Figure 19.1
Normal and polycystic kidneys. A normal kidney is shown on the left and a kidney with a high number of variable-sized cysts that characterize polycystic kidney disease is shown on the right
Other common renal manifestations include hypertension, kidney stones, urinary tract infections, and hematuria. Extrarenal manifestations are highly prevalent, including cysts in the liver, pancreas, seminal vesicles, and arachnoid membranes, cardiac valve abnormalities, and aneurysms. These renal and extrarenal abnormalities are a significant cause of morbidity and mortality. The mean age of onset of hypertension is 20–30 years, usually occurring while renal function is still within the normal range; this is approximately one decade earlier than in the general population with essential hypertension [3]. The prevalence of intracranial aneurysm is about five times higher than in the general population [4]. Approximately 50 % of the ADPKD patients progress to ESRD by the seventh decade [5], and account for approximately 5 % of all patients requiring hemodialysis or kidney transplant [6].
Autosomal recessive PKD (ARPKD) (OMIM#263200) is characterized by non-obstructive dilation of the cortical collecting tubule in utero or during the neonatal period, resulting in massive renal enlargement. However, a subset is increasingly being identified in childhood, adolescence, or even adulthood. These cases with later onset diagnosis usually have milder kidney disease, but more significant complications of liver disease [7]. This is accompanied by a ductal plate malformation of the liver, resulting in congenital hepatic fibrosis. Approximately 2–5 % of ADPKD patients also present during the neonatal period with significant morbidity and mortality [8], and may only be differentiated from ARPKD at that time by histological or genetic analysis.
Many other pediatric disorders are associated with renal cysts or cystic dysplasia as a component of their phenotypes. However, they generally can be distinguished from ARPKD and ADPKD by a detailed physical examination, basic laboratory and imaging studies, and by their extrarenal clinical characteristics [9]. The prevalence of ARPKD has been estimated to be 1 in 20,000-40,000 live births, with a carrier frequency of 1 in 70 [7].
Autosomal Dominant Polycystic Kidney Disease
Molecular Basis of Disease
ADPKD is genetically heterogeneous, caused mainly by mutations in two genes: PKD1 and PKD2, located on chromosome 16p13.3 [10] and 4q21 [11], respectively.
PKD1 is a large gene, with 46 exons, occupying 50 kb of genomic sequence on chromosome 16, and encoding a large transcript with an open reading frame of 12,909 bp. The first 33 exons of PKD1 are located in a region that is duplicated six times on chromosome 16 [12]. These homologous genes, also termed pseudogenes, have 97–99 % homology to PKD1, which complicates genetic testing [13]. PKD2 consists of 15 exons, spanning 68 kb of genomic sequence, and encoding a transcript 2,904 bp [11]. De novo PKD1 and PKD2 mutations are reported to occur in approximately 10 % of affected individuals [14]. The reported prevalence of PKD2 mutations among ADPKD patients is variable. In community-based studies, PKD2 mutations accounted for approximately 29–36 % of cases [15], while in hospital-based studies, PKD2 mutations comprised only 15 % of cases; the remainder had PKD1 mutations. This discrepancy is likely due to a detection bias toward cases with more severe manifestation that characterize patients with PKD1 gene mutations [15].
The PKD1 gene encodes a large integral membrane protein, polycystin-1 (PC1), which has 11 transmembrane domains and an extracellular segment consisting of a variety of domains that occupy approximately 75 % of the entire protein. Overall, PC1 has the structure that is suggestive of a receptor or adhesion molecule and is thought to be a mechanical sensor of fluid flow [16]. The PKD2 gene product, polycystin-2 (PC2), is a member of the transient receptor potential family (i.e., TRPP2) that functions as a nonselective cation channel that transports calcium [17]. PC1 and PC2 localize to the immotile cilia on renal tubule epithelium [18], where PC1 and PC2 interact with each other [19]. A current hypothesis is that the polycystin complex functions as a mechanoreceptor that senses fluid flow in the tubular lumen, triggering Ca2+ influx through TRPP2, consequently affecting the intracellular calcium and cyclic AMP (cAMP) levels [20]. Mutations in either PKD1 or PKD2 result in malfunction of the polycystin complex, leading to abnormal cross talk between the adenylate cyclase and receptor tyrosine kinase pathways. These abnormalities, together with reduced intracellular calcium concentration, promote renal tubular epithelial cell proliferation and fluid secretion, which are key phenotypic features of ADPKD [21].
The renal cysts of ADPKD have two distinctive characteristics: significant size variation and focal development. These features can be explained by a two hit model in which two separate inactivation events in either one of the PKD genes is required for cyst formation [22]. The first hit is a germline mutation, usually inherited from the affected parent, which is necessary but not sufficient for cyst formation. The second hit, a somatic mutation only occurring in an individual renal tubular cell, inactivates either the normal PKD1 or PKD2 allele, thus initiating abnormal, monoclonal proliferation of tubular epithelial cells and cyst formation [23, 24]. The somatic mutation can occur at any given point during the patient’s lifetime, resulting in formation of cysts of different size. The size of the cyst typically correlates with the time the second mutation occurred. Although ADPKD is a dominant disease, the requirement of both a germline and a somatic mutation for cyst formation supports a recessive disease model at the cellular level. In a small number of families, germline mutations can be found in trans in PKD1 and PKD2. This bilineal inheritance demonstrates that co-inheritance of a mutation in both genes is not necessarily lethal during embryogenesis, but is associated with a more severe disease phenotype [25]. Interestingly, somatic PKD2 mutations can be detected in cysts of patients with PKD1 germline mutations and vice versa, further supporting the two-hit model and the dosage effect hypothesis for cyst formation [26, 27].
Various cellular mechanisms have been implicated in the pathogenesis of ADPKD as a consequence of impaired interactions of PC1 and PC2. These can be summarized as negative growth regulation, G protein activation, and Wnt pathway modulation [28]. Increased proliferation of renal tubular epithelial cells has been attributed to loss of inhibition by the mammalian target of rapamycin complex (mTOR) and cyclin-dependent kinases, among others. These pathways have been the targets of treatment strategies [29].
At the population level, ADPKD presents great phenotypic variability, including interfamilial and intrafamilial variability. This phenomenon is best explained by three genetic levels of attributions: genic (gene), allelic, and gene modifier effects. Interfamilial heterogeneity is mainly explained by genic effects; PKD1 gene mutations are associated with more severe disease and earlier age of ESRD onset [30]. The mean age of onset of ESRD is approximately 20 years earlier in patients with PKD1 mutations compared to patients with PKD2 mutations (54.3 years vs 74.0 years) [30]. The more severe phenotype, accounting for the younger age of diagnosis of ADPKD and earlier age of onset of ESRD in the PKD1 population, is likely due to the development of renal cysts at an earlier age, rather than a faster rate of cyst growth [31]. PKD1 patients also have a higher incidence of hypertension and hematuria.
Allelic effects in ADPKD are relatively small compared with genic effects. Patients with mutations in the 5′ region of PKD1 have been reported to reach ESRD slightly earlier than patients with mutations in the 3′ region (53 years vs 56 years) [32]. They also have a higher prevalence of intracranial aneurysms as well as aneurysm rupture [33]. However, in a more recent study, the phenotype was found to be affected by the type of PKD1 mutation, but not the position of the mutation in the gene [34]. No clear correlation has been reported between severity of disease phenotype and the position of the mutation in PKD2. Recently, hypomorphic alleles have been reported in both PKD1 and PKD2, which usually are missense mutations and reduce the levels of gene activity, but do not totally inactivate gene function [35]. Patients with one hypomorphic allele have mild cystic disease, while patients who are homozygous or compound heterozygous for two hypomorphic alleles present with moderate to severe disease, with cyst formation similar to that observed in patients with pathogenic PKD mutations. A hypomorphic allele coexisting with a pathogenic mutation can cause early onset disease [35].
The significant intrafamilial phenotypic variability, both in the rate of progression of chronic kidney disease and in the array of extrarenal manifestations, has been attributed, at least in part, to gene modifier effects [36]. Analysis of the phenotypic variability in renal function between monozygotic twins and siblings supports the involvement of genetic modifiers [37, 38]. Heritable modifying factors were estimated to account for 18–50 % of the variability in disease severity [39]. Recently, a candidate gene approach has identified Dickkopf 3 (DKK3) as marginally associated with ADPKD disease severity; however, replication studies are needed to verify the association reported [40, 41]. The development of high-resolution single nucleotide polymorphism (SNP) arrays enables properly powered genome-wide association studies (GWAS) for mapping modifier genes in an unbiased way; however, the success of such studies is strongly dependent on the availability of clinically well-characterized, large ADPKD cohorts.
Clinical Utility of Testing
Genetic testing for ADPKD has two main clinical applications: (1) clarifying whether ADPKD is present in a young family member without kidney cysts who is considering kidney donation, and (2) identifying the PKD gene mutation in an affected individual for reproductive decision-making (e.g., preimplantation genetic diagnosis). The gold standard method for diagnosis of ADPKD is an age-specific renal phenotype, based on the number of cysts identified by renal ultrasonogram in an individual with a 50 % risk of inheritance, determined by a positive history of ADPKD in a first-degree relative [42]. However, these criteria were suboptimal for patients with PKD2 mutations. A recent revision of the Ravine criteria has improved diagnostic performance for renal ultrasonography in patients with either PKD1 or PKD2 mutations [43]. Accordingly, in families of unknown genotype with an affected first-degree relative, the presence of three or more (unilateral or bilateral) renal cysts is sufficient for establishing the diagnosis of ADPKD in individuals aged 15–39 years; two or more cysts in each kidney is sufficient for individuals aged 40–59 years; and four or more cysts in each kidney is required for individuals 60 years of age or older. Conversely, fewer than two renal cysts in at-risk individuals aged 40 years or older is sufficient to exclude the disease [43]. Diagnostic imaging is sufficient for most patients; however, it is often ambiguous in young patients whose renal sonogram may not be conclusive or when the family history is unknown. A recent, single-center study found that magnetic resonance imaging has high sensitivity and specificity in patients at risk for ADPKD. Specifically, among subjects aged 16–40 years old, the presence of a total of more than 10 renal cysts was found to be sufficient for diagnosis [44].
Recently, Huang et al. proposed an algorithm that incorporates the use of DNA testing and imaging for donors with a family history of ADPKD [45]. They concluded that genetic testing, especially DNA sequencing, is cost effective because it shortens the waiting time for a living related transplant if at least one living donor per year results from every ten linkage or every five DNA sequencing tests performed, given that many individuals are on a transplant list for many years. Although genetic testing of PKD1 and PKD2 can be useful for clarifying the disease status in these individuals, the low sensitivity by the reference commercial laboratory (40–60 %) and high cost (approximately $6,000 per test) limits the use of widespread genotyping at this time [46].
Available Assays
Two methods are available for genetic testing of ADPKD: gene-based mutation screening and DNA linkage analysis. Molecular testing by gene-based mutation screening currently is the predominant method of ADPKD genetic testing. However, the marked allelic heterogeneity of the disease-associated mutations, the vast majority of which are private, and the duplicated structure of PKD1 together with its large size, makes mutation screening a significant technical challenge. Although mutation analysis of the PKD2 gene and the PKD1 single copy region (exons 34–46) is straightforward, analysis of the duplicated 5′ region of PKD1 is more complex because of its 98 % identity to the six PKD1 pseudogenes on chromosome 16.
Several strategies are available for ADPKD genetic testing (Fig. 19.2). Prior to 2001, genetic analysis of ADPKD could be only performed for PKD2 and the 3′ single copy region of PKD1. The first assay for analyzing the entire PKD1, developed in 2001, uses long-range PCR (LR-PCR) to specifically amplify the duplicated exon 1 to exon 33 regions, followed by nested PCR of the individual exons [47]. This method was later commercialized by Athena Diagnostics, Inc., which until recently had the exclusive license for analyzing PKD1 and PKD2 and was the sole provider of clinical ADPKD genotyping in the USA. Garcia-Gonzalez et al. evaluated the clinical utility of this commercially available test by analyzing a cohort of 82 ADPKD patients [48]. Definite pathogenic mutations were detected in 42 % of the patients and the maximal mutation detection rate was 78 %, after including missense, in-frame insertion/deletion, and atypical splice mutations that were likely to be pathogenic [48].
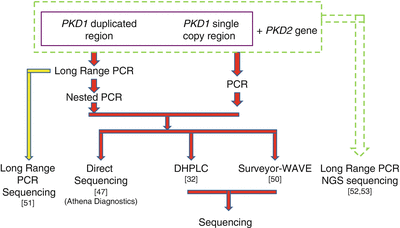
Figure 19.2
Strategies for ADPKD genotyping. NGS next-generation sequencing
Although complete gene sequencing remains the gold standard for ADPKD genetic testing, it is expensive and time-consuming. Thus, several screening-based methods were developed to lower testing costs and minimize turnaround time. Denaturing gradient gel electrophoresis, a traditional gel-based heteroduplex analysis method, as well as single strand conformation polymorphism (SSCP) have been successfully used to screen both the single copy and the duplicated regions of the PKD1 gene [49]. However, these methods suffer from a low mutation detection rate (30–40 %). Rossetti et al. [32] developed a denaturing high-performance liquid chromatography (DHPLC) screening method that was more sensitive than previous assays, with a detection rate of 64 % for definite pathogenic mutations. Further improvement in the utility of DHPLC for PKD genetic testing is the use of CelI endonuclease (SURVEYOR® nuclease, Transgenomic Inc.) and the Transgenomic WAVE® Nucleic Acid High Sensitivity Fragment Analysis System [50]. This method is 100 % sensitive when compared to the complete DNA sequencing method of the entire genes used by a commercial reference laboratory, with a detection rate of 64 % for definite pathogenic mutations, comparable to the rate reported in the literature [32].
An improved method for genetic analysis of ADPKD was developed by Tan et al. [51] using direct sequencing of the PKD1 LR-PCR products. In this strategy, the entire PKD1 coding region is amplified in nine LR-PCR reactions, generating products ranging in length from 2 to 6 kb, followed by column purification and direct sequencing with several pairs of walking primers. When compared with the direct sequencing result of the reference laboratory, this method was highly sensitive (100 %) and specific (98.5 %), circumventing the need for nested PCR of the PKD1 duplicated region, and reducing the risk of PCR amplification carryover contamination that can lead to false-positive results. In addition, this method has decreased the number of required PCR reactions by 80 %, substantially lowering test cost by approximately 20 % and improving turn-around time compared with both the direct sequencing and the SURVEYOR-WAVE screening methods. Another advantage of the LR-PCR sequencing method is that it covers intronic regions extending 200–300 bp beyond the exon-intron junctions, enabling better detection of deep intronic mutations.
Next-generation sequencing (NGS) technology is revolutionizing the field of human genetics and has been recently applied to PKD genetic testing. Rossetti et al. [52] reported a mutation screening strategy for analyzing PKD1 and PKD2 genes using NGS by pooling LR-PCR amplicons and multiplexing barcoded libraries. To increase the throughput, amplicons from four patients and libraries of up to 12 patients were pooled together and analyzed in a single reaction using indexed DNA barcodes. Using this strategy, these authors detected definite and likely pathogenic variants in 115 (63 %) of 183 patients with typical ADPKD. When compared with Sanger sequencing, this approach had a sensitivity of 78 % and a specificity of 100 % for mutation detection. This method also enabled the identification of atypical mutations, including a gene conversion event, and the characterization of deep intronic variations.
A second NGS approach amplifies PKD1 and PKD2 genes in a total of ten LR-PCR reactions, using locus-specific primers [51]. Indexed libraries from up to 25 patients are pooled together and analyzed in a single flow-cell using the MiSeq system (Illumina Inc., San Diego, CA). Sequencing results are sorted according to the barcodes with the FASTX toolkit and aligned against the reference sequence using the BWA program (http://bio-bwa.sourceforge.net/). Sequence variants are called using the GATK software package (The Genome Analysis Toolkit, The Broad Institute), carefully following The Best Practice Guidelines recommended by GATK (Fig. 19.3). With addition of an improved bioinformatics analysis pipeline, this approach has a sensitivity of 100 % in mutation detection when compared with Sanger sequencing, while retaining high specificity [51]. Several typical genetic variations detected by this approach are shown in Fig. 19.4. NGS approaches have the potential to dramatically improve genetic testing for ADPKD by allowing simultaneous detection of point mutations and copy number variations in a single test. Accordingly, these methods can increase the mutation detection rate, particularly of deep intronic mutations, and significantly reduce testing costs and turnaround time.
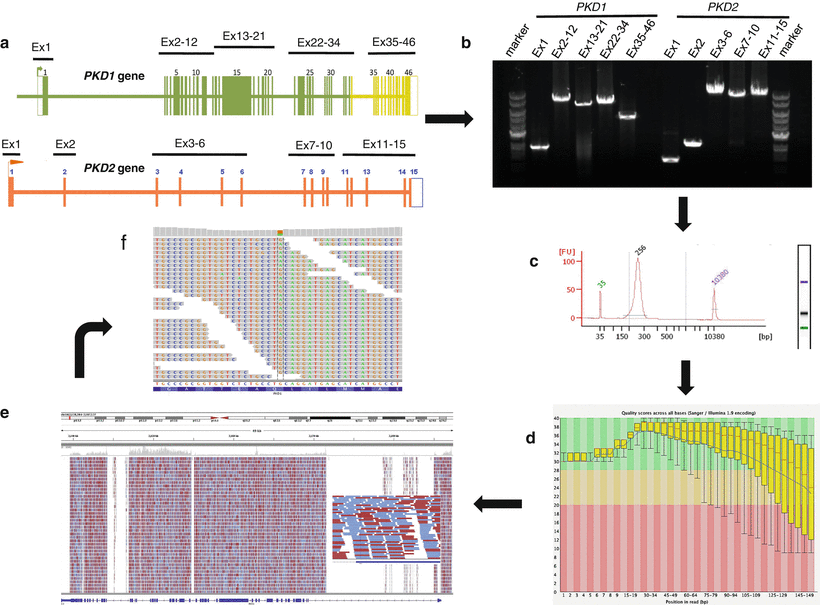
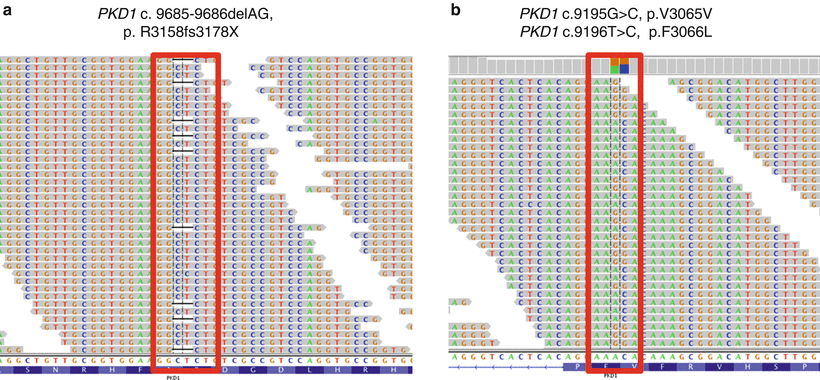
Figure 19.3
Schematic visualization of the NGS testing workflow for ADPKD. The workflow indicated by the arrows is as follows: (a) PKD1 and PKD2 sequences are individually amplified as ten locus-specific long-range PCR products (1.4–10.9 kb in size), covering all coding regions and most intronic regions for a total of ~80 kb. (b) Amplification quality verified using agarose gel electrophoresis with ethidium-bromide staining. (c) Amplicons from each individual sample are batched in equimolar ratios, fragmented, and subjected to library preparation using sample-specific barcodes. The indexed libraries are pooled and analyzed for quality using an Agilent Bioanalyzer. (d) Samples are sequenced on an Illumina MiSeq instrument, and the reads exported as FASTQ files, deconvoluted by bar code, and subjected to quality control analysis before proceeding with the mutation analysis bioinformatics pipeline. The quality score (Phred-like score) is shown at each specific bp location in PKD1. The red line in each box-and-whiskers plot shows the median value of the quality score at each bp position. The yellow box represents the inter-quartile range (25–75 %) of the quality score at each base. The upper and lower whiskers represent the 10 %th and 90 %th percentiles points, respectively. The blue line represents the mean quality score. Very good quality calls (Phred-like score within the green region at >28; chance of error 1:10,000), reasonable quality (Phred-like score within the orange region; chance of error 1:1,000), and calls of poor quality (Phred-like score within the red region; chance of error 1:100). (e) Reads were then mapped back to the genome with the BWA program. In this example, PKD1 sequencing coverage and read depth for a single patient are shown. The x-axis represents PKD1 genomic interval and the y-axis represents the number of reads. Red lines, reads from the plus DNA strands; blue lines, reads from the minus strands. The overall mean coverage of PKD1 sequences was 668-fold. (f) Variant calls made by the GATK software
Figure 19.4
Typical alignment of PKD1 NGS data and gene variation calls. (a) A two-base deletion (c.9685-9686delAG) resulting in a downstream truncation in the protein. (b) A synonymous (c.9195G>C; p.V3065V) variant. Minus strand is called. Colored boxes above each variant indicate the ratios of the two bases at the variant position. Sequence shown in Integrative Genomics Viewer (IGV), Broad Institute, Cambridge, MA
Large DNA rearrangements also play a role in ADPKD causation, but their detection is complicated by the presence of the PKD1 pseudogenes [53]. Field inversion gel electrophoresis followed by Southern blot analysis traditionally has been used to identify large deletions in PKD1. Recently, a multiplex ligation dependent probe assay (MLPA) was developed for detecting large genomic rearrangements in the PKD genes [54]. In this assay, a set of probes were designed to target PKD1, PKD2, and PKD1–TSC2, covering a total of 320 kb genomic region including flanking regions. The amplified products are analyzed using the Luminex FlexMAP technology in a single assay. Using this method, Consugar et al. identified large deletions in 31 % of the previously mutation-negative cases, accounting for approximately 4 % of all patients in a prospectively studied cohort of patients with ADPKD [54]. This method has been recently commercialized by MRC-Holland (MRC-Holland, Amsterdam, The Netherlands) and is available as a clinical test. An example of MLPA analysis results is shown in Fig. 19.5.
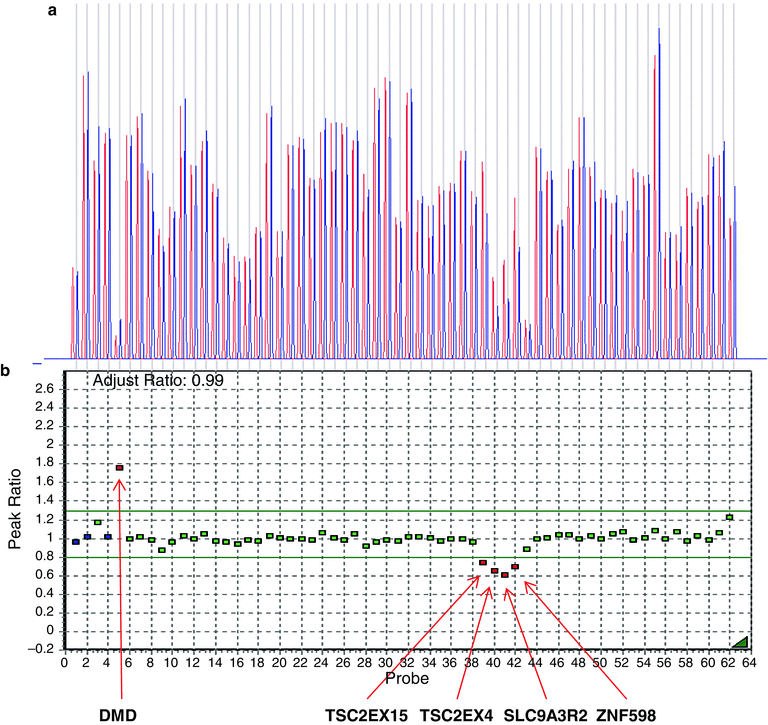
Figure 19.5
Multiplex ligation-dependent probe amplification (MLPA) analysis of a positive control DNA sample (Coriell, Camden, NJ), using probes covering the PKD2 gene on chromosome 4 as well as numerous continuous genes (PKD1, TSC2, SLC9A3, ZNF598, and TBL3) on chromosome 16. Data were deconvoluted according to relative copy number compared to both the internal control genes as well as a normal control male sample using the GeneMarker software (Softgenetics, LLC; State College, PA, USA). (a) The Dosage Histogram displays the ratio of normalized peak intensities between the reference and the sample trace. Sample probes are represented by blue bars and control probes by red bars. (b) The Ratio Plot displays, in graphical form, the ratio of normalized peak intensities between the reference and the sample trace. Each square represents a specific probe. Analysis results show the expected deletions corresponding to one copy of TSC2EX15, TSC2EX4, SLC9A3R2, and ZNF598 genes (red squares and arrows), and two copies of all PKD1 and PKD2 gene probes (green squares) and the two-copy internal controls (blue squares). The X-linked DMD control (upper left side) is indicated in red, demonstrating a patient-control peak ratio of approximately 1.8, consistent with a two-copy female DNA and a single-copy male reference control
Family linkage analysis provides another approach to genetic testing for ADPKD. This strategy employs highly informative microsatellite markers flanking PKD1 and PKD2. Once linkage analysis has been performed, haplotype reconstruction can be used to predict the disease status of other family members. However, this method is suitable in fewer than 50 % of families because of constraints such as an insufficient number of affected family members [55]. Although it is now seldom used for genetic testing in ADPKD, linkage analysis may be considered in situations such as preimplantation genetic diagnosis (PGD), where low amounts of genomic DNA limit the utility of other genotyping approaches. Typing of several markers also is recommended to ensure against confounders such as allele dropout (see below), which can occur when amplifying very low amounts of DNA and/or when evaluating highly polymorphic genes such as PKD1 [56].
Interpretation of Results
Mutations in PKD1 and PKD2 are usually private, highly variable and are distributed throughout the entire gene, with no mutation hot spot. This high level of allelic heterogeneity makes the interpretation of genotyping results challenging. The ADPKD Mutation Database at Mayo Clinic (http://pkdb.mayo.edu/) [57] is the most complete mutation database for ADPKD. Mutation information has been reported for a total of 1,794 families: 1,420 (79 %) have PKD1 mutations and the remaining 21 % have mutations in PKD2 (as of November, 2013).
Genetic variations in the ADPKD Mutation Database are classified into 12 categories according to their characteristics at the DNA or protein level, including frameshift mutations, nonsense mutations, splice-site substitutions, intervening sequence (IVS), variations, silent changes, silent 5′ and 3′ untranslated region changes, synonymous changes, and rearrangements (deletions and duplications). Based on their predicted pathogenicity, those variations have been further classified into six categories: definitely pathogenic, highly likely pathogenic, likely pathogenic, hypomorphic, indeterminate, and likely neutral. A total of 1,923 changes for PKD1 are reported in the ADPKD Mutation Database (Table 19.1), including 48.3 % pathogenic (with 32.2 % definitely pathogenic, 7.5 % highly likely pathogenic, and 8.6 % likely pathogenic), 0.5 % hypomorphic, 9.0 % indeterminate, and 42.2 % neutral. Large rearrangements account for 3 % of the pathogenic mutations reported for PKD1. A total of 241 PKD2 changes are documented in the same database, consisting of 69.2 % pathogenic (with 53.5 % definitely pathogenic, 6.6 % highly likely pathogenic, and 9.1 % likely pathogenic), 0.4 % likely hypomorphic, 6.6 % indeterminate, and 23.7 % neutral (Table 19.1). Figure 19.6 shows the distributions of all known genomic changes in the PKD1 (Fig. 19.6a) and PKD2 (Fig. 19.6b) genes.
Table 19.1
Distribution of PKD1 and PKD2 gene variants by type and pathogenicity (N = 1,794 ADPKD families)
Gene | Mutation type | Definitely pathogenic | Highly likely pathogenic | Likely pathogenic | Likely hypomorphic | Indeterminate | Likely neutral | Total |
|---|---|---|---|---|---|---|---|---|
PKD1 (N = 1,420) | Nonsense | 203 | 0 | 0 | 0 | 0 | 0 | 203 |
Frameshift | 310 | 0 | 0 | 0 | 0 | 0 | 310 | |
Splice | 62 | 10 | 19 | 0 | 0
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|