Pharmacology of GABAergic and Glutamatergic Neurotransmission
Stuart A. Forman, Hua-Jun Feng, Janet Chou, Jianren Mao, and Eng H. Lo
PHARMACOLOGIC CLASSES AND AGENTS AFFECTING GABAERGIC NEUROTRANSMISSION |
PATHOPHYSIOLOGY AND PHARMACOLOGY OF GLUTAMATERGIC NEUROTRANSMISSION |

Inhibitory and excitatory neurotransmitters regulate almost every behavioral process, including consciousness, sleep, learning, memory, and all sensations. Inhibitory and excitatory neurotransmitters are also implicated in pathologic processes such as epilepsy and the neurotoxicity associated with stroke. The interactions among ion channels, the receptors that regulate these channels, and amino acid neurotransmitters in the central nervous system (CNS) constitute the molecular basis for these processes. This chapter discusses the physiology, pathophysiology, and pharmacology of γ-aminobutyric acid (GABA) and glutamate neurotransmission. Together, these molecules are the two most important amino acid neurotransmitters in the CNS.
 SB, a 70-year-old man, is having trouble sleeping. He recalls that his sister has been prescribed phenobarbital, a barbiturate, to control her epileptic seizures, and that barbiturates are sometimes also prescribed as sleeping pills. He decides to take “just a few” with some alcohol to help him sleep. Shortly afterward, SB is rushed to the emergency department after his sister finds him minimally responsive. On examination, he is difficult to arouse and dysarthric, with an unsteady gait and impaired attention and memory. His respiratory rate is approximately six shallow breaths per minute. The patient is subsequently intubated to protect him from aspirating gastric contents. Activated charcoal is administered through a nasogastric tube to limit further absorption of phenobarbital. He also receives intravenous sodium bicarbonate to alkalinize his urine to a pH of 7.5 to facilitate renal drug excretion. Three days later, he has recovered sufficiently to return home.
SB, a 70-year-old man, is having trouble sleeping. He recalls that his sister has been prescribed phenobarbital, a barbiturate, to control her epileptic seizures, and that barbiturates are sometimes also prescribed as sleeping pills. He decides to take “just a few” with some alcohol to help him sleep. Shortly afterward, SB is rushed to the emergency department after his sister finds him minimally responsive. On examination, he is difficult to arouse and dysarthric, with an unsteady gait and impaired attention and memory. His respiratory rate is approximately six shallow breaths per minute. The patient is subsequently intubated to protect him from aspirating gastric contents. Activated charcoal is administered through a nasogastric tube to limit further absorption of phenobarbital. He also receives intravenous sodium bicarbonate to alkalinize his urine to a pH of 7.5 to facilitate renal drug excretion. Three days later, he has recovered sufficiently to return home.
Questions
1. What are the signs of barbiturate poisoning, and how are these signs explained by the drugs’ mechanism of action?
2. How do barbiturates act to control epileptic seizures and to induce sleep?
3. How does the patient’s age affect the extent of CNS depression caused by barbiturates?
4. What is the interaction of barbiturates and ethanol that results in profound CNS and respiratory depression?
 OVERVIEW OF GABAERGIC AND GLUTAMATERGIC NEUROTRANSMISSION
OVERVIEW OF GABAERGIC AND GLUTAMATERGIC NEUROTRANSMISSION
The CNS has high concentrations of certain amino acids that bind to postsynaptic receptors and thereby act as inhibitory or excitatory neurotransmitters. Of the two main classes of neuroactive amino acids, γ-aminobutyric acid (GABA) is the major inhibitory amino acid, and glutamate is the primary excitatory amino acid.
Amino acid neurotransmitters elicit inhibitory or excitatory responses by altering the conductance of one or more ion-selective channels. Inhibitory neurotransmitters induce a net outward current, generally hyperpolarizing the membrane. For example, inhibitory neurotransmitters may open K+ channels or Cl− channels to induce K+ efflux or Cl− influx, respectively. Either type of ion movement—the loss of intracellular cations or the gain of intracellular anions—results in membrane hyperpolarization and decreased membrane resistance (Fig. 13-1), respectively moving the membrane potential further below its threshold value and reducing the ability of inward currents to depolarize the membrane.
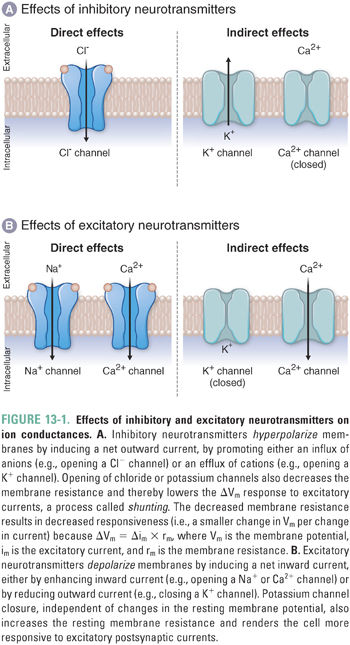
Excitatory amino acid neurotransmitters induce a net inward current, generally depolarizing the membrane. For example, excitatory neurotransmitters may open cation-specific channels, such as a sodium channel, and thereby cause a net influx of sodium ions that depolarizes the membrane. An excitatory (depolarizing) response could also result if a neurotransmitter closes potassium “leak channels” to reduce the outward flow of potassium ions and thereby depolarize the membrane (see Chapter 8, Principles of Cellular Excitability and Electrochemical Transmission).
Pharmacologic agents that modulate GABAergic neurotransmission, including benzodiazepines and barbiturates, are drug classes of major clinical importance. In comparison, pharmacologic agents targeting glutamatergic neurotransmission remain largely experimental. The balance of the discussion is, therefore, addressed at GABAergic physiology and pharmacology; the pathophysiology and pharmacology of glutamatergic neurotransmission is discussed at the end of the chapter.
 PHYSIOLOGY OF GABAERGIC NEUROTRANSMISSION
PHYSIOLOGY OF GABAERGIC NEUROTRANSMISSION
GABA functions as the primary inhibitory neurotransmitter in the mature mammalian CNS. The cell membranes of most vertebrate CNS neurons and astrocytes express GABA receptors, which decrease neuronal excitability through several mechanisms. Because of their widespread distribution, GABA receptors influence many neural circuits and functions. Drugs that modulate GABA receptors affect arousal and attention, memory formation, anxiety, sleep, and muscle tone. Modulation of GABA signaling is also an important mechanism for treatment of focal or widespread neuronal hyperactivity in epilepsy.
The synthesis of GABA is mediated by glutamic acid decarboxylase (GAD), which catalyzes the decarboxylation of glutamate to GABA in GABAergic nerve terminals (Fig. 13-2A). Thus, the amount of GABA in brain tissue correlates with the amount of functional GAD. GAD requires pyridoxal phosphate (vitamin B6) as a cofactor. GABA is packaged into presynaptic vesicles by a vesicular transporter (VGAT). (The same transporter, VGAT, is also expressed in nerve terminals that release glycine, another inhibitory neurotransmitter.) In response to an action potential and the presynaptic elevation of intracellular Ca2+, GABA is released into the synaptic cleft by fusion of GABA-containing vesicles with the presynaptic membrane.
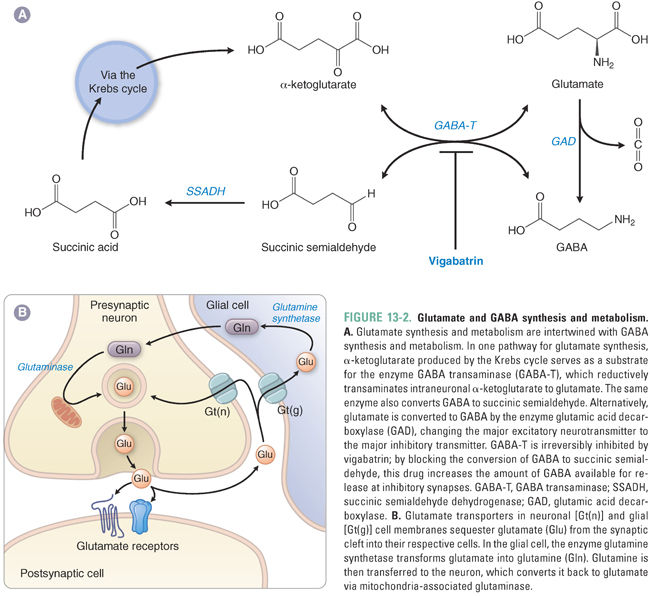
Termination of GABA action at the synapse depends on the removal of GABA from the extracellular space. Neurons and glia take up GABA via specific GABA transporters (GATs) in the cell membrane. Four GATs have been identified, GAT-1 through GAT-4, each with a characteristic distribution in the CNS. Within cells, the widely distributed mitochondrial enzyme GABA transaminase (GABA-T) catalyzes the conversion of GABA to succinic semialdehyde (SSA), which is oxidized to succinic acid by SSA dehydrogenase. Succinic acid enters the Krebs cycle to become α-ketoglutarate, and GABA-T regenerates glutamate from α-ketoglutarate (Fig. 13-2A).
GABA mediates its neurophysiologic effects by binding to GABA receptors. There are two types of GABA receptors. Ionotropic GABA receptors (GABAA and GABAC) are multisubunit membrane proteins that bind GABA and open an intrinsic chloride ion channel. Metabotropic GABA receptors (GABAB) are heterodimeric G protein-coupled receptors that activate neuronal potassium channels through second messengers.
Ionotropic GABA Receptors: GABAA and GABAC
The most abundant GABA receptors in the CNS are ionotropic GABAA receptors, which are members of the superfamily of fast neurotransmitter-gated ion channels. This superfamily includes peripheral and neuronal nicotinic acetylcholine receptors (nAChRs), serotonin type 3A/B (5-HT3A/B) receptors, and glycine receptors. Like other members of this superfamily, GABAA receptors are pentameric transmembrane glycoproteins that are assembled to form a central ion pore surrounded by five subunits, each of which has four membrane-spanning domains (Fig. 13-3A). Sixteen different GABAA receptor subunits are currently known (α1–6, β1–3, γ1–3, δ, ε, π, and θ). The number of pentameric ion channels that could be formed by potential combinations of 16 subunits is very large, but only about 20 different subunit combinations have been identified in native GABAA receptors. Importantly, receptors containing different subunit combinations display distinct distributions at the cellular and tissue levels, and evidence is accumulating that different GABAA receptor subtypes play distinct roles in specific neural circuits. Most synaptic GABAA receptors consist of two α subunits, two β subunits, and one γ subunit. “Extrasynaptic” GABAA receptors have also been identified on dendrites, axons, and neuronal cell bodies. These often contain α5 subunits together with a γ subunit or a δ subunit.
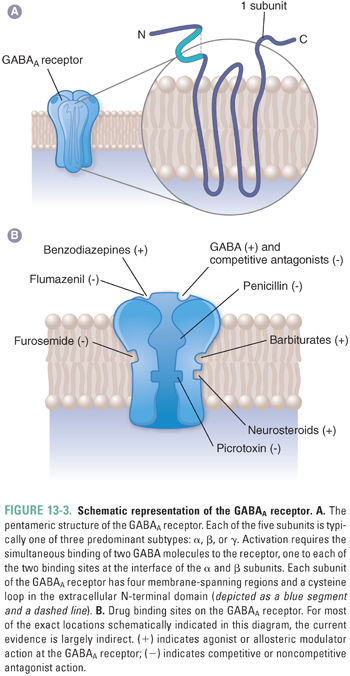
The five subunits of GABAA receptors surround a central chloride-selective ion pore that opens in the presence of GABA. GABA and other agonists bind to two sites, which are located in extracellular portions of the receptor-channel complex at the interface between the α and β subunits. GABAA receptors also contain a number of modulatory sites where other endogenous ligands and/or drugs bind (Fig. 13-3B). In many cases, the presence of these sites and the impact of ligand binding depend on the receptor subunit composition.
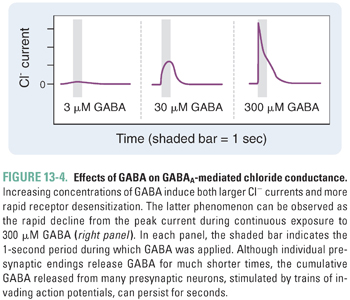
GABAA receptor-channel activation follows the binding of two molecules of GABA, one to each of the receptor’s agonist sites (Fig. 13-3). Fast inhibitory postsynaptic currents (IPSCs) are responses activated by very brief (high-frequency) bursts of GABA release at synapses. Uptake by GAT removes GABA from the synapse in less than 1 ms; IPSCs deactivate over about 12–20 ms, a rate that is determined by both closure of the GABAA receptor ion channel and dissociation of GABA from the receptor. Prolonged occupation of the agonist sites by GABA also leads to GABAA receptor desensitization, a transition to an inactive agonist-bound state (Fig. 13-4). During burst (or “phasic”) firing, the presynaptic nerve membrane releases “quanta” (~1 mM) of GABA by exocytosis of synaptic vesicles, resulting in transient, large-amplitude inhibitory postsynaptic potentials (IPSPs). The diffusion of GABA away from synaptic clefts also results in low concentrations of GABA (up to a few μM) in cerebrospinal fluid and interstitial spaces. Thus, GABA also activates extrasynaptic GABAA receptors and thereby induces a baseline “tonic” inhibitory current in many neurons.
Because the internal chloride concentration [Cl−]in of mature neurons is lower than the extracellular Cl− concentration [Cl−]out, activation of chloride-selective channels (increasing conductance) shifts the neuronal transmembrane voltage toward the Cl− equilibrium potential (ECl ~ −70 mV). This Cl− flux hyperpolarizes or stabilizes the postsynaptic cell near its normal resting membrane potential (Vm ~ −65 mV), reducing the likelihood that excitatory stimuli will initiate action potentials. Open Cl− channels attenuate the change in membrane potential caused by excitatory synaptic currents, an effect called shunting. This is the molecular explanation for the inhibitory effects of GABA signaling via GABAA receptors.
In mature neurons, the chloride gradients are maintained by a potassium-chloride (K+-Cl−) co-transporter (KCC2). In immature neurons of fetal and neonatal brain, the Cl− gradient may be reversed due to a difference in the Cl− transporting pumps (sodium-potassium-chloride [Na+-K+-Cl−] co-transporter, NKCC1). In neurons expressing NKCC1, GABAA receptors may mediate the outward flow of Cl− ions, constituting an inward current and thus depolarizing the neuron. Thus, drugs that activate or potentiate GABAA receptors may have an excitatory action during brain development rather than the inhibitory effect they have in mature neurons.
The molecular role of GABAA receptors in neurons is consistent with their known physiologic roles in CNS disease and with their pharmacology. Drugs that inhibit GABAA receptors produce seizures in animals, and mutations in GABAA receptor subunits that impair activation at the molecular level are associated with inherited human epilepsy syndromes. Conversely, endogenous or exogenous substances that enhance the activation of GABAA receptors reduce neuronal excitability and may impair numerous CNS functions. Recent evidence indicates that GABAA receptors are also expressed in peripheral tissues such as airway epithelium. Activation of these receptors may enhance smooth muscle relaxation (bronchodilation) and could represent a future therapy for asthma.
Certain endogenous substances such as taurine and steroids (also known as neurosteroids) allosterically modulate GABAA receptor activity. The steroid hormones deoxycorticosterone and progesterone are metabolized in the brain to produce pregnenolone, dehydroepiandrosterone (DHEA), 5α-dihydrodeoxycorticosterone (DHDOC), 5α-tetrahydrodeoxycorticosterone (THDOC), and allopregnanolone. Neurosteroids do not act through nuclear receptors like most steroid hormones; instead, they alter GABAA receptor function by binding to allosteric sites on the receptor protein, causing increased GABAA receptor activation. DHDOC and THDOC are thought to modulate brain activity during stress. Menstrual variations in allopregnanolone, a metabolite of progesterone, contribute to perimenstrual (catamenial) epilepsy. Sulfation of pregnenolone and DHEA results in neurosteroids that inhibit GABAA receptors. Another endogenous substance that enhances GABAA receptor activity is oleamide, a fatty acid amide found in the cerebrospinal fluid of sleep-deprived animals. Injection of oleamide into normal animals induces sleep, in part through potentiation of GABAA receptors.
Another group of ionotropic GABA receptors, GABAC, are formed by three subunits that are not found in GABAA receptors (ρ1–3). GABAC receptors are also pentameric ligand-gated chloride channels, but their distribution in the CNS is restricted primarily to the retina. GABAC receptors display distinct pharmacologic properties that differ from those of most GABAA receptors. No drugs currently in use target GABAC receptors.
Metabotropic GABA Receptors: GABAB
GABAB receptors are G protein-coupled receptors that are expressed at lower levels than GABAA receptors and are found principally in the spinal cord (Fig. 13-5). They function as a receptor complex composed of principal GABAB1 and GABAB2 subunits and auxiliary potassium channel tetramerization domain (KCTD) subunits. The principal subunits determine the cell surface expression and distribution of the receptors to axonal and dendritic sites. The auxiliary subunits influence the agonist potency and the kinetics of the receptor response. The GABAB receptor interacts with heterotrimeric G proteins, leading to the dissociation of their βγ subunit, which directly activates K+ channels and inhibits the opening of voltage-gated Ca2+ channels (Fig. 13-5). GABAB receptor activation also leads to inhibition of adenylyl cyclase and concomitant reduction in cAMP, but this seems to have only minor effects on cellular excitability. At GABAergic synapses, GABAB receptors are expressed both presynaptically and postsynaptically. Presynaptic “autoreceptors” modulate neurotransmitter release by reducing Ca2+ influx, while postsynaptic GABAB receptors produce slow IPSPs through activation of G protein-activated “inward rectifier” K+ channels (GIRKs). The slower rates of activation and deactivation of GABAB currents in comparison to GABAA currents are due to the relatively slow second messenger signal transduction mechanisms.
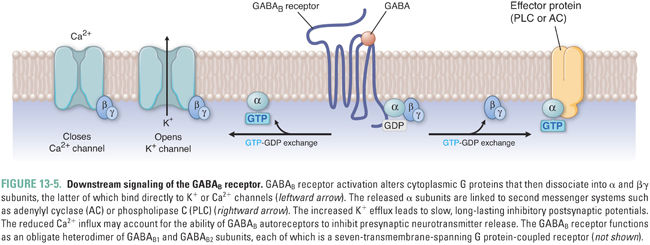
Because K+ has an equilibrium potential near −90 mV, activation of K+ channels by GABAB-coupled G proteins inhibits neuronal firing. Thus, like increased Cl− conductance, increased K+ conductance drives the neuronal transmembrane voltage toward “resting” potentials, reduces the frequency of action potential initiation, and shunts excitatory currents.
 PHARMACOLOGIC CLASSES AND AGENTS AFFECTING GABAERGIC NEUROTRANSMISSION
PHARMACOLOGIC CLASSES AND AGENTS AFFECTING GABAERGIC NEUROTRANSMISSION
Pharmacologic agents acting on GABAergic neurotransmission affect GABA metabolism, transport, or receptor activity. The majority of pharmacologic agents affecting GABAergic neurotransmission act on the ionotropic GABAA receptor. Several drug classes can regulate GABAA receptors by interacting with the GABA binding sites or with allosteric sites (Fig. 13-3). Therapeutic agents that activate GABAA receptors are used for sedation, anxiolysis, hypnosis (general anesthesia), neuroprotection following stroke or head trauma, and control of epilepsy. Other agents that modulate GABAergic transmission are used only for experimental purposes (Table 13-1).
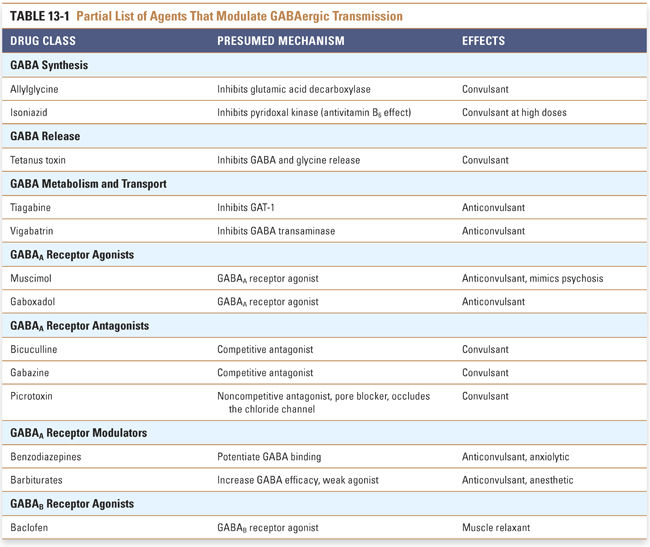
Inhibitors of GABA Metabolism and Transport
Tiagabine is a competitive inhibitor of the GABA transporters in neurons and glia, where it may act selectively on GAT-1. Epilepsy is the major clinical indication for tiagabine. By inhibiting GABA reuptake, tiagabine increases both synaptic and extrasynaptic GABA concentrations. The result is nonspecific agonism of both ionotropic and metabotropic GABA receptors, with the major effects at GABAA receptors.
Tiagabine is an oral medication that is rapidly absorbed with 90% bioavailability and is highly protein bound. Metabolism is hepatic, primarily by CYP3A4. Tiagabine does not induce cytochrome P450 enzymes, but its metabolism is influenced by concomitant use of either inducers or inhibitors of CYP3A4. Adverse effects of tiagabine are those of high GABA activity, including confusion, sedation, amnesia, and ataxia. Tiagabine potentiates the action of GABAA receptor modulators such as ethanol, benzodiazepines, and barbiturates.
γ-Vinyl GABA (vigabatrin) is a “suicide inhibitor” of GABA transaminase (GABA-T, see Fig. 13-2). Administration of this drug blocks the conversion of GABA to succinic semialdehyde, resulting in high intracellular GABA concentrations and increased synaptic GABA release. Like the effect of tiagabine, enhancement of GABA receptor function by γ-vinyl GABA is not selective because GABA concentrations are increased wherever GABA is released, including the retina.
Vigabatrin is used in the treatment of epilepsy, and it is being investigated for treatment of drug addiction, panic disorder, and obsessive-compulsive disorder. Adverse effects of γ-vinyl GABA include drowsiness, confusion, and headache. The drug has been reported to cause bilateral visual field defects associated with irreversible diffuse atrophy of the peripheral retinal nerve fiber layer. This appears to result from accumulation of the drug in retinal nerves.
GABAA Receptor Agonists and Antagonists
Agonists such as muscimol and gaboxadol activate the GABAA receptor by binding directly to the GABA binding site. Muscimol, first derived from hallucinogenic Amanita muscaria mushrooms, is a full agonist at many GABAA receptor subtypes and is used primarily as a research tool. Purified muscimol (as well as other GABAA receptor agonists) does not induce hallucinations, which are probably caused by other factors from Amanita muscaria. Gaboxadol at high concentrations is a partial agonist at synaptic GABAA receptors; at low concentrations, gaboxadol selectively activates extrasynaptic receptors containing α4, β3, and δ subunits. Gaboxadol was initially approved for treatment of epilepsy and anxiety, but therapeutic doses were associated with ataxia and sedation. Lower gaboxadol doses, which activate extrasynaptic receptors, induce slow-wave sleep in laboratory animals. Human trials of gaboxadol for treatment of insomnia were halted in 2007 due to concerns about adverse effects such as hallucinations, disorientation, sleepwalking, and sleep-driving.
Bicuculline and gabazine are competitive antagonists that bind at the GABA sites on GABAA receptors. Picrotoxin is a noncompetitive inhibitor of GABAA receptors that blocks the ion pore. All of these GABAA antagonists induce seizures and are used exclusively for research; they also illustrate the importance of the tonic activity of GABAA receptors in maintaining a state of relatively normal excitability in the CNS.
Benzodiazepines and barbiturates are modulators of GABAA receptors that act at allosteric binding sites to enhance GABAergic neurotransmission (Fig. 13-3B). Benzodiazepines have sedative, hypnotic, muscle relaxant, amnestic, and anxiolytic effects. At high doses, benzodiazepines can cause hypnosis and stupor. However, when used alone, these drugs rarely cause fatal CNS depression. Barbiturates constitute a large group of drugs that were first introduced in the mid-twentieth century and continue to be used, albeit with diminishing frequency, for control of epilepsy, as general anesthetic induction agents, and for control of intracranial hypertension.
Benzodiazepines are high-affinity, highly selective drugs that bind at a single site on GABAA receptors containing α1, α2, α3, or α5 subunits and a γ subunit. In molecular studies, benzodiazepine potency correlates with hydrophobicity. However, benzodiazepines are highly bound to plasma proteins such as albumin, and hydrophobicity enhances protein binding and thereby reduces the drugs’ free concentration and transport across the blood–brain barrier. Therefore, highly protein-bound benzodiazepines may appear less potent in vivo even though they display higher potency in molecular studies. Furthermore, in clinical states associated with low albumin, such as acute hemodilution or liver dysfunction, the clinical potency of benzodiazepines may be dramatically increased.
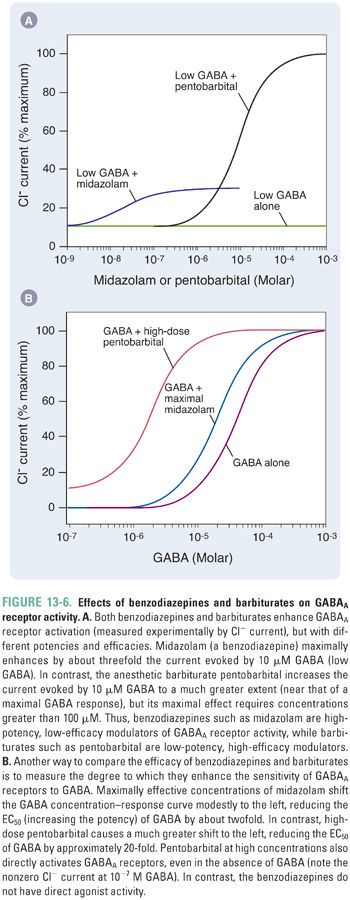
Benzodiazepines act as positive allosteric modulators by enhancing GABAA receptor channel gating in the presence of GABA (Fig. 13-6). Benzodiazepines increase the frequency of channel opening in the presence of low GABA concentrations, and, at GABA concentrations similar to those in synapses, receptor deactivation is slowed. Both actions result in a net increase in Cl− influx. In addition, GABAA receptors in the open state have a higher affinity for GABA than in the closed state, so the ability of benzodiazepines to favor channel openness results, secondarily, in an apparently higher agonist affinity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


