrearranged protein kinase is less active, less highly expressed, and less critical for cell growth and survival (see Chapter 1, Drug–Receptor Interactions). That being said, it is important to recognize that many potential targets that seem attractive remain unexploited because of issues such as unexpected adverse effects, unfavorable pharmacokinetic properties, or prohibitive costs associated with experimental drugs that have been developed to date against these targets.
In the section below, we discuss examples of targets of antimicrobial and antineoplastic drugs that illustrate the principles of selective toxicity.
Unique drug targets include metabolic pathways, enzymes, and other gene products that are present in the pathogen or cancer cell but absent in the host. One attractive target for antibacterial drugs is the biochemical pathway that leads to synthesis of the bacterial peptidoglycan cell wall (see Chapter 35, Pharmacology of Bacterial and Mycobacterial Infections: Cell Wall Synthesis). This structure is both biochemically unique and essential for the survival of growing bacteria. Penicillin and other β-lactam antibiotics inhibit the transpeptidase enzymes that catalyze the final cross-linking step in peptidoglycan synthesis. Without peptidoglycans, bacterial cell wall synthesis is fatally compromised. Because of their unique specificity for bacterial transpeptidase proteins, the penicillins have minimal host toxicity—in fact, allergic hypersensitivity is the major adverse reaction.
Fungi also present a unique target that is exploited by the currently available antifungal drugs. Like the bacterial cell wall, the fungal cell wall is biochemically unique and essential for survival. The echinocandins inhibit the synthesis of β-(1,3)-D-glucan, an essential component of the fungal cell wall. Disruption of cell wall integrity can cause fungal cell lysis. The echinocandins are well tolerated compared to other antifungal drug classes, with adverse effects unrelated to inhibition of their target, but are limited by their lack of oral bioavailability. Thus, drugs that act on unique targets are often highly selective, with high therapeutic indices.
Selective Inhibition of Similar Targets
Many pathogenic organisms and cancer cells have metabolic pathways similar to those of normal human cells but because of evolutionary divergence or mutation possess enzyme or receptor isoforms that differ in sequence and structure from their normal human counterparts. Drugs can take advantage of these differences, even though they are sometimes subtle. However, the resulting therapeutic indices are usually smaller than with unique targets. Examples of this targeting strategy include an inhibitor of the B-Raf protein kinase and inhibitors of bacterial protein synthesis. B-Raf is part of a signaling pathway downstream of cell surface receptors. A mutant form of B-Raf, with a single amino acid substitution, is frequently expressed in the skin cancer melanoma. Despite this very subtle difference in the protein kinase, the drug vemurafenib more potently inhibits the mutant B-Raf than it does the normal enzyme. Because the mutant kinase is required for the growth and survival of many melanomas, vemurafenib treatment can cause dramatic responses in the disease with an acceptable toxicity profile.
In both humans and bacteria, protein synthesis is a multistep process that involves binding of tRNAs and mRNA to the ribosome, decoding of mRNA, synthesis of peptide bonds, translocation of the tRNAs relative to the mRNA, emergence of the polypeptide chain, and release of the polypeptide from the ribosome. The bacterial protein synthesis machinery differs from its human counterpart in the use of a different set of ribosomal RNAs and proteins and, correspondingly, distinct ribosomes. Several drug classes, including the macrolides and aminoglycosides, exploit these differences to selectively inhibit bacterial protein synthesis (see Chapter 34, Pharmacology of Bacterial Infections: DNA Replication, Transcription, and Translation). Macrolide antibiotics such as erythromycin bind to the 50S bacterial ribosomal subunit and block translation by preventing emergence of polypeptides from the ribosome. Aminoglycoside antibiotics such as streptomycin and gentamicin bind to the 30S bacterial ribosomal subunit and disrupt the decoding of mRNA. More generally, the bacterial protein synthesis inhibitors include a wide variety of individual drugs with diverse mechanisms, and the selectivity and dose-limiting toxicities of these drugs are often class- and/or drug-specific. For example, the macrolides rarely cause serious adverse effects, whereas some of the aminoglycosides have dose-limiting ototoxicity and nephrotoxicity. Some adverse effects appear to result from drug binding to human mitochondrial ribosomes in addition to bacterial ribosomes. Thus, selective inhibition of similar targets, as exemplified by mutant B-Raf inhibitors and protein synthesis inhibitors, can result in effects characterized by therapeutic indices that range from low to high, depending on the individual drug or drug class under consideration.
It is often the case that the host and pathogen or cancer share common targets. In these cases, selective toxicity can be achieved when the pathogen or cancer is more affected than the host by inhibition of the target. One striking example is the human chemokine receptor CCR5. This protein is essential for entry of certain strains of HIV into cells but is dispensable for human health as revealed by studies of individuals who, despite frequent exposure to HIV, have remained uninfected due to a deletion in the CCR5 gene. Accordingly, the antiviral drug maraviroc, which binds to CCR5 and prevents HIV entry into cells, can help suppress HIV replication with minimal toxicity to patients (see Chapter 38, Pharmacology of Viral Infections).
The paradigm of common targets is most frequently exemplified by antineoplastic drugs. Because tumor cells arise from transformed normal cells, they share nearly all of the cellular machinery needed for growth and replication. However, tumor cells may be more dependent on certain of these pathways than normal cells and therefore can be more sensitive to their inhibition. Also, some of these pathways can be more sensitized in cancer cells than in normal cells to agents that stress the cells, for example, by damaging DNA. These differences can be subtle, and anticancer drugs that attempt to exploit these differences often have narrow therapeutic indices. Therefore, selective inhibition of cancer cell growth remains a major challenge.
Recent discoveries have identified a number of proteins that are mutant or overexpressed in cancer cells, and selective inhibitors of these proteins are entering clinical use with increasing frequency (see Chapter 40, Pharmacology of Cancer: Signal Transduction, and discussion of vemurafenib above). Nonetheless, it is still the case that the selectivity of many currently used antineoplastic drugs is based not on specific mutant protein targets but rather on variations in cancer cell growth behavior and on the increased susceptibility of cancer cells to induction of apoptosis or senescence. Cancer, as a disease of persistent proliferation, requires continued cell division. Therefore, cytotoxic drugs targeting processes involved in DNA synthesis, mitosis, and cell cycle progression may kill rapidly cycling cancer cells preferentially over their normal relatives. (An important correlate to this statement is that many chemotherapeutic strategies are more successful against rapidly growing than slowly growing cancers.) Antimetabolites such as 5-fluorouracil (5-FU) inhibit DNA synthesis in dividing cells (see Chapter 39, Pharmacology of Cancer: Genome Synthesis, Stability, and Maintenance). 5-FU inhibits thymidylate synthase, the enzyme responsible for converting dUMP to dTMP, a pyrimidine building block of DNA. As a pyrimidine analogue, 5-FU is also incorporated into growing RNA and DNA strands, thereby interrupting the synthesis of these strands. By causing DNA damage, 5-FU induces the cell to activate its apoptotic pathway, resulting in programmed cell death. 5-FU is toxic to all human cells undergoing DNA synthesis and, thus, is particularly toxic both for rapidly cycling tumor cells (therapeutic effect) and for high-turnover host tissues such as the bone marrow and gastrointestinal mucosa (adverse effect).
These examples illustrate the importance of studying the cell biology, molecular biology, and biochemistry of microbes and cancer cells to identify specific targets for selective inhibition. Clinically, an awareness of drug mechanisms and the basis of drug selectivity can help to explain the narrow or broad therapeutic indices that have an impact on drug dosing and treatment strategies. Understanding the selectivity of drugs for their targets is also important in combating drug resistance. Thus, the fundamental pharmacologic principles of drug–receptor interactions, therapeutic and adverse effects, and drug resistance form the basis for selective targeting in antimicrobial and antineoplastic drug therapy.
 PATHOGENS, CANCER CELL BIOLOGY, AND DRUG CLASSES
PATHOGENS, CANCER CELL BIOLOGY, AND DRUG CLASSES
Pharmacologic interventions target specific differences between the host and the microbial pathogen or cancer cell. This section examines some of the distinctive characteristics that evolution has bestowed on organisms and the major drug classes that target these molecular differences among host cells, pathogens, and cancer cells.
Bacteria are organisms that often contain unique targets for pharmacologic intervention. Some of these drug targets have been discussed previously and are illustrated in Figure 33-1. Currently available drugs act to interrupt bacterial DNA replication and repair (this chapter and Chapter 34), transcription and translation (Chapter 34), and cell wall synthesis (Chapter 35).
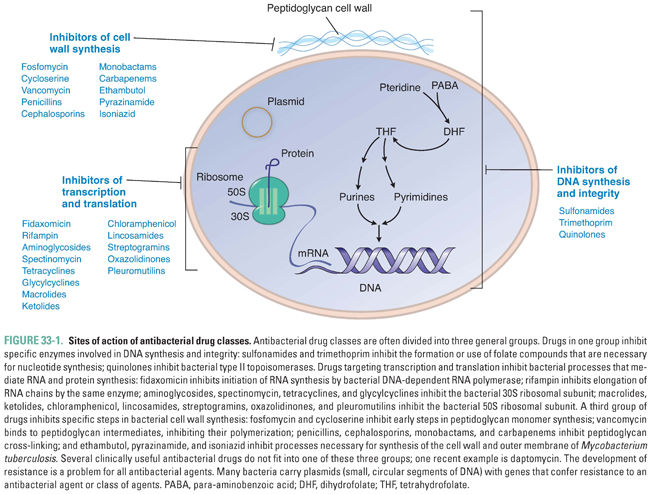
Depending on the role of the drug target in bacterial physiology, antibacterial drugs can produce bacteriostatic or bactericidal effects. Drugs that inhibit the growth of the pathogen without causing cell death are called bacteriostatic. These drugs target metabolic pathways that are necessary for bacterial growth but not for bacterial survival. Most protein synthesis inhibitors have a bacteriostatic effect (aminoglycosides are an important exception). The clinical effectiveness of these drugs relies on an intact host immune system to clear the nongrowing (but viable) bacteria. In contrast, bactericidal drugs kill bacteria. For example, cell wall synthesis inhibitors such as penicillins and cephalosporins cause bacterial lysis when the bacteria grow in hypertonic or hypotonic environments. Bacterial infections in immunocompetent hosts can often be treated with bacteriostatic drugs, whereas the treatment of bacterial infections in immunocompromised hosts often requires bactericidal drugs.
Bacteriostatic and bactericidal effects must be considered when antibiotics are used in combination (see Chapter 41, Principles of Combination Chemotherapy). The combination of a bacteriostatic drug with a bactericidal drug can result in antagonistic effects. For example, the bacteriostatic drug tetracycline inhibits protein synthesis and thereby retards cell growth and division. The action of this drug antagonizes the effects of a cell wall synthesis inhibitor, such as penicillin, which requires bacterial growth in order to be effective. In contrast, the combination of two bactericidal drugs can be synergistic; that is, the effect of the combination is greater than the sum of the effects of each drug alone (at the same doses of the two drugs). For example, a penicillin–aminoglycoside combination can have a synergistic effect because inhibition of bacterial cell wall synthesis by the penicillin allows increased entry of the aminoglycoside. The combination of two bacteriostatic drugs can also be synergistic (see “Synergy of DHFR Inhibitors and Sulfonamides” later in this chapter).
Eukaryotes, which include pathogenic fungi (yeasts and molds) and parasites (protozoa and helminths) as well as all multicellular organisms, are more complex than bacteria. Cells in these organisms contain a nucleus and membrane-bound organelles, as well as a plasma membrane. Eukaryotic cells reproduce by mitotic division rather than binary fission. Because of the similarities among human, fungal, and parasitic cells, infections caused by fungi and parasites can be more difficult to target than bacterial infections. However, the burden of disease from these organisms is vast. Parasitic infections caused by protozoa and helminths (worms) affect some 3 billion people worldwide, especially in less developed countries where the consequences of infection can be devastating. In both developed and less developed parts of the world, increasing numbers of patients are immunocompromised from AIDS, cancer chemotherapy, organ transplants, and old age. Such patients are especially susceptible to fungal and parasitic infections, which are becoming more prominent and will require greater attention in the future.
Currently available antifungal drugs can be divided into four main classes. Polyenes (e.g., amphotericin, nystatin) and azoles (e.g., miconazole, fluconazole) selectively target ergosterol in the fungal cell membrane, and echinocandins (e.g., caspofungin, micafungin) inhibit the synthesis of β-(1,3)-D-glucans in the fungal cell wall. Pyrimidines such as 5-fluorocytosine inhibit DNA synthesis. Another class of miscellaneous antifungals, mostly acids, is used only topically because of unacceptable systemic toxicity. As with antibacterials, antifungals can be fungistatic or fungicidal; this distinction is usually determined empirically. For example, the azoles interfere with fungal cytochrome P450-mediated ergosterol metabolism. Many azoles (e.g., itraconazole and fluconazole) are fungistatic. Newer azole agents (e.g., voriconazole and ravuconazole) may have fungicidal activity against some fungal species. As compared to fungistatic drugs, fungicidal drugs are more efficacious and faster acting and allow more favorable dosing regimens. Antifungal drugs are discussed in further detail in Chapter 36, Pharmacology of Fungal Infections.
Parasites exhibit diverse and complex life cycles and metabolic pathways, and the treatment of parasitic infections utilizes a wide array of drugs (see Chapter 37, Pharmacology of Parasitic Infections). One important protozoal infection is malaria, which is transmitted when the female Anopheles mosquito deposits Plasmodia sporozoites in the human bloodstream. The parasites leave the circulation and develop into tissue schizonts in the liver. The tissue schizonts rupture, releasing merozoites that enter the circulation to infect red blood cells (erythrocytes). The parasites then mature to trophozoites and, finally, to mature schizonts. Crops of mature schizonts are released into the bloodstream when the erythrocytes rupture, causing the typical cyclic fever associated with malaria. Antimalarial drugs target different stages of the protozoal life cycle. Aminoquinolines (such as the previous first-line drug, chloroquine) inhibit the polymerization of heme within the erythrocyte; it is thought that nonpolymerized heme is toxic to intraerythrocytic Plasmodia. Artemisinins are also thought to interfere with heme metabolism in the parasite. Dihydrofolate reductase inhibitors, protein synthesis inhibitors, and other classes of drugs are also used in malaria treatment. The choice of drugs often depends on the local pattern of resistance. As chloroquine resistance is now common and resistance to most antimalarial agents has increased, the WHO recommends against all single-agent treatment regimens in the first line of therapy for malaria. Instead, combination therapies are now recommended as first-line treatments. Resistance to artemisinin and its derivatives has been observed, and the WHO recommends artemisinin-based combination treatments both to increase the efficacy of therapy and to reduce the spread of drug-resistant malaria. Combinations of an artemisinin-based drug with amodiaquine, mefloquine, or sulfadoxine-pyrimethamine are recommended. Malaria is an excellent example of a complex parasite that, while theoretically susceptible to numerous classes of drugs, is becoming resistant to many currently available therapies.
Viruses are noncellular organisms that typically consist of a nucleic acid core of RNA or DNA enclosed in a proteinaceous capsid. Some viruses also possess a host cell-derived lipid envelope containing viral proteins. Viruses lack the capability to synthesize proteins themselves, relying instead on the host cell machinery. Most viruses also encode distinct or even unique proteins not normally produced by human cells, however. Many of these proteins are involved in the viral life cycle, mediating attachment and entry of the virus into the host cell, uncoating of the viral capsid, expression of viral genes, replication of the viral genome, assembly and maturation of the viral particle, and release of viral progeny from the host cell. These virus-specific processes are often targeted by antiviral drugs. A schematic diagram of the general viral life cycle is presented in Figure 33-2 to illustrate the stages of viral replication that can be targeted by antiviral drugs. Because these targets are present only during active viral replication, viruses capable of latency have not been cured by any currently available antiviral drugs.
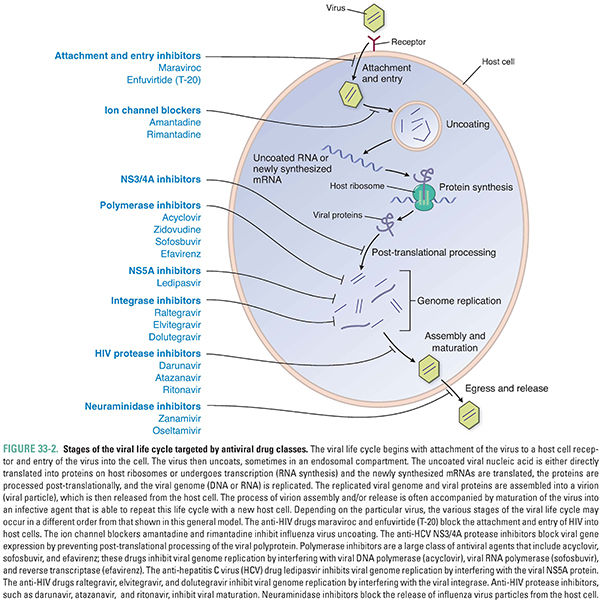
The HIV protease is an excellent example of a viral protein that has served as a fruitful target for drug development. This enzyme cleaves viral precursor proteins to generate the structural proteins and enzymes necessary for virus maturation. Without HIV protease, only immature and noninfective virions (individual virus particles) are produced. HIV protease inhibitors structurally mimic natural substrates of the protease but contain a noncleavable bond. These drugs act as competitive inhibitors at the active site of the enzyme (see Chapter 38). In combination with other classes of anti-HIV drugs, protease inhibitors helped to revolutionize the treatment of patients with HIV/AIDS, converting a nearly invariably fatal disease to a chronic illness.
Influenza viruses offer examples of other classes of proteins that have been successfully targeted. Zanamivir and oseltamivir target a viral neuraminidase that is vital for virion release from host cells. Amantadine and rimantadine act on the influenza virus membrane protein M2 (a proton channel) to inhibit viral uncoating. These anti-influenza drugs are effective inhibitors of their targets. However, at least partly because the immune system ordinarily clears influenza infections in less than a week, these agents have not had as dramatic a clinical impact as anti-HIV drugs have had. This example illustrates the point that even selective inhibitors with high therapeutic indices do not necessarily become highly effective drugs in the clinic.
Currently, the most important class of antiviral drugs is the polymerase inhibitors. Most viruses use a viral polymerase, either an RNA or DNA polymerase, to replicate their genetic material. Polymerase inhibitors are especially effective against human herpesviruses, HIV, hepatitis B virus, and hepatitis C virus. Two types of polymerase inhibitors are the nucleoside analogues and the nonnucleoside polymerase inhibitors. Nucleoside analogues (such as acyclovir, zidovudine, and sofosbuvir) become phosphorylated and thereby activated by viral or cellular kinases (phosphorylating enzymes), at which point they competitively inhibit the viral polymerase and, in some cases, are incorporated into the growing DNA strand. Selectivity is dependent on the relative affinities of the nucleoside analogue for the viral and cellular kinases and polymerases and on the abilities of the enzymes to phosphorylate the drug or incorporate it into DNA, respectively. Nonnucleoside polymerase inhibitors (such as efavirenz) inhibit viral polymerase (in this case, HIV reverse transcriptase), preventing DNA replication, but bind to a different site than the nucleoside analogues do. Mutations in viral polymerase genes are a major mechanism of resistance to polymerase inhibitors. Whether these resistance mutations affect the replicative capacity (fitness) of the virus can play a major role in determining the effectiveness of the therapy.
Chapter 38 provides a detailed discussion of the pharmacology of antiviral drugs.
Cancer is a disease of cell proliferation in which normal cells are transformed into cells with dysregulated growth. Neoplastic cells compete with normal cells for energy and nutrition, resulting in deterioration of normal organ function. Cancers also impinge on vital organs by mass effects. Carcinogenesis, chemotherapy, and the log cell kill model of tumor regression are discussed below to provide an overview of cancer pharmacology. Chapters 39 and 40 should be read with these principles in mind, and Chapter 41 provides integrated examples of the clinical applications of combination antineoplastic chemotherapy.
Carcinogenesis and Cell Proliferation
Carcinogenesis occurs in three main steps—transformation, proliferation, and metastasis. Transformation denotes a change in phenotype from a cell with normal growth controls to a cell with dysregulated growth. Nonlethal genetic changes (mutations) can be inherited in the germ line, can occur spontaneously, or can be caused by environmental agents such as chemicals, radiation, or viruses. If the change is not repaired, the mutated genes (e.g., genes involved in growth regulation and DNA repair) can express altered gene products that allow abnormal cell growth and proliferation. Among other effects, mutations can activate growth-promoting genes, inactivate growth-inhibiting genes, alter apoptosis-regulating genes, confer immortalization, and inactivate DNA repair genes. Additionally, the expression of these genes can be altered by certain heritable changes that do not alter DNA sequence, such as changes in DNA methylation or modifications to histones that package chromosomal DNA. Such epigenetic changes can also promote carcinogenesis. Expression of altered gene products and/or loss of normal gene regulation can cause dysregulated growth. Most cancers are initially clonal (i.e., genetically identical to a single precursor cell) but evolve to heterogeneity as new mutations and epigenetic changes increase the variation among daughter cells. When progeny cells with higher survival capacity are selected, increased cell proliferation ensues, and the tumor progresses to greater and greater heterogeneity. Thus, carcinogenesis, the progression from a normal cell to a malignant tumor, is a multistep process that usually requires an accumulation of multiple alterations. As more is learned about the molecular basis for carcinogenesis, these differences can be targeted for selective drug therapy.
The growth of transformed cells into a tumor requires proliferation, or an increase in the number of cells. Dividing human cells progress through a cell cycle (or mitotic cycle) consisting of distinct phases. The two key events in the cell cycle are the synthesis of DNA during S phase and the division of the parent cell into two daughter cells during mitosis or M phase. The phase between cell division and DNA synthesis is called gap 1 (G1), and the phase between DNA synthesis and mitosis is called gap 2 (G2). Proteins called cyclins and cyclin-dependent kinases (CDKs) govern progression through the phases of the cell cycle. Mutations in cyclin and/or CDK genes can result in neoplastic transformation, and loss of normal cell cycle control can result in genetic instability, augmenting the transformed phenotype.
After a cancer cell divides, a daughter cell has four potential fates: it can become quiescent by entering a resting phase called G0, enter the cell cycle and proliferate, enter a state of cell-cycle arrest called senescence, or die. The ratio of the number of cells that are proliferating to the total number of cells in the tumor is called the growth fraction. An average tumor growth fraction is about 20%, because only one in five cells participates in the cell cycle at any given time. Many antineoplastic drugs are cytotoxic agents that target dividing cells. Hence, tumor cells in a quiescent (G0) state, such as nutrient-starved cells in the center of a large tumor, are not easily killed by cytotoxic chemotherapy. Small or rapidly growing cancers (i.e., cancers with high growth fractions, such as leukemias) often respond more favorably to cytotoxic chemotherapy than do large bulky tumors. Unfortunately, cells in normal tissues characterized by high growth fractions, such as the bone marrow and gastrointestinal mucosa, are also killed by cytotoxic antineoplastic drugs, resulting in dose-limiting toxicities.
While cell-autonomous processes (i.e., processes of the cancer cell itself) are well-understood drivers of the proliferative phenotype in a tumor cell, non-cell-autonomous processes also play vital roles in tumor maintenance and growth. Transformed cancer cells secrete and induce a variety of chemical mediators to create a specialized local environment. These chemical mediators include growth factors such as epidermal growth factor (EGF), and inhibitors of growth factor signaling have been developed for clinical use as targeted cancer chemotherapeutic agents. Some tumors create a protective fibrous connective tissue stroma; for example, this property makes breast cancer nodules palpable. Most solid tumors also require the induction of blood vessel growth (angiogenesis) to deliver nutrients into the center of the tumor; for this reason, angiogenesis inhibitors represent a valuable class of antineoplastic drugs.
Cancer cells may acquire the capability to invade tissues and metastasize throughout the body. In order to metastasize, tumor cells acquire mutations or epigenetic changes that allow invasion into tissues and vessels, seeding of cavities, spread through lymph or blood vessels, and growth in a new environment. Aggressive, rapidly growing primary tumors are generally more likely to metastasize than indolent, slowly growing tumors. In the process of metastasis, tumor cells can also evolve different receptor expression patterns and drug susceptibilities and can even “dedifferentiate” or change cell type (e.g., epidermal to mesenchymal transition). Often, although the primary tumor may respond well to chemotherapy, the metastatic cells respond poorly. Thus, metastatic spread typically represents a poor prognostic sign.
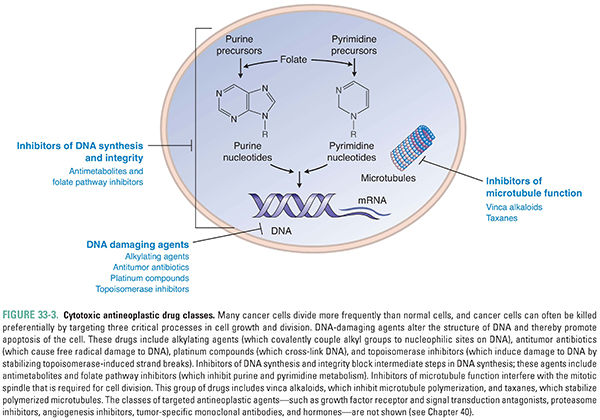
By the time a typical solid tumor is clinically evident, it contains at least 109 cells, has acquired significant genetic heterogeneity, and has developed a distinct surrounding stroma. The tumor may or may not have metastasized from its site of origin (primary site) to one or more secondary sites. These factors can render the cancer difficult to treat pharmacologically. Many cytotoxic (traditional) chemotherapeutic agents interfere with cell proliferation and rely on rapid cell cycling and/or promotion of apoptosis for their relative selectivity against cancer cells (Fig. 33-3). This is because tumors are most sensitive to cytotoxic chemotherapy when they are growing rapidly and progressing through the cell cycle. These metabolically active cells are susceptible to drugs that interfere with cell growth and division (the mitotoxicity hypothesis). Many cytotoxic antineoplastic drugs interfere with the cell cycle at a particular phase; such drugs are called cell-cycle specific. Other cytotoxic antineoplastic drugs act independently of the cell cycle and are called cell-cycle nonspecific (Fig. 33-4). Inhibitors of DNA synthesis, such as antimetabolites, are S-phase specific. Microtubule poisons, such as taxanes and vinca alkaloids, interfere with spindle formation during M phase. Alkylating agents that damage DNA and other cellular macromolecules act during all phases of the cell cycle. These various classes of drugs can be administered in combination, using cell-cycle specific drugs to target mitotically active cells and cell-cycle nonspecific agents to kill both cycling and noncycling tumor cells (see Chapter 41).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


