INTRODUCTION
While the heart provides cardiac output, the blood vessels play a critical role in the distribution of oxygen and nutrients to metabolically active tissues. Blood flow to these tissues is exquisitely controlled by a variety of stimuli that act on vascular smooth muscle cells to regulate vascular tone (i.e., the degree of contraction of vascular smooth muscle). Dysregulation of vascular tone contributes to the pathogenesis of a variety of diseases, including hypertension, coronary artery disease, Raynaud’s phenomenon, and migraine headache. Multiple signal transduction pathways converge on the vascular smooth muscle contractile apparatus, offering numerous targets for pharmacologic intervention. Many successful therapies have already been developed based on a molecular understanding of the regulation of vascular tone. New targets continue to be identified, offering hope that, in the future, even better therapies will be available to treat patients with vascular disease.
 GF, a 63-year-old man with a history of hypertension, diabetes, and hypercholesterolemia, begins to develop episodes of chest pain with exertion. One week after his first episode, a bout of chest pain occurs while he is mowing the lawn. Twenty minutes after the onset of his pain, GF takes two of his wife’s sublingual nitroglycerin tablets. Within a few minutes, he feels much better. GF feels so well that he decides to take one of the sildenafil (Viagra®) pills that a friend had previously offered to him. A few minutes after taking sildenafil, he feels flushed, develops a throbbing headache, and senses his heart racing. Upon standing, GF becomes light-headed and faints. He is taken immediately to the emergency department, where he is found to have severe hypotension. He is quickly placed in a supine position with his legs raised and monitored until he regains consciousness. The physician considers administering an α-adrenergic agonist, such as phenylephrine, but the rapid improvement in GF’s blood pressure after he is placed in a supine position suggests that pharmacologic intervention is unnecessary. After GF recovers, his physician discusses with him the dangers of taking medications without a prescription and, specifically, the risk of concurrent administration of organic nitrates and sildenafil.
GF, a 63-year-old man with a history of hypertension, diabetes, and hypercholesterolemia, begins to develop episodes of chest pain with exertion. One week after his first episode, a bout of chest pain occurs while he is mowing the lawn. Twenty minutes after the onset of his pain, GF takes two of his wife’s sublingual nitroglycerin tablets. Within a few minutes, he feels much better. GF feels so well that he decides to take one of the sildenafil (Viagra®) pills that a friend had previously offered to him. A few minutes after taking sildenafil, he feels flushed, develops a throbbing headache, and senses his heart racing. Upon standing, GF becomes light-headed and faints. He is taken immediately to the emergency department, where he is found to have severe hypotension. He is quickly placed in a supine position with his legs raised and monitored until he regains consciousness. The physician considers administering an α-adrenergic agonist, such as phenylephrine, but the rapid improvement in GF’s blood pressure after he is placed in a supine position suggests that pharmacologic intervention is unnecessary. After GF recovers, his physician discusses with him the dangers of taking medications without a prescription and, specifically, the risk of concurrent administration of organic nitrates and sildenafil.
Questions
1. What is the mechanism by which sublingual nitroglycerin acts so quickly to relieve chest pain?
2. What are the common adverse effects of nitroglycerin?
3. How can sildenafil and organic nitrates interact to precipitate severe hypotension?
4. Are non-nitrate antihypertensives, such as calcium channel blockers, also contraindicated for men taking sildenafil?
5. How can the mechanisms of action of drugs be used to predict possible drug–drug interactions or lack of interactions?

Vascular tone is a key regulator of tissue perfusion, which determines whether tissues receive sufficient oxygen and nutrients to meet their metabolic demands. Blood flow distribution and circulating blood volume are tightly controlled by the tone of resistance arterioles and capacitance veins, respectively. Vascular smooth muscle cells are the functional regulatory unit of vessel tone in these regions, integrating a variety of signals to optimize their contractile state. In general, these regulatory units act through the signal transduction pathways discussed in this chapter, many of which are targets for therapeutic intervention.
Poiseuille’s law approximates flow through blood vessels:

where ΔP is the pressure drop across a length (L) of vessel, r is the vessel radius, and η is blood viscosity. This relationship demonstrates that small changes in the tone of circumferential layers of vascular smooth muscle cells, and thus vessel diameter, can have a significant impact on blood flow. The tone of the arterial portion of the circulation and the tone of the venous portion of the circulation have important yet distinctive effects on the cardiovascular system. Arterial tone directly controls systemic vascular resistance (SVR) and, with cardiac output (CO), is an important determinant of mean arterial blood pressure (MAP):

Perhaps more importantly, these changes in arteriolar resistance regulate blood flow into tissue capillary beds, where increased smooth muscle cell contraction increases vascular resistance and decreases distal perfusion. At the organismal level, a coordinated response of resistance vessels is absolutely required to redirect oxygen and nutrients to the tissues most in need.
Venous tone, by contrast, plays an important role in determining circulating blood volume. Veins are highly compliant (i.e., can accommodate large changes in volume with little change in pressure) and contain approximately 70% of blood volume during rest. Venoconstriction mobilizes these stores to increase the effective circulating blood volume, allowing perfusion of additional vascular beds (i.e., venoconstriction can be thought of as an “autotransfusion”).
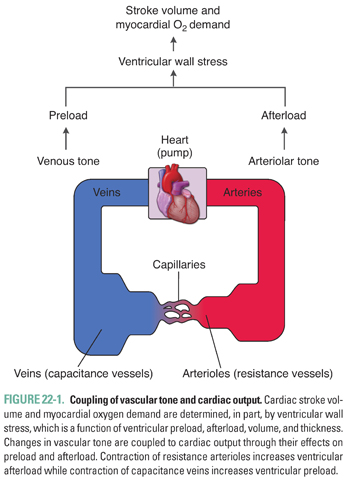
The heart and blood vessels form an integrated and interdependent system, and physiologic or pathophysiologic changes in vascular tone can have a significant impact on tissue perfusion as well as cardiac output. Vasoconstriction and the resulting increase in vascular resistance increases ventricular afterload, or the systolic ventricular wall stress. The volume and thickness of the left ventricle also contribute to the net stress experienced by the contracting ventricle. Venoconstriction and the resulting increase in blood return to the heart increases ventricular preload, defined as end-diastolic ventricular wall stress. These changes directly affect cardiac stroke volume through the Frank-Starling mechanism and indicate the close coupling between vascular and cardiac physiology (Fig. 22-1).
Vascular Smooth Muscle Contraction and Relaxation
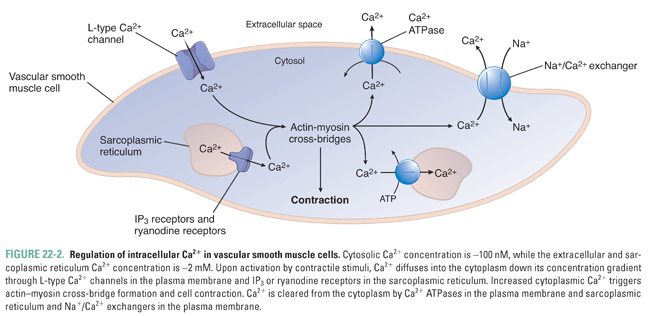
As in cardiac and skeletal muscle cells, vascular smooth muscle cells use cyclic interactions between actin and myosin to generate force. This process is regulated by the intracellular calcium (Ca2+) concentration, which is normally 10,000 times lower than the extracellular concentration (2 mM). The steep transmembrane Ca2+ gradient is maintained by the relative impermeability of the plasma membrane to Ca2+ and by the actions of membrane pumps that actively export Ca2+ from the cell. Contractile stimuli serve to increase intracellular Ca2+ through two mechanisms. First, Ca2+ can diffuse down its concentration gradient into the cell through Ca2+-selective channels in the plasma membrane that can be opened by the activation of cell surface receptors (receptor-operated Ca2+ channels), by mechanical stretch, or by membrane depolarization (voltage-dependent or L-type Ca2+ channels). Second, Ca2+ can be released from intracellular stores by activation of inositol 1,4,5-trisphosphate (IP3) receptors and Ca2+-induced Ca2+ release through ryanodine receptors located in the sarcoplasmic reticulum. Upon termination of the contractile stimulus, Ca2+ is removed from the cytoplasm through active transport by Ca2+ ATPases in the plasma membrane and sarcoplasmic reticulum and Na+/Ca2+ exchangers in the plasma membrane (Fig. 22-2).
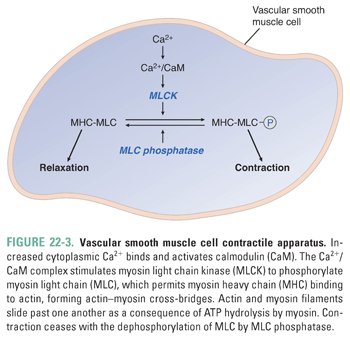
Increases in intracellular Ca2+ are tightly coupled to vasoconstriction. Ca2+ binds to calmodulin (CaM), and the Ca2+/CaM complex binds to and activates myosin light chain kinase (MLCK). Active MLCK phosphorylates myosin light chain (MLC), permitting myosin interactions with actin filaments, leading to cross-bridge cycling and smooth muscle contraction. Vasodilation occurs as a consequence of decreasing intracellular Ca2+ and dephosphorylation of MLC by MLC phosphatase (Fig. 22-3).
Vascular smooth muscle cells integrate a variety of signals to regulate vascular tone, including local environmental factors, endothelium-derived signaling molecules, neurotransmitters, and hormones. Extracellular stimuli generally converge on shared intracellular signal transduction pathways that regulate the smooth muscle contractile apparatus. These signaling cascades are targeted by the drugs discussed in this chapter and, thus, provide the framework for understanding the mechanisms of drug action.
Intracellular signaling pathways are often shared among a variety of extracellular stimuli. These intracellular pathways will be reviewed in this section, and the extracellular signals that activate them will be discussed in subsequent sections (Fig. 22-4).
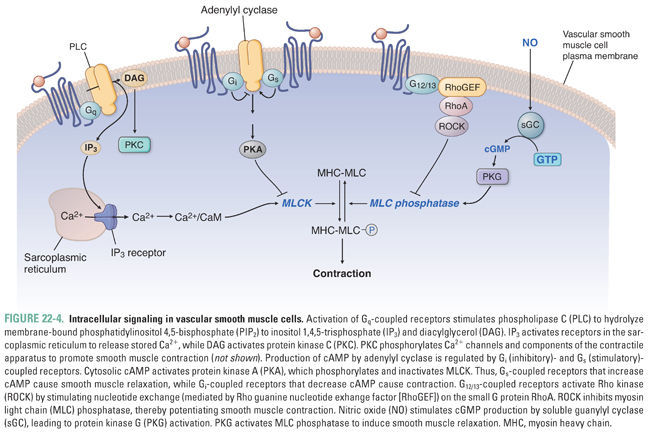
Vasoconstriction is potentiated by three signaling pathways. First, activation of G protein-coupled receptors (GPCR) associated with heterotrimeric Gq proteins activates phospholipase C (PLC) to produce IP3 and diacylglycerol (DAG). IP3 stimulates Ca2+ release from intracellular stores by activating IP3 receptors in the sarcoplasmic reticulum. DAG activates protein kinase C, which also promotes contraction through a variety of phosphorylation events. Second, GPCRs coupled to heterotrimeric G12/13 proteins stimulate nucleotide exchange on the small G protein RhoA. RhoA activates Rho-kinase to phosphorylate and inactivate MLC phosphatase, thereby maintaining MLC phosphorylation. This pathway provides a mechanism to sustain smooth muscle contraction beyond transient increases in intracellular Ca2+. Third, activation of Gi-coupled receptors inhibits adenylyl cyclase and thereby decreases production of cyclic adenosine monophosphate (cAMP). Reduced cAMP decreases protein kinase A (PKA) activity, thereby relieving inhibition of MLCK.
Vasodilation is potentiated by two signaling pathways. First, Gs-coupled receptors stimulate cAMP formation, PKA activation, MLCK inhibition, and ATP-regulated K+ channel (K+ATP) opening. Second, nitric oxide (NO) activates soluble guanylyl cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP). This second messenger activates protein kinase G (i.e., cGMP-dependent protein kinase), which phosphorylates a variety of downstream targets that activate MLC phosphatase to inhibit myosin-actin cycling, inhibit Ca2+ mobilization and increase sequestration, and activate Ca2+-dependent K+ channels. These concerted actions lead to smooth muscle relaxation.
One common feature of these vasodilatory pathways is the opening of plasma membrane K+ channels, leading to membrane hyperpolarization. When K+ channels open, K+ exits the cell down its concentration gradient, moving the Nernst equilibrium potential of the plasma membrane down toward −90 mV (the Nernst potential for K+) from some higher resting value (thereby hyperpolarizing the membrane). This change in potential makes it more difficult for the plasma membrane to depolarize sufficiently for the voltage-gated L-type Ca2+ channels to open, thereby inhibiting smooth muscle cell contraction.
Arteriolar smooth muscle cells coordinate blood flow into the capillary beds of metabolically active tissues. In regions where tissue metabolic demand exceeds supply, increases in H+ (as lactic acid), CO2, K+, and adenosine (from ATP utilization) all lead to vasodilation and increased blood flow. The cerebral circulation is particularly sensitive to fluctuations in pH and CO2, which is why acute hyperventilation (which decreases pH and CO2, leading to vasoconstriction and decreased cerebral blood flow) is one therapy for intracranial hypertension. Increases in extracellular K+ activate inward rectifier K+ channels, thereby causing hyperpolarization of the plasma membrane and inhibiting voltage-gated Ca2+ channel opening. Adenosine activates A2 Gs-coupled receptors. Systemic vessels also respond to decreased O2 by vasodilation, in contrast to pulmonary vessels that vasoconstrict (i.e., hypoxic vasoconstriction) to preserve ventilation–perfusion matching in the lung. The molecular mechanisms mediating these disparate responses remain active areas of research.
In addition to metabolic factors, vascular smooth muscle cells contract in response to stretch through the opening of stretch-activated Ca2+ channels in the cell membrane. This myogenic reflex protects distal capillary beds from high pressures by increasing vascular resistance. In combination with local control by metabolic factors, the myogenic reflex is an important mechanism of vascular autoregulation where the vessels adjust resistance in an attempt to maintain steady blood flow over a range of perfusion pressures (recall that Flow = Pressure/Resistance). Autoregulation is particularly evident in vascular beds that are sensitive to ischemia, such as the brain, heart, and kidneys.
Vascular endothelial cells play a critical role in regulating vascular smooth muscle tone through direct cellular contacts and the elaboration of signaling molecules. Among these signaling molecules, NO, endothelium-derived hyperpolarizing factors, prostacyclin, and endothelin are the most relevant pharmacologically.
The obligatory role of endothelial cells in regulating vascular tone was first recognized with the observation that acetylcholine causes vasoconstriction when applied directly to de-endothelialized blood vessels but causes vasodilation when applied to normally endothelialized vessels. This finding suggested that endothelial cells produce a vasodilatory compound, initially termed endothelium-derived relaxing factor (EDRF) and subsequently proven to be NO.
Nitric oxide is a membrane-permeable gas that reacts with a variety of biomolecules to elicit cellular responses, particularly activation of sGC in vascular smooth muscle cells, leading to cGMP production. Vascular endothelial cells synthesize NO in response to a variety of stimuli, including shear stress, acetylcholine, histamine, bradykinin, sphingosine-1-phosphate, serotonin, substance P, and ATP. These factors increase intracellular Ca2+, thereby stimulating the Ca2+/CaM-activated endothelial NO synthase (eNOS). Endothelial eNOS produces NO from arginine, and NO subsequently diffuses to the vascular smooth muscle cell to initiate downstream signaling events and vasorelaxation (Figs. 22-4 and 22-5).
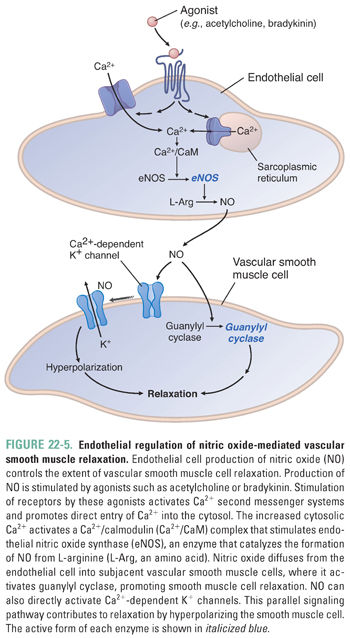
Interestingly, measured levels of NO cannot explain all of the endothelium-dependent responses of vascular smooth muscle cells. Indeed, endothelium-dependent vasodilation can be caused by smooth muscle cell hyperpolarization that is not due to NO. Several molecules have been implicated as endothelium-derived hyperpolarizing factors (EDHFs), including epoxyeicosatrienoic acids (arachidonic acid metabolites), hydrogen peroxide, carbon monoxide, hydrogen sulfide, C-natriuretic peptide, and K+ itself. These mediators open a variety of K+ channels on smooth muscle cells, leading to membrane hyperpolarization and smooth muscle cell relaxation. Hydrogen sulfide has a variety of additional effects that lead to smooth muscle relaxation, largely mediated by covalent S-sulfhydration of target proteins, including not only K+ channels but also Ca2+ channels, and by stimulating release of other EDHFs.
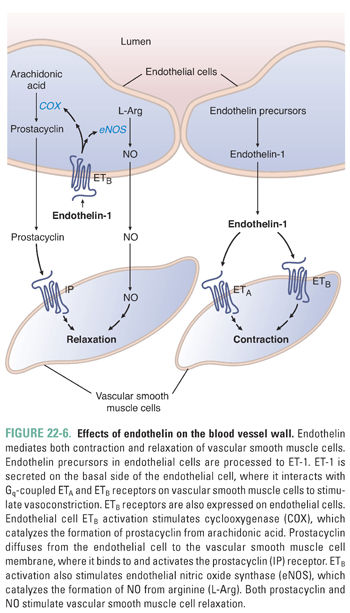
Prostacyclin is also a vasodilatory molecule produced in endothelial cells from arachidonic acid in reactions that involve the cyclooxygenase (COX) enzymes. Prostacyclin activates Gs-coupled receptors on the vascular smooth muscle cells, leading to vasodilation. Since the COX enzymes are inhibited by nonsteroidal anti-inflammatory drugs, such drugs should be used with caution in patients with hypertension because they decrease prostacyclin production.
In contrast to the potent vasodilating effects of the molecules described above, endothelial cells also produce the most potent endogenous vasoconstrictor known, endothelin-1 (ET-1). ET-1 is a 21-amino acid peptide synthesized as preproendothelin, which is cleaved to big endothelin and subsequently to ET-1 by endothelin converting enzyme. ET-1 is released by endothelial cells in response to mechanical stress and vasoactive agents (e.g., vasopressin, angiotensin II), while its release is inhibited by prostacyclin, NO, and atrial natriuretic peptide. ET-1 binds to two receptor subtypes, ETA and ETB, and both are Gq-coupled receptors. Both subtypes are located on vascular smooth muscle cells and mediate vasoconstriction. Interestingly, endothelial cells express ETB receptors, which, when occupied by ET-1, activate eNOS and COX, leading to NO and prostacyclin release. This negative feedback pathway is one mechanism by which endothelial cells assist in modulating vascular tone (Fig. 22-6).
Vascular smooth muscle cells receive input from the sympathetic nervous system, which is an important determinant of vascular tone. Sympathetic nerves innervate vascular smooth muscle cells in both arteries and veins, and sympathetic activation leads to both vasoconstriction and venoconstriction. Sympathetic postganglionic neurons release norepinephrine, which binds to postsynaptic α1– and α2-adrenergic receptors coupled to Gq and Gi, respectively, leading to contraction of the smooth muscle cell (Fig. 22-4). The presynaptic nerve terminal also expresses α2-adrenergic autoreceptors, which inhibit further release of norepinephrine in a negative feedback loop.
Sympathetic activation also induces epinephrine release from the adrenal medulla. In contrast to norepinephrine, epinephrine activates both α- and β2-adrenergic receptors on vascular smooth muscle cells. The β2 receptors activate the Gs signaling pathway, leading to smooth muscle cell relaxation (Fig. 22-4). Thus, the effects of epinephrine on a given vascular bed depend on the dose (β2 receptors have a higher affinity for epinephrine and, thus, are activated at lower epinephrine concentrations than α receptors) and the relative composition of receptors expressed on the target cells. For example, during a “fight or flight” response, blood flow is diverted away from skin and viscera, where α > β2, and toward skeletal muscle, where β2 > α.
While most blood vessels lack parasympathetic innervation, acetylcholine does cause vasodilation through M3-muscarinic receptor-mediated NO release from vascular endothelial cells (Fig. 22-5).
In addition to the autonomic nervous system, several humoral mediators contribute to the regulation of vascular tone and integrate renal and cardiovascular function. Among these, angiotensin II and vasopressin are potent vasoconstrictors through their activation of Gq-coupled AT1 and V1 receptors, respectively (Fig. 22-4). These mediators act to increase both vascular resistance and intravascular volume in response to hypovolemia (i.e., hemorrhagic shock) through coordinated actions in the blood vessels and kidney. Atrial and brain natriuretic peptides are released in response to hypervolemia and induce vasodilation through the activation of membrane-bound guanylyl cyclase receptors and production of cGMP. Histamine and bradykinin also act as vasodilators. Histamine activates Gs-coupled H2 receptors on vascular smooth muscle cells to cause vasodilation (Fig. 22-4) and Gq-coupled H1 receptors on endothelial cells to cause NO generation (Fig. 22-5). Bradykinin also stimulates NO production through the β2 receptor on endothelial cells (Fig. 22-5).
 PHARMACOLOGIC CLASSES AND AGENTS
PHARMACOLOGIC CLASSES AND AGENTS
The pharmacologic agents considered in this chapter are all vasodilators, that is, drugs that act on vascular smooth muscle cells or endothelial cells to decrease vascular tone. This effect can be accomplished either by inhibiting components of contractile signal transduction pathways or by potentiating effects of relaxing signal transduction pathways.
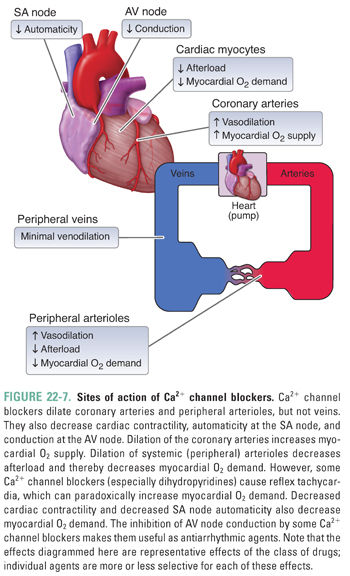
Ca2+ channel blockers are among the most prescribed agents for the management of hypertension and angina owing to their effectiveness and ease of use. These agents are primarily arterial vasodilators, having little impact on the venous system. Importantly, Ca2+ channel blockers bind to both vascular smooth muscle cells and cardiac myocytes, which accounts for their effectiveness as positive lusitropic agents (i.e., agents that relax the myocardium) and in the management of certain cardiac arrhythmias.
Ca2+ influx through L-type Ca2+ channels is an important determinant of vascular tone and cardiac contractility. Ca2+ channel blockers inhibit Ca2+ influx through these channels. In smooth muscle cells, decreased Ca2+ entry keeps intracellular Ca2+ concentrations low, thereby reducing Ca2+/CaM-mediated activation of MLCK, actin–myosin cross-bridge formation, and smooth muscle cell contraction. Vasodilation of resistance arterioles decreases systemic vascular resistance and blood pressure and, consequently, ventricular afterload. Additionally, drug-induced dilation of the coronary arteries augments myocardial oxygen supply, helping to alleviate the symptoms of angina in affected patients (Fig. 22-7).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


