This chapter reviews the physiology and pharmacology of insulin, glucagon, and the other major hormones that regulate glucose homeostasis. Diabetes mellitus is clinically the most common disease of these endocrine axes, and the majority of the chapter is devoted to the physiology and pharmacology of insulin. Type 1 diabetes mellitus is caused by an absolute deficiency of insulin secretion, and type 2 diabetes mellitus is caused by insufficient (or dysfunctional) insulin secretion to overcome insulin resistance in target tissues. Medical students may be interested to note that Charles Best, a fourth-year medical student in Canada, had a significant role in the identification of insulin. Along with his mentor, Frederick Banting, Best isolated a pancreatic extract from dogs that could reduce blood glucose in diabetic dogs and humans. Although the 1923 Nobel Prize in Medicine or Physiology was jointly awarded to surgeon Frederick Banting and physiologist J. J. R. MacLeod, Banting shared his award with Best.
 At her annual check-up, 55-year-old Mrs. S complains of fatigue and frequent urination (polyuria), even at night. She also reports drinking large volumes of fluids (polydipsia) to quench her thirst. These symptoms have worsened during the past 2 years and correlate with a 15-lb weight gain over the same period. Her current body mass index (BMI; the weight in kilograms divided by the square of the height in meters) is 32, thus meeting criteria for class I obesity. She is “frustrated” by her ineffectiveness to curb portion sizes and maintain a routine of physical activity. She denies other urinary symptoms; her kidney function is essentially normal. Her past medical history is remarkable for hyperlipidemia for the past 10 years. Both of her parents died of coronary heart disease in their early 60s.
At her annual check-up, 55-year-old Mrs. S complains of fatigue and frequent urination (polyuria), even at night. She also reports drinking large volumes of fluids (polydipsia) to quench her thirst. These symptoms have worsened during the past 2 years and correlate with a 15-lb weight gain over the same period. Her current body mass index (BMI; the weight in kilograms divided by the square of the height in meters) is 32, thus meeting criteria for class I obesity. She is “frustrated” by her ineffectiveness to curb portion sizes and maintain a routine of physical activity. She denies other urinary symptoms; her kidney function is essentially normal. Her past medical history is remarkable for hyperlipidemia for the past 10 years. Both of her parents died of coronary heart disease in their early 60s.
On physical examination, Mrs. S is moderately obese but otherwise appears normal. Glucose is detected in her urine, but proteins and ketones are not. Her blood tests are significant for elevated glucose (210 mg/dL), total cholesterol (340 mg/dL), and HbA1c (a measure of glucose covalently bound to hemoglobin; 8.2%) levels. The physician explains to Mrs. S that she most likely has type 2 diabetes mellitus. In this disease, the body fails to respond normally to insulin (insulin resistance) and cannot produce a sufficient amount of insulin to overcome this resistance.
The physician discusses that lifestyle interventions (adhering to a balanced diet with controlled total calorie intake as well as increasing her exercise) are the cornerstone of diabetes treatment. The physician also discusses the benefits and risks of antidiabetic medications and prescribes metformin (a biguanide) for her diabetes.
Questions
1. What are the cellular and molecular actions of insulin?
2. What is the etiology of diabetes mellitus, and how is type 1 diabetes mellitus different from type 2 diabetes mellitus?
3. In addition to alleviating her polyuria and polydipsia, why is it important to control Mrs. S’s diabetes (i.e., what acute and chronic complications could arise)?
4. What do the blood glucose and HbA1c levels indicate about Mrs. S’s diabetes? Are there circumstances under which one parameter could be elevated and the other could be normal?
5. What are the effects of antidiabetic medications on body weight? Why might Mrs. S’s physician have chosen metformin to treat her diabetes?

The pancreas is a glandular organ that contains both exocrine and endocrine tissue. The exocrine portion—which constitutes 99% of the pancreatic mass—secretes bicarbonate and digestive enzymes into the gastrointestinal (GI) tract. Scattered within the exocrine tissue are nearly one million small islands of endocrine tissue that secrete hormones directly into the blood. These tiny endocrine glands, collectively called islets of Langerhans, include several different cell types that secrete different hormones: α-cells release glucagon; β-cells release insulin and amylin; δ-cells release somatostatin and gastrin; and PP cells release pancreatic polypeptide.
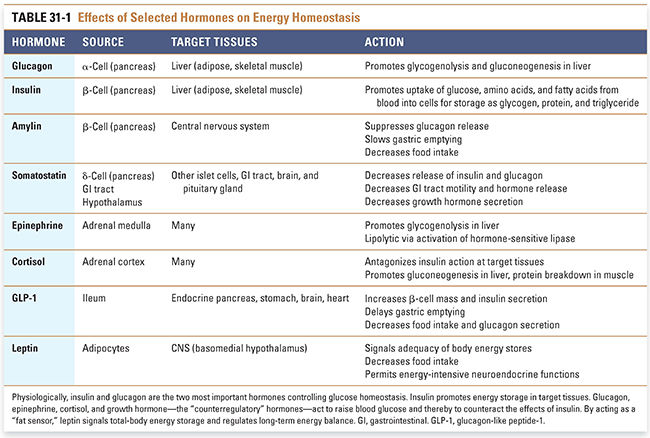
The storage and subsequent release of nutrients provides a homeostatic mechanism for sustained cellular nutrition in the absence of continuous feeding. Multiple hormones are involved in controlling the uptake, utilization, storage, and release of nutrients. Insulin promotes the uptake and storage of glucose and other small, energy-containing molecules. The “counterregulatory” hormones oppose the actions of insulin and promote glucose release (Table 31-1). These hormones include (1) glucagon from islet β-cells, (2) the catecholamines norepinephrine and epinephrine from the sympathetic nervous system and adrenal medulla, (3) the glucocorticoid cortisol from the adrenal cortex, and (4) growth hormone from the pituitary gland. Glucagon-like peptide-1 (GLP-1) from the GI tract enhances insulin release in response to an ingested meal, while amylin suppresses endogenous production of glucose in the liver.
Blood glucose is easily measured and provides an accurate guide to the current balance of insulin and the counterregulatory hormones. This balance normally keeps blood glucose levels within a narrow range (70–120 mg/dL) regardless of recent food intake. Blood glucose excursions are much greater in patients with diabetes, potentially rising higher than 400 mg/dL in instances of poor control. The drugs used to treat diabetes, particularly insulin and the insulin secretagogues (sulfonylureas and glinides), may reduce blood glucose values below the normal range of 70 mg/dL. Severe hypoglycemia, which is defined by the patient requiring assistance to administer glucose or glucagon to restore normal blood glucose levels, is dangerous because organs—particularly the brain—depend on a constant supply of glucose for proper functioning. Conversely, chronic hyperglycemia is toxic to many cells and tissues.
Energy Repletion and the Fed State
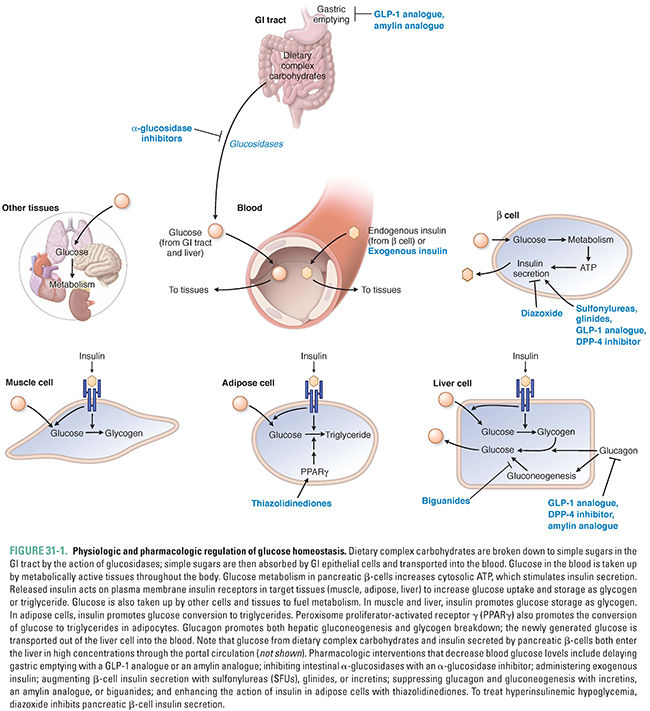
After a meal, complex carbohydrates are broken down to monosaccharides (e.g., glucose, galactose, and fructose) in the lumen of the GI tract and transported into GI epithelial cells by a combination of active and passive apical membrane transporters. Sugars are then transported by basal membrane transporters from the epithelial cell cytosol to intercellular spaces, from which the sugars continue into the bloodstream. Elevated glucose in the blood is the signal for pancreatic β-cells to release insulin, which enters the portal vein. The liver, therefore, receives the highest concentrations of insulin concurrently with the nutrients absorbed from the digestive tract. The liver and the other energy-storing tissues, such as skeletal muscle and adipose tissue, are the primary target tissues for insulin (Fig. 31-1). Insulin also acts on pancreatic α-cells to suppress the secretion of glucagon.
The hormone leptin is not directly involved in nutrient regulation, but it plays an important role in the neuroendocrine response to energy storage and long-term energy balance. Leptin is secreted by adipocytes and detected by receptors in the hypothalamus. Its concentration in the plasma is proportional to total fat mass. Thus, leptin signals from the periphery to the central nervous system that energy stores (in the form of adipose tissue) are replete. Leptin also suppresses appetite, which switches the body from an energy-accumulating state to a state of energy utilization. This allows growth and reproduction to be switched on, whereas the lack of leptin, as occurs in prolonged starvation, results in persistently increased appetite and suppression of energy-utilizing functions.
A key intracellular mediator of energy storage is the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ). PPARγ is a transcription factor that has roles in both adipose cell differentiation and lipid metabolism. PPARη activation decreases serum free fatty acid levels and increases lipogenesis in adipose tissue, which increases the storage of fatty acids in adipose tissue. This allows other tissues—such as the liver—to lower their fat content, lower their glucose production, and increase their insulin sensitivity. PPARγ is the target for the thiazolidinedione (TZD) class of diabetes drugs.
Like many small hydrophilic molecules, glucose is freely filtered at the glomerulus but is normally reabsorbed almost completely by the sodium-glucose co-transporter 2 (SGLT-2) in the proximal convoluted tubule. Therefore, the glucose content of the urine is normally negligible. However, the transport maximum (Tmax) for SGLT-2 is reached at glucose concentrations of 180–200 mg/dL, so, in diabetes, when blood glucose levels are over 200 mg/dL and SGLT-2 is saturated, glucose does appear in the urine. (Urinary glucose “dipsticks” were used by patients with diabetes to monitor glycemic control long before “finger stick” measures of blood glucose came into use.) The principle of losing glucose in the urine has been capitalized on with the development of selective inhibitors of SGLT-2, which cause enough glucose to remain in the urine that blood glucose levels, HbA1c levels, and even body weight are lowered.
As the blood glucose concentration decreases, pancreatic α-cells release increasing amounts of glucagon and pancreatic β-cells secrete decreasing amounts of insulin. In contrast to insulin, which promotes the cellular uptake of glucose in the fed state, glucagon mobilizes glucose from the liver by stimulating gluconeogenesis and glycogenolysis. As fasting continues, catecholamine and glucocorticoid levels also increase, promoting the release of fatty acids from adipose tissue and the breakdown of protein to amino acids in muscle.
In low-energy (low adenosine triphosphate [ATP]) states, the enzyme adenosine 5′-monophosphate-activated protein kinase (AMPK) also triggers a shift from anabolic to catabolic activities. AMPK is present in tissues throughout the body, and it helps to regulate energy metabolism at both the cellular and organismal levels. Exercise activates AMPK, which increases muscle uptake of glucose. Activated AMPK also decreases glucose production and the synthesis of lipids and proteins by the liver. The pharmacologic effects of metformin and other biguanides are incompletely understood despite intense investigation but are contributed to by both AMPK-dependent and AMPK-independent mechanisms.
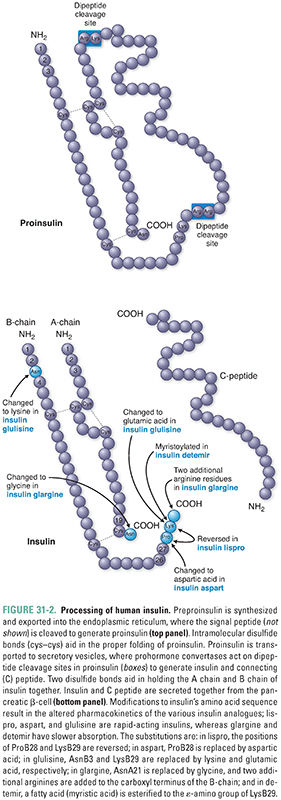
Insulin is a 51-amino acid protein composed of two peptide chains that are linked by two disulfide bridges. Its name comes from the Latin insula (meaning “island,” after the islets of Langerhans). The human pancreas contains approximately 8 mg of insulin, of which 0.5 to 1.0 mg is secreted and replenished daily. Insulin is initially synthesized in pancreatic β-cells as preproinsulin, which is processed first to proinsulin and then insulin and free connecting (C) peptide (Fig. 31-2).
Resting pancreatic β-cells are poised to secrete insulin, which is preformed and stored in secretory vesicles at the plasma membrane. The low basal rate of insulin secretion is increased dramatically upon exposure of β-cells to glucose.
Glucose enters β-cells via GLUT2, a specific plasma membrane transporter. As blood glucose rises (e.g., after feeding), more enters the cell, where it is phosphorylated to glucose-6-phosphate and enters the glycolytic pathway and the citric acid cycle. ATP is produced and ADP consumed by these processes, and the ATP/ADP ratio in the β-cell rises. This modulates the activity of a membrane-spanning ATP-sensitive K+ channel (K+/ATP channel). When open, the K+/ATP channel hyperpolarizes the cell by allowing an outward flux of K+, and insulin release is inhibited. When closed, the cell depolarizes and insulin is released. Because ATP inhibits the channel and ADP activates the channel, a high intracellular ATP/ADP ratio closes the K+/ATP channel. Depolarization of the β-cell activates voltage-gated Ca2+ channels to promote the influx of extracellular Ca2+ and the fusion of insulin-containing vesicles with the plasma membrane, which increases insulin release into the circulation (Fig. 31-3).
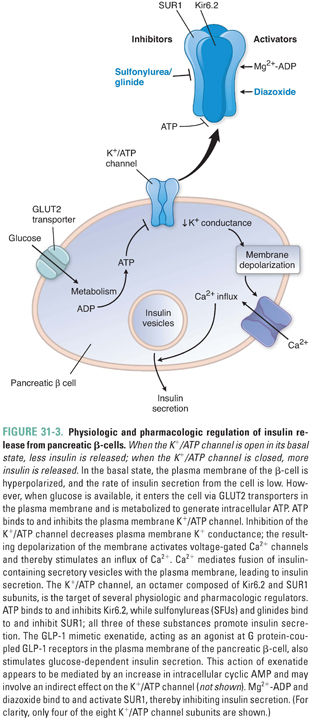
β-cell K+/ATP channels are octameric structures containing four subunits of Kir6.2 and four subunits of the sulfonylurea receptor, SUR1. The Kir6.2 tetramer forms the pore of the K+/ATP channel, while the associated SUR1 proteins regulate the channel’s sensitivity to ADP and pharmacologic agents, including sulfonylurea and related insulin secretagogue drugs. Mutations in Kir6.2 or SUR1 can result in hyperinsulinemic hypoglycemia because the channel remains closed and the β-cell remains continually depolarized even when the extracellular glucose concentration and the intracellular ATP/ADP ratio are low.
In addition to blood glucose, G protein-mediated pathways, parasympathetic activity, and the GI hormones GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) also inhibit K+/ATP channel activity and stimulate insulin secretion. β-Cell exposure to nutrients promotes insulin transcription, translation, processing, and packaging in addition to insulin secretion.
Insulin binds receptors on the surface of target cells. Although virtually all tissues express insulin receptors, the energy-storing tissues (liver, muscle, and adipose) express higher levels of the receptor and have greater responses to insulin and thus constitute the main insulin target tissues. The insulin receptor (Fig. 31-4) is a glycoprotein consisting of four disulfide-linked subunits and two intracellular tyrosine kinase domains. The binding of insulin to the extracellular portion of the insulin receptor activates the intracellular tyrosine kinase, which phosphorylates itself and the intracellular insulin receptor substrate proteins (IRS). Tyrosine-phosphorylated IRS-1 recruits second messenger proteins that contain phosphotyrosine-binding src homology 2 (SH2) domains. Type IA phosphatidylinositol 3-kinase (PI3-kinase) is one such SH2 domain-containing protein that generates second messenger products important for many aspects of insulin action.
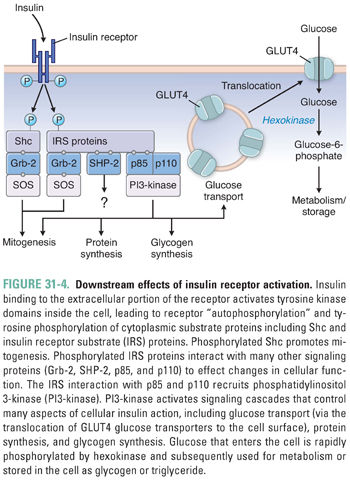
Although the details linking the insulin receptor second messengers to the metabolic effects of insulin remain open to investigation, the metabolic effects of insulin action are well understood: insulin is the classic anabolic (energy storing) hormone (Fig. 31-1). In the liver, insulin increases glucokinase activity, thereby mediating the phosphorylation and trapping of glucose in hepatocytes and fueling glycogen synthesis, glycolysis, and fatty acid synthesis.
In skeletal muscle and adipose tissue, insulin stimulates translocation of the insulin-responsive glucose transporter, GLUT4, from intracellular vesicles to the cell surface, which facilitates the transport of glucose into the cell (Fig. 31-4). In muscle, insulin also increases amino acid uptake, stimulates the ribosomal protein synthesis machinery, and promotes glycogen synthase activity and subsequent glycogen storage. In adipose tissue, insulin promotes the expression of lipoprotein lipase, which hydrolyzes triglycerides from circulating lipoproteins for uptake into fat cells. Once inside the fat cell, glucose and fatty acids are stored predominantly as triglycerides. This process is enhanced by the deactivation of hormone-sensitive lipase, which degrades intracellular triglyceride. Insulin is rapidly degraded in the liver and kidney, with a circulating half-life of 6 minutes.
Glucagon—a single-chain polypeptide of 29 amino acids—is a catabolic (energy releasing) hormone secreted by pancreatic β-cells. When plasma glucose levels are low, glucagon mobilizes glucose, fat, and protein from storage as energy sources. Besides low glucose and high insulin levels, stimuli for glucagon secretion include sympathetic nervous system activity, stress, exercise, and high plasma levels of amino acids (because the latter indicates a state of starvation). Glucagon binding to its G protein-coupled receptor on the plasma membrane of hepatocytes increases intracellular cAMP and activates protein kinase A, a serine/threonine kinase. Glucagon promotes hepatic glycogenolysis and gluconeogenesis (Fig. 31-1). Glucagon also promotes lipolysis in adipose tissue. The liver and kidneys degrade glucagon; like insulin, its circulating half-life is about 6 minutes.
Amylin is a 37-amino acid protein that is packaged together with insulin in secretory granules of the β-cell. Insulin and amylin are co-secreted following a meal. Amylin binds receptors within the central nervous system, and its glucose-regulatory actions complement those of insulin. Specifically, amylin suppresses glucagon release, slows gastric emptying, and decreases food intake. Together, these actions favor gradual entry of glucose into the circulation following a meal. Amylin is cleared by the kidney; its half-life is approximately 10 minutes.
Somatostatin has 14- and 28-amino acid forms that are selectively produced in pancreatic ε-cells, the gastrointestinal tract, and the hypothalamus. Primary functions of somatostatins are to inhibit (1) the release of pituitary growth hormone and thyroid-stimulating hormone (see Chapter 27, Pharmacology of the Hypothalamus and Pituitary Gland), (2) the secretion of pancreatic insulin and glucagon, and (3) GI motility and the release of various GI hormones. The stimuli for pancreatic somatostatin release are similar to those for insulin (i.e., high plasma levels of glucose, amino acids, and fatty acids). Local somatostatin release allows the hormone to act in a paracrine fashion. The circulating half-life of somatostatin is only 2 minutes.
Glucagon-like peptide-1 (GLP-1) and glucagon are both encoded by the glucagon gene, but their sequences are different due to differential mRNA splicing in the enteroendocrine L cells of the distal small intestine and in pancreatic β-cells, respectively. Bioactive forms of GLP-1 are 29 or 30 amino acids in length. Blood levels of GLP-1 are low during fasting and rise after a meal. GLP-1 acts on G protein-coupled receptors located on islet α- and β-cells and in the central and peripheral nervous systems, heart, kidney, lung, and GI tract. At the pancreatic β-cell, GLP-1 augments insulin secretion in response to an oral glucose load (hence GLP-1’s “incretin” effect). At the pancreatic α-cell, GLP-1 suppresses glucagon secretion. GLP-1 acts in the stomach to delay gastric emptying and at the hypothalamus to decrease appetite. GLP-1 has a short half-life in the circulation (1–2 minutes) due to enzymatic degradation by dipeptidyl peptidase-4 (DPP-4).

As early as AD 200, the Greek physician Aretaeus observed patients who had excessive thirst and urination. He named this condition diabetes, which is Greek for “to siphon, or pass through.” Later, physicians added mellitus (Latin for “honeyed, sweet”) to the disease name after noticing that diabetic patients produce urine that contains sugar. The designation diabetes mellitus also distinguishes this disorder from diabetes insipidus (see Chapter 27), in which either the secretion of antidiuretic hormone (ADH) or the response to ADH action in the kidney is dysregulated, such that water is not reabsorbed in the collecting duct of the nephron and copious amounts of dilute urine are produced.
The syndrome of diabetes mellitus results from a heterogeneous group of metabolic disorders that have hyperglycemia in common (Table 31-2). Hyperglycemia can result from an absolute lack of insulin (type 1 diabetes mellitus) or from a relative insufficiency of insulin production in the face of insulin resistance (type 2 diabetes mellitus). The pathophysiology of diabetes that occurs during pregnancy or is induced by other causes (e.g., pancreatitis) is beyond the scope of this chapter.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


