Rajesh Garg and Gail K. Adler

Like the pituitary gland, the adrenal gland consists of two organs fused together during embryologic development. The outer adrenal cortex originates from mesoderm, and the inner adrenal medulla is derived from neural crest cells. The adrenal cortex synthesizes and secretes steroid hormones essential for salt balance, intermediary metabolism, and, in females, androgenic actions. The adrenal medulla synthesizes and secretes the catecholamine epinephrine, which is important, although not essential, for maintaining sympathetic tone. This chapter focuses on the adrenal cortex; because of its importance in neuropharmacology, the adrenal medulla is discussed in Chapter 11, Adrenergic Pharmacology.
The therapeutic utility of adrenocortical hormones spans almost every area of medicine. This is largely because of the usefulness of glucocorticoid analogues as efficacious and potent anti-inflammatory agents. Unfortunately, long-term systemic glucocorticoid therapy also produces a substantial number of predictable but undesirable adverse effects. The physiology of mineralocorticoids has been studied in the etiology of hypertension, cardiovascular disease, and renal disease, and there is considerable interest in the use of mineralocorticoid receptor antagonists as therapies for these disorders. Adrenal androgens, although lacking a definitive therapeutic indication at present, are the focus of investigation for use in female sexual dysfunction. Both deficiency and excess of adrenocortical hormones can cause human disease. Deficiency states are treated by replacing the hormones in the form of therapeutic agents, while inhibitors of adrenocortical biosynthetic enzymes and antagonists at adrenocortical hormone receptors can be used to treat hormone excess.
 Eight-year-old Johnny finds that he can barely catch his breath at times, especially while exercising. His asthma comes and goes, but no therapy seems to stop the asthma attacks completely. Although his doctor is concerned that it could stunt Johnny’s growth, she eventually prescribes oral prednisone (a glucocorticoid analogue) and tells Johnny’s parents to make sure he takes the medication every day. After a few weeks, Johnny’s asthma attacks subside, and he is able to have a fairly normal childhood. During this time, the doctor pays close attention to Johnny’s linear growth. Two years later, Johnny’s doctor decides that a new inhaled glucocorticoid could be a safer medication for him. Johnny switches to the inhaled glucocorticoid and discontinues oral prednisone. Three days later, he develops a respiratory infection and is brought to the emergency department with low blood pressure and a temperature of 103°F. Based on his history of prednisone use, Johnny is immediately given hydrocortisone (cortisol) intravenously, as well as a saline infusion. Johnny recovers and for the next 6 months slowly tapers his oral prednisone dose with continued use of the inhaled glucocorticoid. Eventually, he is able to take the inhaled glucocorticoid alone as an effective therapy for his asthma.
Eight-year-old Johnny finds that he can barely catch his breath at times, especially while exercising. His asthma comes and goes, but no therapy seems to stop the asthma attacks completely. Although his doctor is concerned that it could stunt Johnny’s growth, she eventually prescribes oral prednisone (a glucocorticoid analogue) and tells Johnny’s parents to make sure he takes the medication every day. After a few weeks, Johnny’s asthma attacks subside, and he is able to have a fairly normal childhood. During this time, the doctor pays close attention to Johnny’s linear growth. Two years later, Johnny’s doctor decides that a new inhaled glucocorticoid could be a safer medication for him. Johnny switches to the inhaled glucocorticoid and discontinues oral prednisone. Three days later, he develops a respiratory infection and is brought to the emergency department with low blood pressure and a temperature of 103°F. Based on his history of prednisone use, Johnny is immediately given hydrocortisone (cortisol) intravenously, as well as a saline infusion. Johnny recovers and for the next 6 months slowly tapers his oral prednisone dose with continued use of the inhaled glucocorticoid. Eventually, he is able to take the inhaled glucocorticoid alone as an effective therapy for his asthma.
Questions
1. Why are cortisol analogues such as prednisone used for treating asthma?
2. Why did abrupt cessation of oral prednisone precipitate Johnny’s clinical presentation in the emergency department?
3. Why are inhaled glucocorticoids safer than oral glucocorticoids for long-term treatment of asthma?
4. Why did the doctor monitor Johnny’s linear growth?
 OVERVIEW OF THE ADRENAL CORTEX
OVERVIEW OF THE ADRENAL CORTEX
The adrenal cortex synthesizes three classes of hormones: mineralocorticoids, glucocorticoids, and androgens. Histologically, the adrenal cortex is divided into three zones. Moving from the capsule toward the medulla, these regions are the zona glomerulosa, zona fasciculata, and zona reticularis (Fig. 29-1). The glomerulosa is responsible for mineralocorticoid production and is under the control of angiotensin II, blood potassium concentration, and, to a lesser extent, adrenocorticotropic hormone (ACTH). The fasciculata and reticularis synthesize glucocorticoids and androgens, respectively. Both the fasciculata and reticularis are mainly under the control of ACTH, which, in turn, is regulated by corticotropin-releasing hormone (CRH), vasopressin, and cortisol (see Chapter 27, Pharmacology of the Hypothalamus and Pituitary Gland).
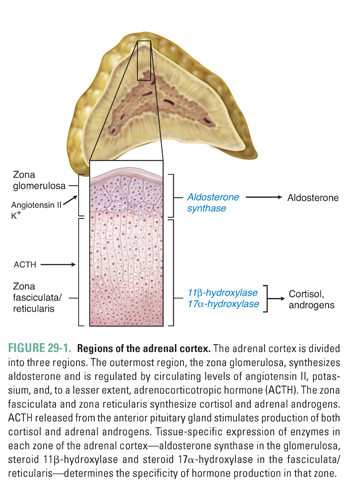
Through its mineralocorticoid, glucocorticoid, and adrenal androgen products, the adrenal cortex plays a role in diverse aspects of homeostasis. The following discussion considers the physiology, pathophysiology, and pharmacology of each class of adrenal hormones. The glucocorticoids are discussed first, followed by the mineralocorticoids and the adrenal androgens.

Cortisol, the endogenous glucocorticoid, is synthesized from cholesterol. Its synthesis begins with the rate-limiting conversion of cholesterol to pregnenolone, a reaction catalyzed by side-chain cleavage enzyme (Fig. 29-2). This first step converts the 27-carbon cholesterol into a 21-carbon precursor common to all adrenocortical hormones. From this precursor, steroid metabolism can proceed down three different pathways to generate mineralocorticoids, glucocorticoids, or adrenal androgens.
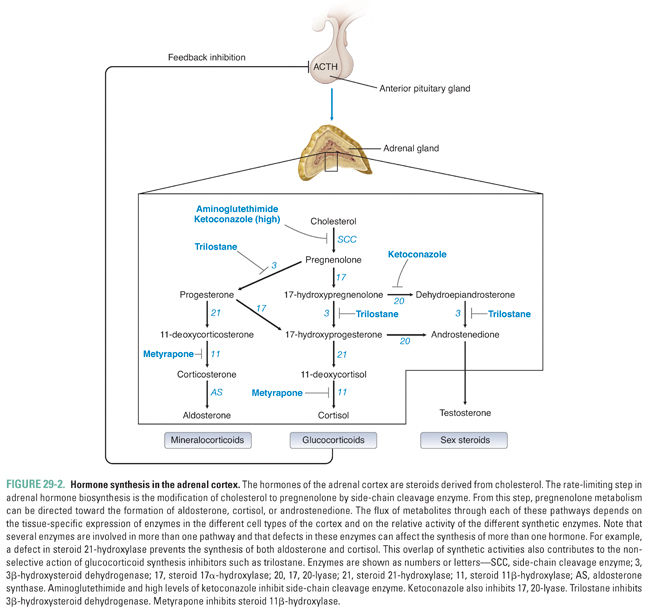
An oxidase enzyme catalyzes each step in the pathway of adrenocortical hormone synthesis. The oxidase enzymes are mitochondrial cytochromes, similar to the liver cytochrome P450 oxidase system. Tissue-specific expression of particular oxidase enzymes in each zone of the adrenal cortex provides the biochemical basis for the differences among the hormonal end products of the different zones of the cortex. For example, the zona fasciculata synthesizes cortisol but not aldosterone or androgens (Fig. 29-1). This is because enzymes required uniquely for cortisol synthesis, such as steroid 11β-hydroxylase, are expressed in the zona fasciculata, whereas enzymes required for aldosterone and androgen synthesis are not. The human zona glomerulosa does not express steroid 17α-hydroxylase, which is required for production of cortisol and androgens but is not required for production of aldosterone (Fig. 29-2).
Approximately 90% of circulating cortisol is bound to plasma proteins, the most important of which are corticosteroid-binding globulin (CBG, also referred to as transcortin) and albumin. CBG has high affinity for cortisol but low overall capacity, whereas albumin has low cortisol affinity but high overall capacity. Only molecules of cortisol that are unbound to protein (the so-called free fraction) are bioavailable, that is, available to diffuse through plasma membranes into cells. Thus, the affinity and capacity of plasma binding proteins regulate the availability of active hormone and, consequently, hormone activity.
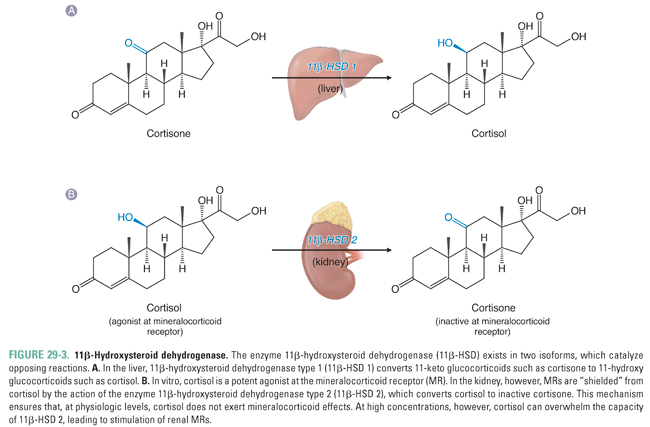
The liver and kidneys are the primary sites of peripheral cortisol metabolism. Through reduction and subsequent conjugation to glucuronic acid, the liver is responsible for inactivating cortisol in the plasma. The conjugation reaction makes cortisol more water soluble, thus enabling renal excretion. Importantly, the liver and kidneys express different isoforms of the enzyme 11β-hydroxysteroid dehydrogenase, a regulator of cortisol activity. The two isoforms catalyze opposing reactions. In distal collecting duct cells of the kidney, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD 2) converts cortisol to the biologically inactive compound cortisone, which (unlike cortisol) does not bind to the mineralocorticoid receptor (see below; Fig. 29-3B). Expression of 11β-HSD 2 protects the mineralocorticoid receptor from activation by cortisol in a variety of cell types, including endothelial cells and vascular smooth muscle cells. In contrast, cortisone can be converted back to cortisol (also referred to as hydrocortisone) in the liver by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD 1, Fig. 29-3A). The interplay between these opposing reactions determines overall glucocorticoid activity. In addition, as discussed below, the activity of these enzymes is important in glucocorticoid pharmacology.
Like other steroid hormones, unbound cortisol diffuses across the plasma membrane into the cytosol of target cells, where the hormone binds to a cytosolic receptor. There are two types of glucocorticoid receptors: the Type I (mineralocorticoid) and Type II glucocorticoid receptors. The Type I receptor is expressed in the organs of excretion (kidney, colon, salivary glands, sweat glands) and other tissues including the hippocampus, vasculature, heart, adipose tissue, and peripheral blood cells. The Type II receptor has a broader tissue distribution. The Type I glucocorticoid receptor is synonymous with the mineralocorticoid receptor. The nomenclature is unfortunate, and this chapter hereafter refers to the Type I receptor as the “mineralocorticoid receptor.”
Once cortisol binds to its cytosolic receptor and forms a hormone–receptor complex, the complex is transported into the nucleus. In the case of cortisol, a homodimerized hormone–receptor complex binds to gene promoter elements referred to as glucocorticoid response elements (GREs) or negative GREs, which enhance or inhibit the expression of specific genes, respectively. The glucocorticoid receptor also regulates transcription through direct and indirect interaction with co-activator molecules. Cortisol has profound effects on mRNA expression; about 10% of all human genes are estimated to contain GREs. Because the expression of such a large number of genes is affected by activation of GREs, cortisol has physiologic actions in most tissues. These actions can be divided generally into metabolic effects and anti-inflammatory effects.
Metabolic effects of cortisol increase nutrient availability by raising blood glucose, amino acid, and triglyceride levels. Cortisol increases blood glucose by antagonizing insulin action and by promoting gluconeogenesis in the fasting state. Cortisol also increases muscle protein catabolism, leading to release of amino acids that can be utilized by the liver as fuels for gluconeogenesis. By potentiating growth hormone action on adipocytes, cortisol increases the activity of hormone-sensitive lipase and the subsequent release of free fatty acids (lipolysis). Free fatty acids further increase insulin resistance. Cortisol levels increase as a component of stress responses induced by a wide range of stimuli, such as vigorous exercise, psychological stress, acute trauma, surgery, fear, severe infection, hypoglycemia, and pain. By increasing blood glucose, the physiologic effects of glucocorticoids maintain energy homeostasis during the stress response, thus ensuring that critical organs such as the brain continue to receive nutrients.
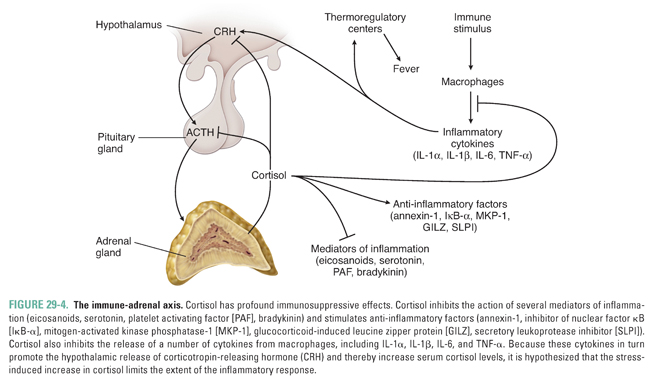
Cortisol also has multiple anti-inflammatory actions. Cortisol negatively regulates cytokine release from cells of the immune system by inhibiting nuclear factor κB (NF-κB); this action may be an important mechanism to limit the extent of immune responses and to regulate the inflammatory response. In turn, certain cytokines, including IL-1, IL-2, IL-6, and TNF-α, can stimulate hypothalamic release of CRH, which stimulates ACTH and cortisol release. This series of stimulatory and inhibitory effects creates a feedback loop in which inflammatory cytokines and cortisol are coordinately regulated to control immune and inflammatory responses (Fig. 29-4). Glucocorticoid-mediated suppression of the inflammatory response also has important pharmacologic implications for clinical conditions such as organ transplantation, rheumatoid arthritis, and asthma. Indeed, the introductory case demonstrates that glucocorticoids are an effective therapy for asthma. The exact mechanisms by which glucocorticoids act to ameliorate the symptoms of asthma are unknown but are thought to be related to the ability of glucocorticoids to reduce inflammation in the airways (see below and Chapter 48, Integrative Inflammation Pharmacology: Asthma).
The hypothalamic-pituitary unit coordinates the production of cortisol (refer to Chapter 27 for an overview). In response to central circadian rhythms and to stress, neurons in the paraventricular nucleus of the hypothalamus synthesize and secrete corticotropin-releasing hormone (CRH), a peptide hormone. CRH then travels through the hypothalamic-pituitary portal system and binds to G protein-coupled receptors on the surface of corticotroph cells in the anterior pituitary gland. CRH binding stimulates the corticotrophs to synthesize proopiomelanocortin (POMC), a precursor polypeptide that is cleaved into multiple peptide hormones including ACTH. Neurons in the paraventricular nucleus can also respond to stress by synthesizing and secreting arginine vasopressin. This vasopressin is released into the hypothalamic-pituitary portal system together with CRH, and it synergizes with CRH to increase the release of ACTH by the anterior pituitary gland. Interestingly, the stress-responsive parvocellular neurons that secrete CRH and vasopressin into the hypothalamic-pituitary portal system are different from the osmolality-responsive magnocellular neurons that synthesize vasopressin and transport this hormone to the posterior pituitary gland (see Chapter 27), even though both types of neurons are located in the paraventricular nucleus of the hypothalamus.
Proteolytic cleavage of POMC yields not only ACTH but also γ-melanocyte-stimulating hormone (MSH), lipotropin, and β-endorphin. MSH binds to receptors on skin melanocytes, promoting melanogenesis and thereby increasing skin pigmentation. Because of the similarities between the ACTH and MSH peptide sequences, high concentrations of ACTH can also bind to and activate MSH receptors. This action becomes apparent in primary hypoadrenalism (see below), in which increased ACTH levels result in increased skin pigmentation. The role of lipotropin in human physiology is uncertain but is thought to involve control of lipolysis. β-Endorphin is an endogenous opioid that is important for pain modulation and for regulation of reproductive physiology.
Because steroid hormones are able to diffuse freely across cell membranes and because the adrenal gland stores little cortisol, ACTH regulates cortisol production by promoting synthesis of the hormone. ACTH also has a trophic effect on the zona fasciculata and zona reticularis of the adrenal cortex, and hypertrophy of the cortex can occur in response to chronically elevated levels of ACTH.
As in other endocrine axes, the hormone (cortisol) produced by the target organ (adrenal cortex) exerts negative feedback regulation at the level of both the hypothalamus and the anterior pituitary gland. High cortisol levels decrease both synthesis and release of CRH and ACTH. Because ACTH has important trophic effects on the adrenal cortex, the absence of ACTH leads to atrophy of the cortisol-producing zona fasciculata and the androgen-producing zona reticularis. However, the aldosterone-producing zona glomerulosa cells continue to function in the absence of ACTH because angiotensin II and blood potassium continue to stimulate the production of aldosterone.
Diseases affecting glucocorticoid physiology can be divided into disorders of hormone deficiency and disorders of hormone excess. Addison’s disease is the classic example of adrenocortical insufficiency, while Cushing’s syndrome exemplifies cortisol excess.
Addison’s disease is an example of a primary adrenal insufficiency in which the adrenal cortex is selectively destroyed, most commonly due to a T cell-mediated autoimmune reaction but alternatively due to infection, infiltration, cancer, or hemorrhage. Destruction of the cortex results in decreased synthesis of all classes of adrenocortical hormones. By comparison, secondary adrenal insufficiency is caused by hypothalamic or pituitary disorders or by prolonged administration of exogenous glucocorticoids. In secondary adrenal insufficiency, the decrease in ACTH levels causes decreased synthesis of sex hormones and cortisol but does not alter aldosterone synthesis (see earlier discussion).
Regardless of the underlying cause, adrenal insufficiency has serious consequences and can be life threatening if left untreated in the setting of stress. Patients with adrenal insufficiency frequently experience fatigue, loss of appetite, weight loss, dizziness on standing, and nausea. Hyperkalemia is common in primary adrenal insufficiency due to the lack of aldosterone. If adrenal insufficiency is the result of high-dose, prolonged therapy with exogenous glucocorticoids, then the dose of glucocorticoid should be tapered slowly to allow the hypothalamic-pituitary-adrenal (HPA) axis time to regain full activity. Importantly, it can take up to 1 year for the HPA axis to recover function after discontinuation of exogenous glucocorticoid treatment.
In the introductory case, Johnny was switched from an oral glucocorticoid to an inhaled glucocorticoid that delivered a much lower systemic concentration of glucocorticoid. His adrenal cortex had atrophied because he had been maintained for 2 years on chronically high doses of prednisone; therefore, he was unable to produce a sufficient amount of cortisol in response to the stress of a respiratory infection. As a result, he arrived in the emergency department with acute adrenal insufficiency and required intravenous therapy with saline and hydrocortisone.
Cushing’s syndrome refers to a number of underlying pathophysiologies, all of which increase cortisol production. The term Cushing’s disease is reserved for ACTH-secreting pituitary adenomas that lead to increased cortisol production (Fig. 27-5C). Other causes of Cushing’s syndrome include ectopic secretion of ACTH, most commonly by small cell carcinomas of the lung (Fig. 27-5D), and (rarely) ectopic CRH production. Cushing’s syndrome can also result from cortisol-secreting tumors (adenoma or carcinoma) of the adrenal cortex (Fig. 27-5B). However, iatrogenic Cushing’s syndrome—secondary to pharmacologic treatment with exogenous glucocorticoids—is by far the most common cause of Cushing’s syndrome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


