INTRODUCTION
Fungi are free-living microorganisms that exist as yeasts (single-cell, round fungi), molds (multicellular filamentous fungi), or a combination of the two (so-called dimorphic fungi). All fungi are eukaryotic organisms. Because of their phylogenetic similarity, fungi and humans have homologous metabolic pathways for energy production, protein synthesis, and cell division. Consequently, there is greater difficulty in developing selective antifungal agents than in developing selective antibacterial agents. The success of many antibacterial agents has resulted from the identification of unique molecular targets in bacteria, emphasizing the necessity for identifying unique fungal targets that can be exploited.
Certain patient populations are particularly susceptible to fungal infections (mycoses). These populations include surgical and intensive care unit (ICU) patients, patients with prostheses, and patients with compromised immune defenses. In the past three to four decades, the extensive use of broad-spectrum antibiotics, the wider use of long-term intravenous catheters, and infection with human immunodeficiency virus (HIV) have correlated with an increasing incidence of opportunistic and systemic mycoses. Additionally, the successes of organ transplantation, immunosuppressive therapy, and cancer chemotherapy have contributed to an increasing number of chronically immunosuppressed patients, who are particularly susceptible to fungal infections.
Traditionally, the diagnosis of fungal infections has relied on culture-based methods and direct examination of specimens under light microscopy. However, the indolent growth of fungi makes culturing inefficient, while direct microscopic examination may not be reliable or provide definitive speciation. These disadvantages have important clinical implications because prognosis often correlates inversely with the duration of time from clinical presentation to accurate diagnosis. Consequently, one major focus of modern mycology is the development of rapid, nonculture-based methods of early diagnosis. New diagnostic techniques rely on the polymerase chain reaction (PCR), western blot, antigen detection, and identification of fungal metabolites. Because many of these techniques are still investigational, they must be performed in parallel with traditional culture-based methods.
The treatment options for opportunistic and systemic fungal infections were once thought to be limited. These options are now expanding, however. Fungal processes that have been exploited in the development of antifungal agents include nucleic acid synthesis, mitosis, and membrane synthesis and stability. Traditional antifungal agents, such as azoles and polyenes, are directed against molecular targets involved in the synthesis and stability of the fungal membrane. The echinocandins, a relatively newer class of antifungal agents, target an enzyme complex involved in the synthesis of the fungal cell wall. As the emergence of resistant fungi increases, it will become increasingly important to identify and exploit new molecular targets for antifungal therapy.
 James F, a 31-year-old HIV-positive man, presents to his physician with a 3-week history of fever, cough, and chest pain after touring Southern California. His history is notable for past intravenous drug use. Clinical evaluation and chest x-ray reveal a left lower lobe infiltrate and left paratracheal adenopathy. Sputum cultures are positive for Coccidioides immitis, and blood tests are notable for an elevated titer of antibodies directed against this fungal pathogen. The physician makes a preliminary diagnosis of pulmonary coccidioidomycosis and prescribes a course of amphotericin B.
James F, a 31-year-old HIV-positive man, presents to his physician with a 3-week history of fever, cough, and chest pain after touring Southern California. His history is notable for past intravenous drug use. Clinical evaluation and chest x-ray reveal a left lower lobe infiltrate and left paratracheal adenopathy. Sputum cultures are positive for Coccidioides immitis, and blood tests are notable for an elevated titer of antibodies directed against this fungal pathogen. The physician makes a preliminary diagnosis of pulmonary coccidioidomycosis and prescribes a course of amphotericin B.
Over the next several days, however, Mr. F does not improve. He goes to the emergency department with fever, chills, sweats, cough, fatigue, and headaches. His temperature is 100°F, but he shows no evidence of meningitis or peripheral adenopathy. Lung examination reveals diffuse wheezing over the left lung fields, noted on both inspiration and expiration. Bronchoscopy shows narrowing of the tracheal lumen by numerous mucosal granulomas from the left mainstem bronchus to the level of the midtrachea. Fungal culture grows Coccidioides immitis, a definitive diagnosis of chronic pulmonary coccidioidomycosis is made, the granulomas are bronchoscopically removed, and amphotericin B is continued. A week later, Mr. F’s symptoms begin to subside, amphotericin B is discontinued, and a course of fluconazole is initiated.
Questions
1. What factors predisposed Mr. F to fungal infection?
2. What are the mechanisms of action of amphotericin B and fluconazole?
3. What adverse effects could Mr. F experience as a consequence of treatment with amphotericin B and fluconazole?
 BIOCHEMISTRY OF THE FUNGAL MEMBRANE AND CELL WALL
BIOCHEMISTRY OF THE FUNGAL MEMBRANE AND CELL WALL
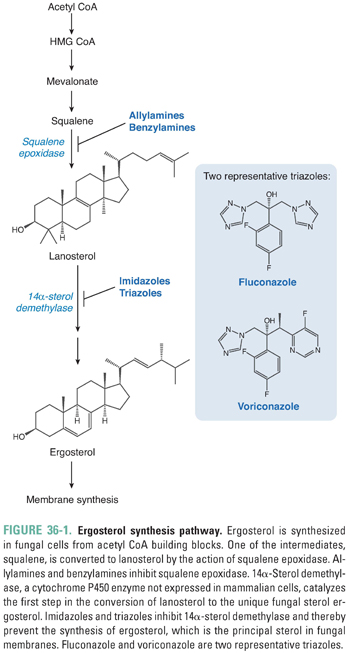
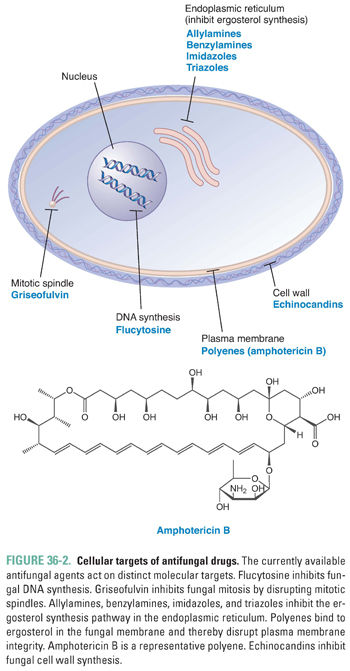
Although fungi have a cellular ultrastructure similar to that of animal cells, there are a number of unique biochemical differences that have been exploited in the development of antifungal drugs. To date, the most important biochemical difference lies in the principal sterol used to maintain plasma membrane structure and function. Mammalian cells use cholesterol for this purpose, whereas fungal cells use the structurally distinct sterol ergosterol. The biosynthesis of ergosterol involves a series of steps, two of which are targeted by currently available antifungal drugs (Fig. 36-1). The enzymes that catalyze ergosterol synthesis are localized in fungal microsomes, which contain an electron transport system nearly identical to that found in mammalian liver microsomes. The first targeted step, the conversion of squalene to lanosterol, is catalyzed by the enzyme squalene epoxidase. This enzyme is the molecular target of the allylamine and benzylamine antifungal agents. The fungus-specific cytochrome P450 enzyme 14α-sterol demethylase mediates the key reaction in the second targeted step, the conversion of lanosterol to ergosterol. Imidazole and triazole antifungal agents inhibit 14α-sterol demethylase. Therefore, allylamine, benzylamine, imidazole, and triazole antifungal agents all inhibit the biosynthesis of ergosterol. Because ergosterol is necessary for the maintenance of plasma membrane structure and function, these agents compromise fungal membrane integrity. Ergosterol synthesis inhibitors suppress fungal cell growth under most circumstances (fungistatic effect), although they can sometimes cause fungal cell death (fungicidal effect).
Fungal cells are surrounded by a cell wall, a rigid structure that has been studied intensively as a new and important target for antifungal therapy. The major components of the fungal cell wall are chitin, β-(1,3)-D-glucan, β-(1,6)-D-glucan, and cell wall glycoproteins (especially proteins containing complex mannose chains, or mannoproteins). Chitin is a linear polysaccharide consisting of more than 2,000 N-acetylglucosamine units joined by β-(1,4) linkages; these chains are bundled into microfibrils that form the fundamental scaffold of the cell wall. β-(1,3)-D-glucan and β-(1,6)-D-glucan, which are polymers of glucose units joined by β-(1,3) and β-(1,6) glycosidic linkages, respectively, are the most abundant components of the cell wall. These glucan polymers are covalently linked to the chitin scaffold. The cell wall glycoproteins comprise a diverse group of proteins that are noncovalently associated with other cell wall components or are covalently linked to chitin, glucan, or other cell wall proteins. Because mammalian cells do not have cell walls, drugs directed against the fungal cell wall would be expected to have a high therapeutic index. Echinocandin antifungal agents target β-(1,3)-D-glucan synthase, the enzyme that adds glucose residues from the donor molecule UDP-glucose to the growing polysaccharide chain. By inhibiting cell wall biosynthesis, echinocandins disrupt fungal cell wall integrity. Echinocandins often have fungicidal activity, although these agents are fungistatic under some circumstances (see “Suggested Reading”).
Fungal adhesion represents a third potential target for antifungal drugs. Adhesion to host cells is mediated by the binding of fungal adhesins to host cell receptors. In yeasts, for example, aspartyl proteases and phospholipases mediate adhesion. Compounds that block adhesive interactions between fungal cells and mammalian cells are currently under development.
 PATHOPHYSIOLOGY OF FUNGAL INFECTIONS
PATHOPHYSIOLOGY OF FUNGAL INFECTIONS
Mycoses (fungal infections) can be divided into superficial, cutaneous, subcutaneous, systemic or primary, and opportunistic infections. Few fungi possess sufficient virulence to be considered primary pathogens capable of initiating serious infections in immunocompetent hosts. However, immunocompromised hosts can develop serious systemic infections with fungi that are not pathogenic in normal individuals. In the introductory case, Mr. F’s HIV infection likely increased his risk of infection with Coccidioides immitis. Thus, the pathogenesis of fungal infections is based on the interplay between a host’s immune system and the pathogenicity of the particular fungal organism. Polymorphonuclear leukocytes, cell-mediated immunity, and humoral immunity are all important components of the host immune defense against fungal pathogens.
The pathogenesis of fungal infections is only partly understood, and different fungi possess distinct virulence factors that are unique to the pathogen. Adhesion is an initial step in the early stages of infection. Adhesion and localization can occur on skin, mucosal, and prosthetic device surfaces. For example, Candida species adhere to a variety of surfaces via a combination of specific ligand–receptor interactions as well as nonspecific forces such as van der Waals and electrostatic interactions. Virulent pathogens are subsequently able to invade the colonized surface and proliferate in deep tissue, sometimes reaching the systemic circulation. Systemic dissemination can be accelerated by local tissue injury, such as that caused by cancer chemotherapy, ischemia, or the presence of a prosthetic device. In addition, some pathogens secrete lytic enzymes to enable invasive growth and systemic dissemination. C. immitis breaches the respiratory mucosa by producing an alkaline proteinase capable of digesting structural proteins in lung tissue. C. immitis also produces a 36-kDa extracellular proteinase capable of degrading human elastin, collagen, immunoglobulins, and hemoglobin.
Fungal cell wall composition plays an important role in the pathogenesis of fungal infections. Pathogens such as Blastomyces dermatitidis, Histoplasma capsulatum, and Paracoccidioides brasiliensis modulate the complement of glycoproteins in their cell walls in response to host immune system interactions. For example, the cell wall of B. dermatitidis contains a 120-kDa glycoprotein, BAD-1 (formerly WI-1), which elicits a potent humoral and cellular immune response. Avirulent strains of B. dermatitidis have increased expression of BAD-1, which is recognized by the host immune system and leads to elimination of the pathogen through phagocytosis. In contrast, the cell wall of virulent strains of B. dermatitidis contains high levels of α-(1,3)-glucan, which is inversely correlated with the amount of BAD-1 detectable on the cell surface. It is speculated that the increased amount of α-(1,3)-glucan in the cell wall effectively masks the BAD-1 surface glycoprotein, thereby allowing the virulent strains to evade host immune detection and destruction.
The ability of a fungal pathogen to change from one morphotype to another is termed phenotype switching. By responding to changes in the microenvironment, Candida species are capable of undergoing yeast-to-hyphae transformation. The hyphal forms of Candida species possess a “sense of touch” that allows them to grow in crevices and pores, thereby increasing their infiltrative potential. Similarly, B. dermatitidis undergoes transformation from conidia (small, asexual reproductive structures) to the larger yeast forms. The larger forms offer an important survival advantage because they are capable of resisting the phagocytic action of neutrophils and macrophages.
 PHARMACOLOGIC CLASSES AND AGENTS
PHARMACOLOGIC CLASSES AND AGENTS
The ideal antifungal agent would possess four characteristics: broad spectrum of action against a variety of fungal pathogens, low drug toxicity, multiple routes of administration, and excellent penetration into the cerebrospinal fluid (CSF), urine, and bone. With the recent expansion in identifying novel targets of antifungal therapy, treatment options for superficial and deep fungal infections are improving. Some antifungal agents can be used to treat both superficial and deep mycoses (in some cases, using different formulations), while others are restricted to narrower indications. In this section, the currently available antifungal drugs are categorized according to their molecular targets and mechanisms of action. The primary molecular targets for antifungal therapy are enzymes and other molecules involved in fungal DNA synthesis, mitosis, plasma membrane synthesis, and cell wall synthesis (Fig. 36-2). Because the clinical trials used to support regulatory approval of new drugs often exclude children and women of childbearing potential (see Chapter 52, Clinical Drug Evaluation and Regulatory Approval), the safety of some of the newer antifungal agents is not precisely determined in these patient populations. The treating physician must therefore weigh the risks of treatment against the expected benefits.
Inhibitor of Fungal Nucleic Acid Synthesis: Flucytosine
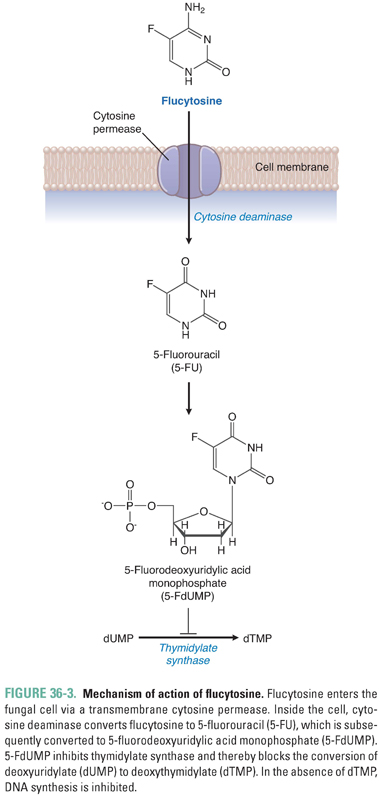
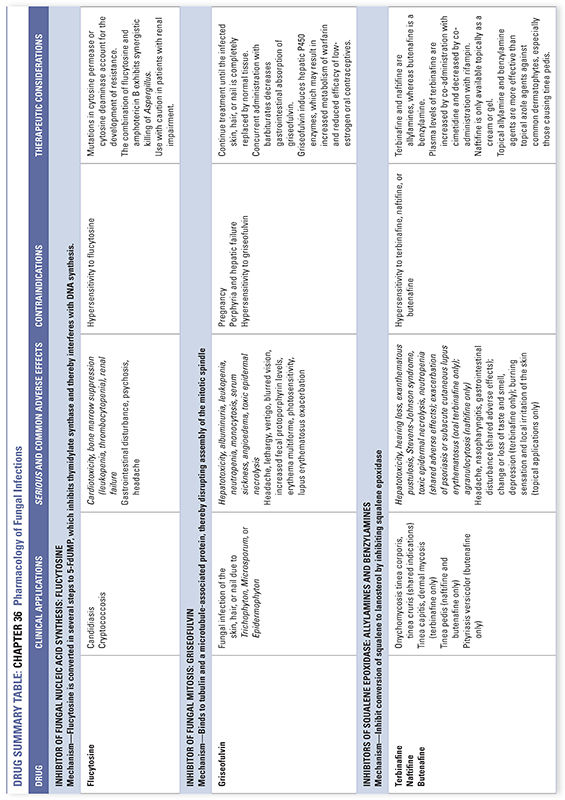
Flucytosine is the name of the fluorinated pyrimidine 5-fluorocytosine. Flucytosine is selectively taken up by fungal cells via cytosine-specific permeases that are expressed only in fungal membranes. Lacking these transporters, mammalian cells are protected. Inside the fungal cell, the enzyme cytosine deaminase converts flucytosine to 5-fluorouracil (5-FU). (5-FU is itself an antimetabolite that is used in cancer chemotherapy; see Chapter 39, Pharmacology of Cancer: Genome Synthesis, Stability, and Maintenance.) Subsequent reactions convert 5-FU to 5-fluorodeoxyuridylic acid monophosphate (5-FdUMP), which is a potent inhibitor of thymidylate synthase. Inhibition of thymidylate synthase results in inhibition of DNA synthesis and cell division (Fig. 36-3). Flucytosine appears to be fungistatic under most circumstances. Although mammalian cells lack cytosine-specific permeases and cytosine deaminase, fungi and bacteria in the intestine can convert flucytosine to 5-fluorouracil, which can cause adverse effects in host cells.
Flucytosine is typically used in combination with amphotericin B to treat systemic mycoses; when the drug is used as a single agent, resistance emerges rapidly due to mutations in fungal cytosine permease or cytosine deaminase. Although flucytosine has no intrinsic activity against Aspergillus, synergistic killing of Aspergillus by the combination of flucytosine and amphotericin B can be demonstrated experimentally. The mechanism of this synergistic interaction appears to involve enhancement of flucytosine uptake by fungal cells due to amphotericin-induced damage to the fungal plasma membrane. The spectrum of activity of flucytosine as a single agent is limited to candidiasis, cryptococcosis, and chromomycosis. Combination treatment with amphotericin B is recommended in acute cryptococcal meningitis in HIV-infected adults. A pharmacokinetic advantage of flucytosine is its large volume of distribution, with excellent penetration into the central nervous system (CNS), eyes, and urinary tract. Dose-dependent adverse effects include bone marrow suppression leading to leukopenia and thrombocytopenia, nausea, vomiting, diarrhea, and hepatic dysfunction. Flucytosine is contraindicated during pregnancy.
Inhibitor of Fungal Mitosis: Griseofulvin
Derived from Penicillium griseofulvum in the 1950s, griseofulvin inhibits fungal mitosis by binding to tubulin and a microtubule-associated protein and thereby disrupting assembly of the mitotic spindle. The drug is also reported to inhibit fungal RNA and DNA synthesis. Griseofulvin accumulates in keratin precursor cells and binds tightly to keratin in differentiated cells. The prolonged and tight association of griseofulvin with keratin allows new growth of skin, hair, or nail to be free of dermatophyte infection. Griseofulvin appears to be fungistatic under most circumstances.
The therapeutic use of oral griseofulvin is currently limited, due to the availability of topical antifungal medications as well as other oral antifungal agents with fewer adverse effects. Griseofulvin can be used to treat fungal infection of the skin, hair, and nail due to Trichophyton, Microsporum, and Epidermophyton. The drug is not effective against yeast (such as Pityrosporum) and dimorphic fungi. Doses should be taken at 6-hour intervals because blood levels of griseofulvin can be variable; absorption is enhanced if the drug is taken with a fatty meal. It is important to continue treatment until the infected skin, hair, or nail is completely replaced by normal tissue.
Griseofulvin use is not associated with a high incidence of serious adverse effects. A relatively common (up to 15%) adverse effect is headache, which tends to disappear as therapy continues. Other nervous system effects include lethargy, vertigo, and blurred vision; these adverse effects can be exacerbated by the consumption of alcohol. Occasionally, hepatotoxicity or albuminuria without renal insufficiency can be observed. Hematologic adverse effects—including leukopenia, neutropenia, and monocytosis—can occur during the first month of therapy. Serum sickness, angioedema, exfoliative dermatitis, and toxic epidermal necrolysis are extremely rare but potentially life-threatening adverse effects. Chronic use can sometimes result in increased fecal protoporphyrin levels. Concurrent administration with barbiturates decreases the gastrointestinal absorption of griseofulvin. Because griseofulvin induces hepatic cytochrome P450 enzymes, it can increase the metabolism of warfarin and potentially reduce the efficacy of low-estrogen oral contraceptive medications. Griseofulvin should be avoided during pregnancy, since fetal abnormalities have been reported.
Inhibitors of the Ergosterol Synthesis Pathway
Inhibitors of Squalene Epoxidase: Allylamines and Benzylamines
In the ergosterol synthesis pathway (Fig. 36-1), squalene is converted to lanosterol by the action of squalene epoxidase. Inhibitors of squalene epoxidase prevent the formation of lanosterol, which is a precursor for ergosterol. These drugs also promote accumulation of the toxic metabolite squalene in the fungal cell, making them fungicidal under most circumstances. The antifungal agents that inhibit squalene epoxidase can be divided into allylamines and benzylamines based on their chemical structures: terbinafine and naftifine are allylamines, whereas butenafine is a benzylamine.
Terbinafine is available in both oral and topical formulations. When taken orally, the drug is 99% protein-bound in the plasma, and it undergoes first-pass metabolism in the liver. Because of this first-pass metabolism, the oral bioavailability of terbinafine is 40%. The drug’s elimination half-life is extremely long, approximately 300 hours, because terbinafine accumulates extensively in the skin, nails, and fat. The oral form of terbinafine is used in the treatment of onychomycosis, tinea corporis, tinea cruris, tinea pedis, and tinea capitis. Terbinafine is not recommended in patients with renal or hepatic failure or in pregnant women. Very rarely, the oral form of terbinafine can lead to hepatotoxicity, Stevens-Johnson syndrome, neutropenia, and exacerbation of psoriasis or subacute cutaneous lupus erythematosus. Liver function enzymes should be monitored during the treatment course. Plasma levels of terbinafine are increased by co-administration with cimetidine (a cytochrome P450 inhibitor) and decreased by co-administration with rifampin (a cytochrome P450 inducer). Topical terbinafine is available in cream or spray form and is indicated for tinea pedis, tinea cruris, and tinea corporis.
Similar to terbinafine, naftifine is a squalene epoxidase inhibitor that has broad-spectrum antifungal activity. Naftifine is only available topically as a cream or gel; it is effective in tinea corporis, tinea cruris, and tinea pedis.
Butenafine, a benzylamine, is a topical antifungal agent with a mechanism of action and spectrum of antifungal activity similar to that of the allylamines. Topical allylamines and benzylamines are more effective than topical azole agents against common dermatophytes, especially those causing tinea pedis. However, topical terbinafine and butenafine are less effective than topical azoles against Candida skin infections (see below).
Inhibitors of 14α-Sterol Demethylase: Imidazoles and Triazoles
Another important molecular target in the ergosterol synthesis pathway is 14α-sterol demethylase, a microsomal cytochrome P450 enzyme that catalyzes the first step in the conversion of lanosterol to ergosterol. The azoles are antifungal agents that inhibit fungal 14α-sterol demethylase. The resulting decrease in ergosterol synthesis and accumulation of 14α-methyl sterols disrupt the tightly packed acyl chains of the phospholipids in fungal membranes. Destabilization of the fungal membrane leads to dysfunction of membrane-associated enzymes, including those in the electron transport chain, and may ultimately lead to cell death. Azoles are not completely selective for the fungal P450 enzyme, however, and they can also inhibit hepatic P450 enzymes. While the extent of hepatic P450 enzyme inhibition varies among the azoles, drug–drug interactions are an important consideration whenever an azole antifungal agent is prescribed. For example, cyclosporine is an immunosuppressive drug used to prevent graft rejection in recipients of allogeneic kidney, liver, and heart transplants. It is metabolized by hepatic P450 enzymes and excreted in the bile. To minimize the risk of cyclosporine-associated nephrotoxicity and hepatotoxicity, patients concomitantly receiving an azole antifungal agent should be treated with lower doses of cyclosporine.
As a group, the azoles have a wide range of antifungal activity and are clinically useful against B. dermatitidis, Cryptococcus neoformans, H. capsulatum, Coccidioides species, P. brasiliensis, dermatophytes, and most Candida species. Azoles have intermediate clinical activity against Fusarium, Sporothrix schenckii, Scedosporium apiospermum, and Aspergillus species. Pathogens mediating zygomycosis (invasive fungal infections caused by Zygomycetes species) and Candida krusei are resistant to azoles. The azoles are generally fungistatic rather than fungicidal against susceptible organisms.
The azole antifungal agents can be categorized into two broad classes, imidazoles and triazoles, which share the same mechanism of action and similar antifungal spectrum. Because systemically administered triazoles tend to have less effect than systemically administered imidazoles on human sterol synthesis, recent drug development has focused primarily on triazoles.
The imidazole antifungal class includes ketoconazole, clotrimazole, luliconazole, miconazole, econazole, butoconazole, oxiconazole, sertaconazole, sulconazole, and tioconazole. Ketoconazole was introduced in 1977 as the prototypic drug in this class. Ketoconazole is available in both oral and topical formulations. Its broad spectrum of action includes C. immitis, C. neoformans, Candida species, H. capsulatum, B. dermatitidis, and a variety of dermatophytes. The pharmacokinetic and adverse effect profiles of ketoconazole limit its clinical utility. (In fact, oral ketoconazole has been replaced by itraconazole for the treatment of many mycoses; see discussion below.) Gastrointestinal absorption of oral ketoconazole depends on conversion of the drug to a salt in the acidic environment of the stomach. Thus, ketoconazole cannot be used if the patient has achlorhydria or is receiving bicarbonate, antacids, H2-blockers, or proton pump inhibitors. Ketoconazole has little penetration into the CSF and urine, which limits its efficacy in CNS and urinary tract infections. In approximately 20% of patients, the drug causes nausea, vomiting, or anorexia; hepatic dysfunction occurs in 1–2% of patients.
Ketoconazole potently inhibits hepatic P450 enzymes and therefore affects the metabolism of many other drugs. At therapeutic doses, it also inhibits the P450 enzymes 17, 20-lyase and side-chain cleavage enzyme in the adrenal gland and gonads, thereby decreasing steroid hormone synthesis. Persistent adrenal insufficiency has been reported in association with ketoconazole therapy; at high doses of the drug, significant inhibition of androgen synthesis can result in gynecomastia and impotence. This dose-dependent adverse effect has been exploited therapeutically by some clinicians, who prescribe ketoconazole to inhibit androgen production in patients with advanced prostate cancer and to inhibit corticosteroid synthesis in patients with advanced adrenal cancer.
Topical ketoconazole is widely used to treat common dermatophyte infections and seborrheic dermatitis. Topical ketoconazole has been shown to have anti-inflammatory activity comparable to that of hydrocortisone. The cream formulation contains sulfites and therefore should be avoided in patients with sulfite hypersensitivity.
Clotrimazole, luliconazole, miconazole, econazole, butoconazole, oxiconazole, sertaconazole, sulconazole, and tioconazole are topical imidazole antifungal agents used to treat superficial fungal infections of the stratum corneum, squamous mucosa, and cornea. All of these agents are comparable to one another in efficacy. In addition to inhibiting 14α-sterol demethylase, miconazole affects fatty acid synthesis and inhibits fungal oxidative and peroxidase enzymes. The currently available topical azoles are generally not effective against hair or nail fungal infections, and topical azoles should not be used to treat subcutaneous or systemic mycoses. Topical azole agents are available for cutaneous and vaginal application, and selection of a particular agent should be based on cost and availability. Rare adverse effects of these agents include itching, burning, and sensitization.
The triazole class of antifungal agents includes itraconazole, fluconazole, voriconazole, terconazole, posaconazole, and isavuconazole; one additional member of this class, ravuconazole, is currently in clinical trials. Itraconazole is available in both oral and intravenous formulations. Given its broad spectrum of activity, itraconazole has largely replaced oral ketoconazole for the treatment of many mycoses. The absorption of oral itraconazole is maximized in an acidic gastric environment. However, because the oral bioavailability of itraconazole is unpredictable, intravenous administration is sometimes preferred. Itraconazole is oxidized in the liver to the active metabolite hydroxyitraconazole, which is more than 90% bound to plasma protein. Hydroxyitraconazole inhibits fungal 14α-sterol demethylase. Compared to ketoconazole and fluconazole, itraconazole shows increased activity in aspergillosis, blastomycosis, and histoplasmosis. Itraconazole is not efficiently transported into the CSF, urine, or saliva; however, itraconazole can be used in certain meningeal fungal infections due to the high drug levels achieved in the meninges. Hepatotoxicity is the major adverse effect associated with itraconazole therapy. Other adverse effects include nausea, vomiting, abdominal pain, diarrhea, hypokalemia, pedal edema, and hair loss.
Posaconazole is an oral triazole developed from itraconazole. Posaconazole is fungistatic against most species of Candida, Cryptococcus, Trichosporon, and some species of Fusarium. It is also active against multidrug-resistant Candida, Aspergillus, and Zygomycetes isolates. Posaconazole is used primarily in the prophylaxis and treatment of invasive fungal infections. The most common adverse effects are nausea, vomiting, diarrhea, rash, hypokalemia, thrombocytopenia, and abnormal liver function tests. Interactions may occur with co-administration of cimetidine, rifabutin, and phenytoin, and these drugs should be avoided in patients on posaconazole. Moreover, cyclosporine, tacrolimus, and midazolam dosages should be reduced in patients taking posaconazole.
Fluconazole is currently the most widely used antifungal drug. Fluconazole is a hydrophilic triazole that is available in both oral and intravenous formulations. The bioavailability of oral fluconazole is nearly 100%, and, unlike ketoconazole and itraconazole, its absorption is not influenced by gastric pH. Once absorbed, fluconazole diffuses freely into CSF, sputum, urine, and saliva. Fluconazole is excreted primarily by the kidneys.
Its relatively low adverse effect profile (see below) and excellent CSF penetration make fluconazole the drug of choice for systemic candidiasis and cryptococcal meningitis. Due to the morbidity associated with intrathecal amphotericin B administration, fluconazole is also the drug of choice for coccidioidal meningitis. While fluconazole is active against blastomycosis, histoplasmosis, and sporotrichosis, it is less effective than itraconazole against these infections. Fluconazole is not effective against aspergillosis.
Fungal resistance to fluconazole develops readily, and Candida species are the most notable pathogens to develop resistance (e.g., C. glabrata). Mechanisms of drug resistance include mutation of fungal P450 enzymes and overexpression of multidrug efflux transporter proteins.
Numerous drug interactions have been noted with fluconazole. As examples, fluconazole can increase the levels of amitriptyline, cyclosporine, phenytoin, and warfarin, and the levels and effects of fluconazole can be decreased by carbamazepine, isoniazid, and phenobarbital. Adverse effects of fluconazole include nausea, vomiting, abdominal pain, and diarrhea in about 10% of patients, as well as reversible alopecia with prolonged oral therapy. Rare cases of Stevens-Johnson syndrome and hepatic failure have been reported.
Ravuconazole, a fluconazole derivative that is currently in clinical trials, demonstrates an expanded spectrum of antifungal activity in vitro against multiple fungal species, including Aspergillus and the relatively resistant Candida species C. krusei and C. glabrata.
Voriconazole is a triazole antifungal agent that is available in both oral and parenteral forms. It is the drug of choice in the treatment of invasive aspergillosis and other molds such as Fusarium and Scedosporium. Voriconazole is fungicidal against essentially all species of Aspergillus, and its spectrum of activity also includes Candida species (including C. krusei and C. glabrata) and a number of newly emerging fungi. It is ineffective in the treatment of zygomycosis. Compared to amphotericin, voriconazole is associated with significantly better outcomes, particularly in difficult-to-treat cases such as allogeneic bone marrow transplant recipients, patients with CNS infections, and patients with disseminated infections. Voriconazole inhibits hepatic P450 enzymes to a significant extent, and lower doses of cyclosporine or tacrolimus are used when these drugs are combined with voriconazole. Due to accelerated voriconazole metabolism, co-administration with ritonavir, rifampin, and rifabutin is contraindicated. The intravenous formulation of voriconazole should not be used in patients with renal failure because the cyclodextrin excipient accumulates and can cause CNS toxicity. Hepatic toxicity is common but can usually be managed by decreasing the dose. Unusual visual symptoms (photophobia and colored lights) can occur at peak plasma concentrations of voriconazole; typically, these symptoms last for 30–60 minutes.
Isavuconazole is a triazole antifungal agent approved by the US Food and Drug Administration (FDA) in March 2015 for the treatment of invasive aspergillosis and invasive mucormycosis. It is available in oral and parenteral formulations with equivalent bioavailability.
Terconazole is a topical triazole used to treat vaginal candidiasis. Its mechanism of action and spectrum of antifungal activity are similar to those of the other topical azoles. Terconazole is available as a vaginal suppository that is inserted at bedtime.
Inhibitors of Fungal Membrane Stability: Polyenes
Amphotericin B, nystatin, and natamycin are polyene macrolide antifungal agents. These drugs act by binding to ergosterol and disrupting fungal membrane stability. The three agents are natural products derived from Streptomyces species. For decades, amphotericin B provided the only effective treatment for systemic mycoses, including candidiasis, cryptococcal meningitis, invasive aspergillosis, zygomycosis, coccidioidomycosis, blastomycosis, and histoplasmosis. Both its therapeutic effect and its toxicity are related to its affinity for plasma membrane sterols. Fortunately, the affinity of amphotericin B for ergosterol is 500 times greater than its affinity for cholesterol. The binding of amphotericin B to ergosterol produces channels or pores that alter fungal membrane permeability and allow for leakage of essential cellular contents, leading ultimately to cell death. The concentration of membrane-associated ergosterol in a given fungal species determines whether amphotericin B is fungicidal or fungistatic for that species. Resistance to amphotericin B, although less frequent than with other antifungal agents, is attributable to a decrease in the ergosterol content of the fungal membrane. In addition to its pore-forming activity, amphotericin B appears to destabilize fungal membranes by generating toxic free radicals upon oxidation of the drug.
Because amphotericin B is highly insoluble, it is supplied as a buffered deoxycholate colloidal suspension. This suspension is poorly absorbed from the gastrointestinal tract and must be administered intravenously. Once in the bloodstream, more than 90% of the drug binds rapidly to tissue sites, while the remainder binds to plasma proteins. Penetration of amphotericin B into the CSF is extremely low (2–4%). Hence, intrathecal therapy may be necessary for treatment of serious meningeal disease. The drug also diffuses poorly into vitreous humor and amniotic fluid.
The toxicity of amphotericin B limits its clinical use. Adverse effects are divided into three groups: immediate systemic reactions, renal effects, and hematologic effects. Systemic reactions can include cytokine storm, in which amphotericin B elicits release of tumor necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1) from cells of the host immune system. In turn, TNF-α and IL-1 cause fever, chills, rigors, and hypotension within the first several hours after drug administration. These responses can usually be minimized by decreasing the rate of drug administration or by pretreatment with antipyretic agents (e.g., acetaminophen, nonsteroidal anti-inflammatory drugs [NSAIDs], or hydrocortisone).
Renal toxicity of amphotericin B is a serious adverse effect. The mechanism of renal toxicity is unknown but may be related to amphotericin-mediated vasoconstriction of afferent arterioles, leading to renal ischemia. Renal toxicity is often the limiting factor in determining the extent of the therapeutic response to amphotericin B. It may be necessary to discontinue therapy temporarily if the blood urea nitrogen (BUN) exceeds 50 mg/dL or the serum creatinine exceeds 3 mg/dL. (BUN and creatinine are surrogate measures of renal function.) Renal tubular acidosis, cylindruria (the presence of renal cell casts in the urine), and hypokalemia can occur to such a degree that electrolyte replacement is required. In the introductory case, treatment with amphotericin B was discontinued as soon as Mr. F’s acute symptoms resolved in order to prevent renal toxicity.
Hematologic toxicity of amphotericin B is also common; anemia is probably secondary to decreased production of erythropoietin. The renal and hematologic toxicities of amphotericin B are cumulative and dose-related. Therapeutic measures that can minimize these toxicities include avoidance of other nephrotoxic drugs, such as aminoglycosides and cyclosporine, and maintenance of euvolemia to provide adequate renal perfusion.
Attempts to reduce nephrotoxicity have also led to the development of lipid formulations of amphotericin B. The strategy is to package amphotericin B in liposomes or other lipid carriers, with the goal of preventing high drug exposure to the proximal tubule of the nephron. Amphotec®, Abelcet®, and AmBisome® are all FDA-approved lipid-containing preparations of amphotericin B. They are equal in efficacy to each other and to native amphotericin deoxycholate. These formulations are less toxic than the native compound but more expensive.
Nystatin, a structural relative of amphotericin B, is a polyene antifungal agent that also acts by binding ergosterol and causing pore formation in fungal cell membranes. The drug is used topically to treat candidiasis involving the skin, vaginal mucosa, and oral mucosa. Nystatin is not absorbed systemically from the skin, vagina, or gastrointestinal tract.
Natamycin, another polyene antifungal agent that binds ergosterol in the fungal cell membrane, is primarily used to treat Aspergillus or Fusarium corneal infections. It is also indicated in the treatment of blepharitis and conjunctivitis. Natamycin accumulates in the corneal stroma but not in intraocular fluid and is effective at low concentrations.
Inhibitors of Fungal Wall Synthesis: Echinocandins
The key components of the fungal cell wall are chitin, β-(1,3)-D-glucan, β-(1,6)-D-glucan, and cell wall glycoproteins. Because human cells do not have a cell wall, fungal cell wall components represent unique targets for antifungal therapy, and antifungal agents directed at these targets are likely to be relatively nontoxic. Echinocandins are a class of antifungal agents that target fungal cell wall synthesis by noncompetitively inhibiting the synthesis of β-(1,3)-D-glucans. Disruption of cell wall integrity results in osmotic stress, lysis of the fungal cell, and ultimately fungal cell death. The three antifungal agents in the echinocandin class are caspofungin, micafungin, and anidulafungin; all are semisynthetic lipopeptides derived from natural products. The echinocandins have in vitro and in vivo antifungal activity against Candida and Aspergillus species. All three echinocandins are fungicidal against Candida species, including C. krusei and C. glabrata, and fungistatic against Aspergillus species. They have poor activity against zygomycetes. All three agents are available only in parenteral form because they are insufficiently bioavailable for oral use.
Caspofungin was the first echinocandin to be approved. The drug is used as primary therapy for esophageal candidiasis and candidemia, as salvage therapy for Aspergillus infections, and as empiric therapy for febrile neutropenia. Like the other echinocandins, caspofungin is highly protein-bound (97%) in the plasma; it is metabolized in the liver via peptide bond hydrolysis and N-acetylation; and it penetrates poorly into the CSF (although animal data indicate that the echinocandins do have some activity in the CNS). Caspofungin does not require dose adjustment for renal insufficiency, but dose adjustment is required for patients with moderate hepatic dysfunction. Because co-administration with cyclosporine significantly increases plasma concentrations of caspofungin and elevates liver function enzymes, this drug combination is generally not recommended unless the expected benefits outweigh the risks. Similarly, co-administration with tacrolimus significantly increases plasma concentrations of tacrolimus. To achieve therapeutic plasma concentrations, caspofungin dosing may need to be increased in patients receiving nelfinavir, efavirenz, phenytoin, rifampin, carbamazepine, or dexamethasone.
Micafungin is approved for the treatment of esophageal candidiasis and as antifungal prophylaxis for recipients of hematopoietic stem cell transplants. It is also effective against candidemia and pulmonary aspergillosis. Anidulafungin is approved for the treatment of esophageal candidiasis and candidemia. Several small case series have reported the use of echinocandins in combination with amphotericin B, flucytosine, itraconazole, or voriconazole in patients with refractory fungal infections. Aminocandin is an investigational echinocandin with a spectrum of activity similar to that of the other echinocandins. It has a half-life threefold to fourfold greater than that of the other echinocandins, thus permitting less frequent administration.
Echinocandins are generally well tolerated; their adverse-effect profile is comparable to that of fluconazole. Because echinocandins contain a peptide backbone, symptoms related to histamine release can be observed (see “Suggested Reading”). Other adverse effects include headache, fever (more common with caspofungin), rash, abnormal liver function tests, and, rarely, hemolysis.
Chelator of Polyvalent Cations: Ciclopirox
Ciclopirox is a synthetic hydroxypyridone antifungal agent whose mechanism of action is poorly understood. In experimental studies, the drug chelates the polyvalent cations Fe+3 and Al+3. Chelation of these ions inhibits numerous metal-dependent enzymes responsible for electron transport, DNA and RNA synthesis, energy production, catalase activity, and peroxide degradation within fungal cells. Ciclopirox also demonstrates mild anti-inflammatory properties that may be due to inhibition of 5-lipoxygenase and cyclooxygenase.
Ciclopirox is approved for the treatment of seborrheic dermatitis, tinea versicolor, tinea corporis, tinea pedis, cutaneous candidiasis, and onychomycosis. It is administered as a topical cream, gel, lotion, shampoo, or lacquer. No serious adverse effects have been associated with topical use of ciclopirox. Common adverse effects include a burning sensation with application, pruritus, and contact dermatitis.
 CONCLUSION AND FUTURE DIRECTIONS
CONCLUSION AND FUTURE DIRECTIONS
The development of antifungal agents has progressed significantly since the introduction of amphotericin B. As the population of immunocompromised patients increases, opportunistic fungal infections that are resistant to conventional antifungal therapy pose new challenges to researchers and clinicians. For example, new antifungal therapy is greatly needed in the treatment of zygomycosis. Effective topical antifungal agents are eagerly sought for the treatment of nail and hair dermatophytosis, because oral therapies for these superficial fungal infections carry risks such as hepatotoxicity. The development of protease inhibitors and phospholipase inhibitors represent new frontiers in the treatment of Candida and Cryptococcal species, respectively. As novel and unique molecular targets are identified in fungal pathogens, newer antifungal agents will be developed with the goal of minimizing mechanism-based (“on-target”) toxicity while expanding antifungal spectrum of action.
Acknowledgment
We thank Lorne W. Murray, Robert H. Rubin, Charles R. Taylor, and Ali Alikhan for their valuable contributions to this chapter in the First, Second, and Third Editions of Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy.
Gauwerky K, Borelli C, Korting HC. Targeting virulence: a new paradigm for antifungals. Drug Discov Today 2009;14:214–222. (Discusses virulence factors of fungi and their inhibitors, with an emphasis on new options for antifungal development, including inhibitors of the secreted aspartyl protease of C. albicans.)
Miceli MH, Kauffman CA. Isavuconazole: a new broad-spectrum triazole antifungal agent. Clin Infect Dis 2015;61:1558–1565. (Discusses the use of this recently approved agent.)
Naeger-Murphy N, Pile JC. Clinical indications for newer antifungal agents. J Hosp Med 2008;4:102–111. (Discusses the use of echinocandins and triazoles in several common and/or important clinical situations.)
Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH. An insight into the antifungal pipeline: selected new molecules and beyond. Nat Rev Drug Discov 2010;9:719–727. (Discusses development of polyenes, azoles, echinocandins, and investigational antifungal drugs, including vaccines and antibody-based immunotherapy.)
Patterson TF. Advances and challenges in management of invasive mycosis. Lancet 2005;366:1013–1025. (Focused discussion of fungal pathogens that occur in immunocompromised hosts and management strategies for these opportunistic pathogens.)
Ruiz-Herrera J, Elorza MV, Valentin E, Sentandreu R. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res 2006;6:14–29. (Comprehensive review of the fungal cell wall.)
Scher RK, Nakamura N, Tavakkol A. Luliconazole: a review of a new antifungal agent for the topical treatment of onychomycosis. Mycoses 2014;57:389–393. (Discusses the development and therapeutic potential of luliconazole, the latest FDA-approved antifungal agent.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


